

Abstract
Chronic use of inhaled beta-agonists by asthmatics is associated with a loss of bronchoprotective effect and deterioration of asthma control. Beta-agonist-promoted desensitization of airway smooth muscle beta-2-adrenergic receptors, mediated by G protein-coupled receptor kinases and arrestins, is presumed to underlie these effects, but such a mechanism has never been demonstrated. Using in vitro, ex vivo, and in vivo murine models, we demonstrate that beta-arrestin-2 gene ablation augments beta-agonist-mediated airway smooth muscle relaxation, while augmenting beta-agonist-stimulated cyclic adenosine monophosphate production. In cultures of human airway smooth muscle, small interfering RNA-mediated knockdown of arrestins also augments beta-agonist-stimulated cyclic adenosine monophosphate production. Interestingly, signaling and function mediated by m2/m3 muscarinic acetylcholine receptors or prostaglandin E2 receptors were not affected by either beta-arrestin-2 knockout or arrestin knockdown. Thus, arrestins are selective regulators of beta-2-adrenergic receptor signaling and function in airway smooth muscle. These results and our previous findings, which demonstrate a role for arrestins in the development of allergic inflammation in the lung, identify arrestins as potentially important therapeutic targets for obstructive airway diseases.—Deshpande, D. A., Theriot, B. S., Penn, R. B., Walker, J. K. L. β-Arrestins specifically constrain β2-adrenergic receptor signaling and function in airway smooth muscle.
Beta-agonists, via activation of the beta-2-adrenergic receptor (β2AR), antagonize airway smooth muscle (ASM) contraction and are important therapeutic agents in the management of obstructive airway diseases such as asthma (1). However, many asthmatics are not sufficiently managed by beta-agonist therapy, and continuous use of inhaled beta-agonist has been shown to result in a loss of bronchoprotective effect (2–4) and deterioration of asthma control (5).
One of the potential mechanisms that limit the therapeutic efficacy of beta-agonists is desensitization of ASM β2ARs. β2AR desensitization has been shown to occur in numerous cell types following either acute or chronic exposure to beta-agonists. Studies examining recombinant β2ARs expressed in cell lines, as well as a handful of studies examining endogenous β2ARs in primary cell types, have demonstrated important roles for G protein-coupled receptor (GPCR) kinases (GRKs) and arrestins in mediating β2AR desensitization to beta-agonists (as reviewed in refs. 1, 6, 7). GRKs phosphorylate the β2AR when it is occupied by beta-agonists, and arrestins subsequently bind the phosphorylated receptor, effectively blocking receptor interaction with, and activation of, the heterotrimeric G protein Gs. This quenching or “arresting” of β2AR signaling occurs rapidly and also initiates a process of receptor internalization. With chronic exposure to beta-agonists, arrestins help to direct internalized β2ARs to lysosomes, where they are degraded, such that new β2AR synthesis is required to ultimately recover from chronic desensitization (8).
The extent to which GRK- and arrestin-dependent β2AR desensitization occurs in ASM and affects the functional response to beta-agonists is unclear. We have previously determined that overexpression of β-arrestin (βarr) -1 or βarr-2 (also termed arrestin2 and arrestin3, respectively) in human ASM cultures causes β2AR desensitization and attenuation of beta-agonist-stimulated signaling (9). In a murine model of ovalbumin-induced allergic inflammation, we have also observed that ablation of the βarr-2 gene causes a significant inhibition of inflammatory cell influx into allergen-challenged murine airway and prevents airway hyperresponsiveness (10). Whether manipulation of arrestin expression within physiological ranges affects β2AR ASM signaling or function in either cell-based or in vivo assays has yet to be determined.
In the current study, we examined the selectivity and efficacy of βarr-1/2 knockdown and specific βarr-2 knockout in regulating GPCR function in in vitro, ex vivo, and in vivo analyses of ASM. Results demonstrate that arrestins are selective and efficacious modulators of beta-agonist-mediated signaling and function in ASM, thus identifying them as a potentially important therapeutic target for obstructive airway diseases.
MATERIALS AND METHODS
Generation of βarr-2-knockout (βarr-2-KO) mice
βarr-2-KO mice were generated as described previously (11) and backcrossed for six generations onto the C57BL/6 background. βarr-2-KO and wild-type (WT) sibling pairs aged 10–16 wk were used for in vivo measurement of lung function, ex vivo measurement of tracheal contraction, and isolation of ASM cells under protocols approved by the Animal Care and Use Committees at Duke University and Wake Forest University Health Sciences. βarr-2 gene ablation was confirmed by triplex polymerase chain reaction (PCR).
Immunoblotting
Mouse lung was pulverized in liquid nitrogen and solubilized in radioimmunoprecipitation assay (RIPA) buffer. Tissue lysates were buffered with Laemmli solution and sonicated, and equal amounts of total protein (as measured by a DC protein assay read at 750 nm; Bio-Rad, Hercules, CA, USA) were loaded and separated on 10% Tris-glycine polyacryl-amide gels (Invitrogen, Carlsbad, CA, USA). Proteins were transferred to nitrocellulose membranes for immunoblotting. β-Arrestins were detected with the polyclonal Ab A2CT (generous gift from Robert J. Lefkowitz, Duke University, Durham, NC, USA). A2CT preferentially recognizes βarr-2 over βarr-1. Chemiluminescent detection was performed with horseradish peroxidase-coupled secondary Ab (Amersham Biosciences, Pittsburgh, PA, USA) and SuperSignal West Femto reagent (Pierce, Rockford, IL, USA). Chemiluminescence was quantified by a charge-coupled device camera (Syngene, Frederick, MD, USA ); representative images are shown as inverted grayscale. For immunoblots of lysates from cultured human ASM cells, blots were probed with Arr178 antibody (generous gift from Jeffrey L. Benovic, Thomas Jefferson University, Philadelphia, PA, USA) and an IRDye 800 secondary antibody (Rockland Immunochemicals, Gilbertsville, PA, USA) conjugated with an infrared fluorophore. Bands were visualized and signals (infrared emission) quantified directly using the Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE, USA), as described previously (12).
Mouse lung and ASM membrane preparation
To prepare ASM membranes, tracheae and bronchi were dissected free of loose connective tissue and lung parenchyma, and the epithelium was denuded. The tissue was placed in ice-cold buffer (25 mM Tris-HCl, pH 7.4, and 5 mM EDTA, pH 8.0) containing protease inhibitors and homogenized using a Polytron tissue homogenizer (Brinkmann Instruments, Inc., Westbury, NY) at setting 8 for 3 × 10 s periods. The resulting homogenate was spun at 250 g for 10 min at 4°C, the low-speed pellet was discarded, and the supernatant was centrifuged at 18,000 rpm for 20 min at 4°C. The pellet was resuspended in binding buffer (75 mM Tris-HCl, 2 mM EDTA, 12.5 mM MgCl2) at a concentration of 0.5 to 1.0 mg protein/ml (protein concentration was determined by the method of Bradford, using bovine serum albumin as the standard). Membranes from peripheral lung were similarly prepared after dissecting away major airways and blood vessels.
β-AR radioligand binding
β-AR radioligand binding studies were performed in fresh lung and ASM membranes by use of the nonselective β-AR antagonist [125I]-labeled cyanopindolol (125I-CYP), as described previously (13). Maximal binding (Bmax) was measured by use of a saturating amount of 125I-CYP on 20 μg of membrane protein, incubating 40 min at 37°C. Inclusion of 25 μM propranolol defined nonspecific binding. Assays were stopped by dilution with 5 ml of ice-cold binding buffer and rapid filtration over glass fiber filters, followed by four 5-ml washes, before counting in a gamma counter. Total binding represented <10% of 125I-CYP counts added. Specific binding, which was calculated as the difference between triplicate averages of total and nonspecific counts bound to the filters, comprised on average 64% and 96% of total binding for ASM and lung, respectively.
Lung function measurements
Contractile and relaxation responsiveness of airways to methacholine and albuterol were determined as described previously (14). Briefly, mice were anesthetized with an intraperitoneal injection of pentobarbital sodium (60 mg/kg) diluted 50% with saline, then surgically prepared with a tracheal cannula and a jugular vein catheter. Mice were paralyzed with pancuronium bromide (0.25 mg/kg) and ventilated with 100% oxygen at a constant volume of 6–8 ml/kg and a frequency of 165 breaths/min. These ventilator settings resulted in an average resting peak tracheal pressure of 6.5 ± 0.1 cm H2O and have been previously shown to provide normal arterial blood gases. Airway pressure was measured at a side port of the tracheal cannula connected to a Validyne differential pressure transducer (Validyne Engineering, Northridge, CA, USA). The time-integrated change in peak tracheal pressure, or airway pressure time index (APTI) (15, 16), was calculated for a 60 s period from the point at which tracheal pressure began to increase in response to methacholine (MCh).
Anesthetized and paralyzed mice were intravenously administered 50 μg/kg MCh as a contractile priming dose. This was followed by two consecutive intravenous administrations of 100 μg/kg MCh alone and administration of a combined preparation of either 100 μg/kg MCh and 30 μg/kg albuterol or 100 μg/kg MCh and 5 μg/kg prostaglandin E2 (PGE2). Each drug administration was separated by a 5-min interval. The response to MCh in the presence of albuterol or PGE2 was reduced compared to the contractile response to MCh alone, and the percent change was calculated and used to determine the net decrease in the MCh-induced airway response.
Ex vivo ASM tension development
Murine tracheae excised after euthanasia by CO2 inhalation were cleaned of surrounding connective tissue and mounted into a multiwire myograph chamber in Krebs-Henseleit solution (pH 7.40 –7.45) maintained at 37°C with 5% CO2 and 95% O2, with frequent changing of the solution. The chambers were mounted onto the Myo-Interface (model 610 M) and connected to the transducer and PowerLab (ADInstruments, Colorado Springs, CO, USA) for data transferring and recording. Chart5 software for Windows (ADInstruments) was used to collect and analyze the data. A basal tension of 0.5 g was set, and the tracheal rings were stimulated with 10 μM MCh for 5 min. The rings were washed, and preload was reset to 0.5 g for 30 min. After stabilization, the tracheal rings were stimulated with 10 μM MCh and allowed to contract for 10 min. Stimulation of the rings with 10 μM MCh produces ~80% maximum tension in the rings, as determined previously (15). Subsequently, the precontracted rings were stimulated with increasing concentrations of isoproterenol (ISO) or PGE2 for 5 min and tension was recorded. In a select set of experiments, the precontracted rings were stimulated with 10 μM forskolin (FSK). In another set of experiments, the rings were contracted with 60 mM KCl for 5 min and subsequently stimulated with 1 μM MCh for 5 min. The net contractile responses were determined by subtracting the basal tension from that of the peak tension on agonist stimulation, and the change in the tension was normalized to the wet weight of the tracheal ring segment. Relaxation responses were calculated as percentage change in MCh-induced tension on stimulation with different concentrations of relaxant agents.
Generation of murine and human ASM cultures
Human ASM cell cultures were generated from tissue obtained at autopsy, as described previously (13), and cultures in passages 5–7 were examined. For generation of murine ASM cultures, tracheae harvested from 5 mice were cleaned of surrounding tissues and pooled to isolate ASM cells using the above procedure for human ASM cultures with minor modifications, as described recently (17). ASM cells were characterized by immunostaining using anti-α-actin and anti-smooth muscle myosin heavy-chain antibodies.
Small interfering (si)RNA-mediated knockdown of β-arrestins in ASM
siRNA oligos were designed based on human βarr-1/2 nucleotide sequence (sense, 5′ ACC UGC GCC UUC CGC UAU GTT 3′ antisense, 5′ CAU AGC GGA AGG CGC AGG UTT 3′), as described previously (18). Control (scrambled) or βarr-1/2 siRNA oligos were annealed at 37°C for 1 h, and 5 μg of the annealed oligo mixtures was used to transfect HASM cells by electroporation (Amaxa Biosystems, Gaithersburg, MD, USA), as described previously (13). Twenty-four hours after transfection, the cells were replated for subsequent assessment of βarr-1/2 protein expression by immunoblot analysis.
Measurement of intracellular cAMP accumulation, phosphoinositide hydrolysis
For analyses of regulation of intracellular cAMP accumulation, murine or human ASM cells plated in 24-well plates were grown to near confluence and serum starved for 24 h. The cells were stimulated with various concentrations of ISO, PGE2, or FSK in phosphate-buffered saline containing 300 μM ascorbic acid and 1 mM RO-20–1724 (phosphodiesterase inhibitor) at 37°C for 0–10 min. cAMP was extracted and quantified by RIA using 125I-cAMP and anti-cAMP antibody, as described previously (13). All conditions were assayed in triplicate wells and duplicate RIA tubes. Analysis of agonist-stimulated phosphoinositide (PI) hydrolysis in murine ASM cultures was performed as described previously (17). Cells were plated and serum starved as above and loaded with 2 μCi/ml myo-[3H]inositol for 18 h. After washing with phosphate-buffered saline, the cells were incubated with DMEM containing 5 mM LiCl for 10 min, then stimulated with vehicle or the muscarinic receptor agonist carbachol (CCh; 10 μM) for 30 min. Reactions were quenched with 20 mM formic acid, and inositol and phosphoinositide (PI) fractions were separated by anion exchange chromatography. PI production was calculated by dividing PI fraction by total (PI + inositol) inositol fraction, and the data were reported as fold basal (agonist-stimulated/vehicle). PI hydrolysis was not examined in human ASM cells due to the rapid loss of m3 mAChRs that occurs with culture (19).
Statistical analysis
Data analysis was performed using GraphPad Prism (San Diego, CA, USA), and data are expressed as means ± se. Group comparisons were performed using 2-way ANOVA or Student's t test where appropriate, with a value of P < 0.05 sufficient to reject the null hypothesis.
RESULTS
Arrestin knockout and knockdown in ASM cells enhances β-AR signaling
To assess arrestin-dependent regulation of β-AR signaling in ASM cells, ASM cultures derived from βarr-2-KO mice tracheae and human ASM cultures derived from multiple donors were generated. As shown in Fig. 1A, βarr-1 and βarr-2 are both expressed in WT mouse lung, whereas βarr-2-KO mice lack the faster migrating (~46.3 kDa predicted and 55 kDa observed, βarr-2) band in immunoblots. Relative to that observed for ASM cultures derived from WT mice, cAMP accumulation in βarr-2-KO ASM cultures in response to ISO stimulation was significantly greater at all concentrations ≥10 nM tested (Fig. 1B, C). FSK-stimulated cAMP accumulation was unaffected by βarr-2 knockout, suggesting differences in ISO-stimulated cAMP accumulation were a result of regulation of the β2AR locus. Conversely, PGE2 stimulated a low level of cAMP accumulation that was similar in both WT and βarr-2-KO ASM cultures (data not shown), and βarr-2 knockout also failed to affect phosphoinositide hydrolysis stimulated by mAChR activation (Fig. 1D).
Figure 1.

Effects of βarr-2 knockout on GPCR signaling in murine ASM cultures. A) Immunoblot analysis of βarr-1 and βarr-2 expression in mouse lung from WT and βarr-2-KO mice. Arrestin expression was detected using the A2CT Ab. βarr-2 knockout was confirmed by genotyping; faint reactivity below βarr-1 band in lysates from βarr-2-KO mice is nonspecific. Positive control lanes for βarr-1 and βarr-2 contained 2.5 and 0.25 ng of pure rat arrestins, respectively. B, C) ASM cultures derived from WT and βarr-2-KO mice were generated and cAMP accumulation stimulated by 10−9–10−6 ISO was assessed as described in Materials and Methods. D) CCh-stimulated phosphoinositide generation in ASM cultures derived from WT and βarr-2-KO mice. FSK-stimulated cAMP accumulation did not differ between groups (WT, 41±2 pmol/well; βarr-2-KO, 44±4 pmol/well). Values are means ± se from 3 (B) and 6 (C, D) experiments. *P < 0.05; 2-way ANOVA (B), t test for unpaired samples (C).
To determine whether β2AR signaling was similarly regulated by arrestins in human ASM, five distinct cultures derived from five different donor tracheae were transfected with siRNA oligonucleotides targeting βarr-1 and -2. Immunoblot analysis of culture lysates harvested 96 h after transfection reveals significant knockdown of arrestin expression relative to that in control cells transfected with control oligos (Fig. 2A). βarr-1 expression is readily detected in human ASM cultures and is significantly reduced (73±2%, n=5) by siRNA-mediated knockdown. βarr-2 expression is either absent or at the limit of detection in human ASM cultures; a faint, faster migrating band (relative to the βarr-1 band) is suggested in some blots but is not consistently recognized whether blots are probed with Arr178 Ab or more βarr-2-selective antibodies (not shown). cAMP accumulation stimulated by submaximal concentrations of beta-agonist was not significantly affected by arrestin knockdown (not shown) but that stimulated by 1 μM ISO was significantly increased (Fig. 2B). Conversely, PGE2-stimulated cAMP accumulation in human ASM cultures was slightly inhibited by arrestin knockdown, and FSK-stimulated cAMP accumulation was unaffected (Fig. 2B).
Figure 2.

Effects of βarr knockdown on GPCR signaling in human ASM cultures. A) Immunoblot analysis of arrestin expression in five distinct cultures transfected with scrambled (CON) or βarr-1/2 (ARR) siRNA oligonucleotides. B) Time-dependent cAMP accumulation in human ASM cultures stimulated with 1 μM ISO (left panel) or 1 μM PGE2 (right panel). cAMP accumulation stimulated by 50 nM ISO (CON siRNA, 7.7±1.5 pmol/well; ARR siRNA, 6.8±1.1 pmol/well) or FSK (CON siRNA, 46±5 pmol/well; ARR siRNA, 42±4 pmol/well) did not differ between groups. Values represent mean ± se values from 5 experiments. *P < 0.05; ISO-stimulated cAMP accumulation CON vs. ARR siRNA, 2-way ANOVA.
Arrestin knockout augments relaxant effect of beta-agonist on contracted ASM ex vivo and in vivo
To assess arrestin-dependent regulation of β2AR-mediated function in ASM, regulation of ASM contraction in excised tracheal rings was characterized. Tension generation stimulated by MCh was similar in rings from WT and βarr-2-KO mice, as was KCl-mediated contraction (Fig. 3A). However, ISO-mediated relaxation of MCh-induced contraction was significantly greater in rings from βarr-2-KO mice compared to that in WT rings (Fig. 3B). Conversely, PGE2- and FSK-mediated relaxation was not significantly different between groups.
Figure 3.

Effects of βarr-2 knockout on tracheal ASM contraction and relaxation ex vivo. Tracheal rings excised from WT and βarr-2-KO mice were mounted in a bath and contractile properties were assessed as described in Materials and Methods. A) Active tension development stimulated by 10 μM MCh (left panel) or 60 mM KCl (right panel) was compared between WT and βarr-2-KO groups. B) The relaxant effect of increasing concentrations of ISO (left) or PGE2 (right) on rings precontracted with 10 μM MCh was compared between groups. FSK-mediated relaxation did not differ among groups (78±7% and 80±9% reductions in WT and βarr-2-KO groups, respectively). Values are means ± se from 7 experiments. *P < 0.05; ISO effect on MCh-stimulated tension development WT and βarr-2-KO, 2-way ANOVA.
Regulation of β2AR function in vivo was characterized by assessing albuterol-mediated changes in MCh-stimulated airway responsiveness. MCh challenge increased airway responsiveness (APTI) to a similar degree in both WT and βarr-2-KO mice (52.1±5.5 vs. 48.4±3.0 cm H2O × s in WT and βarr-2-KO groups, respectively). Albuterol (30 μg/kg) treatment significantly inhibited MCh-induced increases in airway responsiveness for both WT and βarr-2-KO mice. However, this reduction was significantly greater in βarr-2-KO mice compared to that in WT controls (Fig. 4). Conversely, PGE2 treatment significantly reduced MCh-induced airway responsiveness, but βarr-2 knockout did not alter this effect.
Figure 4.
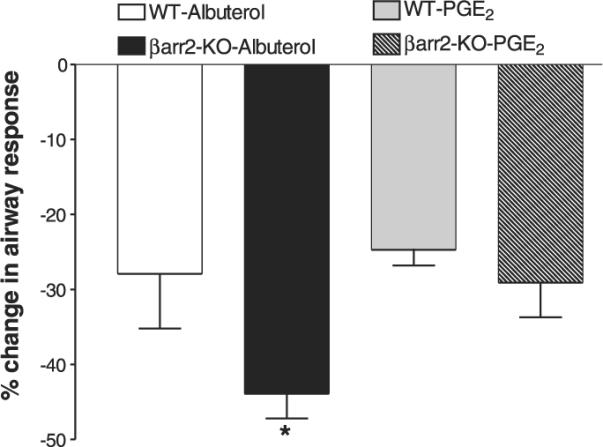
Effects of βarr-2 knockout on airway responsiveness in vivo. To measure the effect of albuterol or PGE2 on airway responsiveness, the magnitude of the APTI response to 100 μg/kg MCh was compared to that of combined 100 μg/kg MCh and 30 μg/kg albuterol or 100 μg/kg MCh and 5 μg/kg PGE2. The percentage change in airway response with albuterol administration is significantly greater in βarr-2-KO mice than in WT mice. Loss of βarr-2 does not alter bronchodilation in response to PGE2 relative to WT mice. Values are means ± se from 6 to 8 mice per group. *P < 0.05; unpaired t test.
Arrestin knockout does not alter lung or ASM β-AR density
We examined total β-AR density (Table 1). The maximum concentration of β-AR binding sites (Bmax) to tracheal smooth muscle membranes was not different between WT and βarr-2-KO mice (80.3±25.2 and 88.9±13.8 fmol/mg protein, respectively). Similarly, WT and βarr-2-KO mice expressed similar levels of β-AR in whole lung membranes (444±56 and 471±38 fmol/mg protein, respectively).
TABLE 1.
Effect of βarr-2 knockout on lung and ASM β-AR density
| WT ASM | βarr-2-KO ASM | WT lung | βarr-2-KO lung | |
|---|---|---|---|---|
| β-AR density | 80.3 ± 25.2 | 88.9 ± 13.8 | 444 ± 56 | 471 ± 38 |
Membranes from WT and βarr-2-KO mice were prepared as described in Materials and Methods. β-AR density was measured as radioligand binding (fmol/mg protein). Results are expressed as means ± sd of 2 (lung) or 3 (ASM) experiments.
DISCUSSION
This study is the first to demonstrate the efficacy and selectivity of arrestins in regulating GPCR function in ASM. In both in vivo and ex vivo murine models of ASM contractile regulation, βarr-2 is shown to constrain beta-agonist-mediated (β2AR-dependent) antagonism of ASM contraction, while not affecting antagonism mediated by PGE2 (EP2/4R-dependent) or the contrac-tile effect of methacholine (m3 mAChR-dependent). These in vivo and ex vivo data are consistent with in vitro cell data, which demonstrate increased β-agonist-mediated cAMP accumulation in cells lacking βarr-2. Taken together, these data demonstrate that the classic desensitization of β2AR signaling involving β-arrestins is functionally relevant with respect to bronchorelaxation in the whole animal.
The “arresting” function of β-arrestin was first demonstrated for the β2AR by Benovic et al. (20). The canonical β2AR signaling pathway involves agonist-induced Gαs activation, stimulation of adenylyl cyclase (AC), cAMP accumulation, and PKA activation. In the case of ASM, PKA phosphorylates several target proteins to promote ASM relaxation (reviewed in ref. 1). This pathway is desensitized by β-arrestin binding to the agonist-activated, GRK-phosphorylated receptor, which sterically interdicts further receptor-Gαs coupling (21). In the present study, we measured cAMP accumulation as an indicator of receptor-Gαs–AC signaling. Using primary murine ASM cultures, we demonstrate that isoproterenol-, but not PGE2- or forskolin-induced cAMP accumulation is increased in cells lacking βarr-2 relative to WT cells. Similar results were obtained in human ASM cells (which express predominantly βarr-1) when βarr-1 and βarr-2 levels were knocked down by transfection of siRNA. Because forskolin raises cAMP levels in ASM through receptor-independent activation of AC (22), the lack of effect of arrestin knockout or knockdown of forskolin-stimulated cAMP accumulation suggests β-arrestins regulate signaling at the receptor locus. The lack of effect on PGE2-mediated signaling and function further suggests that neither Gαs nor the EP2 receptor (EP2R) is regulated by β-arrestins in ASM.
These results, demonstrating the lack of effect of arrestin knockout/knockdown on PGE2 effects on ASM, are in agreement with our previous in vitro data that has demonstrated that the EP2R is the functionally dominant EP receptor expressed in ASM and that β-arrestins do not translocate to activated EP2R, arrest EP2R-Gαs signaling, or promote EP2R internalization (9). Thus, the EP2R is an example of one GPCR whose signaling and internalization are not regulated by β-arrestins, presumably a consequence of the EP2R being a poor substrate for GRKs.
Our finding that MCh-induced airway responsiveness and ASM contraction ex vivo were not affected by βarr-2 knockout identifies the m3 mAChR (the principal effector of ASM contraction mediated by mAChR agonists in the mouse; ref. 23) as an additional GPCR whose function is not regulated by arrestins. Consistent with these data, we also failed to observe differences in MCh-mediated phosphoinositide hydrolysis between cultures of ASM from βarr-2-KO and WT mice. Although these are the first data assessing arrestin-dependent regulation of endogenously expressed m3 mAChR, previous studies have demonstrated that although GRKs can phosphorylate purified, reconstituted m3 mAChR (24), agonist-induced internalization of overexpressed m3 mAChRs in human embryonic kidney cells is not affected by overexpressed or dominant-negative arrestins (25).
The aforementioned actions of β-arrestins occur within seconds to minutes of agonist-receptor binding and demonstrate a physiological mechanism by which β2AR-mediated relaxation of ASM is innately limited. Steric inhibition of β2AR-Gαs coupling by β-arrestins, relieved by β-arrestin knockout or knockdown, is known to mediate rapid β2AR desensitization. In numerous cell-based studies, a more delayed β-arrestin-mediated regulation of the β2AR is known to occur as a result of the dependence of receptor internalization on β-arrestin function (6). β-arrestin acts as an adaptor protein that links the β2AR to clathrin-mediated internalization. Once internalized, β2ARs are directed to acidic endosomes, where they are dephosphorylated and recycled as competent receptors to the cell membrane or directed to lysosomes where they are degraded and thus, down-regulated (8, 26). β-arrestin regulation of internalization and/or receptor recycling did not likely contribute to the beta-agonist effects observed in this study, since experiments assessing cAMP accumulation and airway contractile responsiveness were of short duration. Moreover, the similarity in lung and ASM β-AR density between WT and βarr-2-KO mice suggests that chronic up-regulation of β-ARs does not account for the effect of arrestins on beta-agonist regulation of airway responsiveness. Future studies examining the effects of chronic beta-agonist treatment should help elucidate the role of arrestin-dependent β2AR trafficking in modulating the β2AR function in ASM.
The ability of arrestin knockout or knockdown to augment signaling and functional consequences of β2AR activation occurs against a backdrop of other regulatory mechanisms also capable of desensitizing β2AR signaling. GRKs have been shown sufficient to mediate agonist-induced β2AR desensitization (21, 27), and PKA-mediated phosphorylation of the β2AR (28) has also been shown to be an important feedback regulator of the β2AR in many cell types. Because these mechanisms are known to be active in ASM (ref. 13 and unpublished results), our findings suggest that they cannot fully compensate for a loss of arrestins and suggest a clear physiological role for arrestins in ASM.
The observation that knockout of βarr-2 in the mouse is sufficient to augment β2AR-mediated signaling and function in ASM is consistent with the findings of Oakley et al. (29), which demonstrate that the β2AR binds to βarr-2 more readily and to a greater extent than it does βarr-1. That βarr-1 is unable to fully compensate for a loss of βarr-2 suggests that these differences in binding kinetics and affinity are of physiological relevance. However, in human ASM cell cultures, which express βarr-1 but have at most extremely low levels of βarr-2, knockdown of βarr-1/2 was able to significantly augment β2AR signaling. This finding suggests that βarr-1-dependent regulation of the β2AR is also of physiological consequence in ASM. Because knockout of both βarr-1 and βarr-2 is lethal (reviewed in ref. 30), we are unable to judge the relative importance of each arrestin on our experimental outcomes.
Nevertheless, our findings point to β-arrestins as important regulators of ASM function and implicate agonist-specific (homologous) desensitization of ASM β2ARs as important in airway physiology, and possibly, airway disease. Although the significance of endogenous catecholamines on ASM contraction is unclear, most asthmatics control their symptoms through the use of inhaled beta-agonists. Both short- and long-acting β-agonists are prescribed with caution as it is generally accepted that their chronic use is associated with a loss of bronchoprotection and a loss of asthma control associated with increased risk of exacerbations and death (31–35). The mechanisms underlying worsening of asthma severity with chronic use of beta-agonists have not been clearly established; however, agonist-specific desensitization of the ASM β2AR is a prime and logical candidate. Not only would GRK- and arrestin-mediated desensitization of the β2AR be expected to occur as a consequence of chronic inhaled beta-agonist treatment, a recent study by Mak et al. (36) suggests that the homologous desensitization mechanism per se may be upregulated by the allergic inflammation of the airway. Given our current findings that demonstrate the relevance of arrestins in regulating ASM function and airway physiology, the dots appear to be connected to suggest that relief of β2AR desensitization by targeting arrestins has therapeutic potential. Drug delivery strategies that target arrestin expression or function could serve as a means of improving the efficacy of inhaled beta-agonist in bronchodilation, minimize lung inflammatory cell influx and its pathogenic effect (10), and thereby possibly thwart the loss of asthma control that occurs in the subset of asthmatics managed with chronic beta-agonists.
Acknowledgments
This work was funded by HL-58506 and AI059755 (R.B.P.) and the U.S. Department of Veterans Affairs and HL-084123 (J.K.L.W). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. D.A.D. is supported by Pathway to Independence award K99 HL-087560. We thank Sudha K. Shenoy for guidance with the binding assay.
REFERENCES
- 1.Deshpande DA, Penn RB. Targeting G protein-coupled receptor signaling in asthma. Cell Signal. 2006;18:2105–2120. doi: 10.1016/j.cellsig.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Bhagat R, Kalra S, Swystun VA, Cockcroft DW. Rapid onset of tolerance to the bronchoprotective effect of salmeterol. Chest. 1995;108:1235–1239. doi: 10.1378/chest.108.5.1235. [DOI] [PubMed] [Google Scholar]
- 3.Cheung D, Timmers MC, Zwinderman AH, Bel EH, Dijkman JH, Sterk PJ. Long-term effects of a long-acting beta 2-adrenoceptor agonist, salmeterol, on airway hyperresponsiveness in patients with mild asthma [see comments] N. Engl. J. Med. 1992;327:1198–1203. doi: 10.1056/NEJM199210223271703. [DOI] [PubMed] [Google Scholar]
- 4.Peters SP, Fish JE. Prior use of long-acting beta-agonists: friend or foe in the emergency department? Am. J. Med. 1999;107:283–285. doi: 10.1016/s0002-9343(99)00228-4. [DOI] [PubMed] [Google Scholar]
- 5.Israel E, Chinchilli VM, Ford JG, Boushey HA, Cherniack R, Craig TJ, Deykin A, Fagan JK, Fahy JV, Fish J, Kraft M, Kunselman SJ, Lazarus SC, Lemanske RF, Jr., Liggett SB, Martin RJ, Mitra N, Peters SP, Silverman E, Sorkness CA, Szefler SJ, Wechsler ME, Weiss ST, Drazen JM. Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomised, placebo-controlled cross-over trial. Lancet. 2004;364:1505–1512. doi: 10.1016/S0140-6736(04)17273-5. [DOI] [PubMed] [Google Scholar]
- 6.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 7.Penn RB, Benovic JL. Regulation of G protein-coupled receptors. In: Conn PM, editor. Handbook of Physiology. Oxford University Press; Oxford, UK: 1998. pp. 125–164. [Google Scholar]
- 8.Shenoy SK, Lefkowitz RJ. Trafficking patterns of beta-arrestin and G protein-coupled receptors determined by the kinetics of beta -arrestin deubiquitination. J. Biol. Chem. 2003;278:14498–14506. doi: 10.1074/jbc.M209626200. [DOI] [PubMed] [Google Scholar]
- 9.Penn RB, Pascual RM, Kim Y-M, Mundell SJ, Krymskaya VP, Panettieri RA, Jr., Benovic JL. Arrestin specificity for G protein-coupled receptors in human airway smooth muscle. J. Biol. Chem. 2001;276:32648–32656. doi: 10.1074/jbc.M104143200. [DOI] [PubMed] [Google Scholar]
- 10.Walker JK, Fong AM, Lawson BL, Savov JD, Patel DD, Schwartz DA, Lefkowitz RJ. Beta-arrestin-2 regulates the development of allergic asthma. J. Clin. Invest. 2003;112:566–574. doi: 10.1172/JCI17265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 12.Billington CK, Kong KC, Bhattacharyya R, Wedegaertner PB, Panettieri RA, Chan TO, Penn RB. Cooperative regulation of p70S6 kinase by receptor tyrosine kinases and G protein-coupled receptors augments airway smooth muscle growth. Biochemistry. 2005;44:14595–14605. doi: 10.1021/bi0510734. [DOI] [PubMed] [Google Scholar]
- 13.Guo M, Pascual RM, Wang S, Fontana MF, Valancius CA, Panettieri RA, Jr., Tilley SL, Penn RB. Cytokines regulate beta-2-adrenergic receptor responsiveness in airway smooth muscle via multiple PKA- and EP2 receptor-dependent mechanisms. Biochemistry. 2005;44:13771–13782. doi: 10.1021/bi051255y. [DOI] [PubMed] [Google Scholar]
- 14.Walker JK, Peppel K, Lefkowitz RJ, Caron MG, Fisher JT. Altered airway and cardiac responses in mice lacking G protein-coupled receptor kinase 3. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1999;276:R1214–R1221. doi: 10.1152/ajpregu.1999.276.4.R1214. [DOI] [PubMed] [Google Scholar]
- 15.Gavett SH, Wills-Karp M. Elevated lung G protein levels and muscarinic receptor affinity in a mouse model of airway hyperreactivity. Am. J. Physiol. Lung Physiol. 1993;265:L493–L500. doi: 10.1152/ajplung.1993.265.5.L493. [DOI] [PubMed] [Google Scholar]
- 16.Levitt RC, Mitzner W. Expression of airway hyperreactivity to acetylcholine as a simple autosomal recessive trait in mice. FASEB J. 1988;2:2605–2608. doi: 10.1096/fasebj.2.10.3384240. [DOI] [PubMed] [Google Scholar]
- 17.Deshpande DA, Pascual RM, Wang SW, Eckman DM, Riemer EC, Funk CD, Penn RB. PKC-dependent regulation of the receptor locus dominates functional consequences of cysteinyl leukotriene type 1 receptor activation. FASEB J. 2007;21:2335–2342. doi: 10.1096/fj.06-8060com. [DOI] [PubMed] [Google Scholar]
- 18.Ahn S, Nelson CD, Garrison TR, Miller WE, Lefkowitz RJ. Desensitization, internalization, and signaling functions of beta-arrestins demonstrated by RNA interference. Proc. Natl. Acad. Sci. U. S. A. 2003;100:1740–1744. doi: 10.1073/pnas.262789099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Widdop S, Daykin K, Hall IP. Expression of muscarinic M2 receptors in cultured human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 1993;9:541–546. doi: 10.1165/ajrcmb/9.5.541. [DOI] [PubMed] [Google Scholar]
- 20.Benovic JL, Kuhn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein) Proc. Natl. Acad. Sci. U. S. A. 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lohse MJ, Benovic JL, Caron MG, Lefkowitz RJ. β-Arrestin: A protein that regulates β-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 22.Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc. Natl. Acad. Sci. U. S. A. 1981;78:3363–3367. doi: 10.1073/pnas.78.6.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Struckmann N, Schwering S, Wiegand S, Gschnell A, Yamada M, Kummer W, Wess J, Haberberger RV. Role of muscarinic receptor subtypes in the constriction of peripheral airways: studies on receptor-deficient mice. Mol. Pharmacol. 2003;64:1444–1451. doi: 10.1124/mol.64.6.1444. [DOI] [PubMed] [Google Scholar]
- 24.Hosey MM, Benovic JL, DebBurman SK, Richardson RM. Multiple mechanisms involving protein phosphorylation are linked to desensitization of muscarinic receptors. Life Sci. 1995;56:951–955. doi: 10.1016/0024-3205(95)00033-3. [DOI] [PubMed] [Google Scholar]
- 25.Lee KB, Pals-Rylaarsdam R, Benovic JL, Hosey MM. Arrestin-independent internalization of the m1, m3, and m4 subtypes of muscarinic cholinergic receptors. J. Biol. Chem. 1998;273:12967–12972. doi: 10.1074/jbc.273.21.12967. [DOI] [PubMed] [Google Scholar]
- 26.Krueger KM, Daaka Y, Pitcher JA, Lefkowitz RJ. The role of sequestration in G protein-coupled receptor resensitization. Regulation of β2-adrenergic receptor dephosphorylation by vesicular acidification. J. Biol. Chem. 1997;272:5–8. doi: 10.1074/jbc.272.1.5. [DOI] [PubMed] [Google Scholar]
- 27.Benovic JL, Kuhn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Functional desensitization of the isolated β-adrenergic receptor by the β-adrenergic receptor kinase: Potential role of an analog of the retinal protein arrestin (48 kDa protein) Proc. Natl. Acad. Sci. U. S. A. 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran TM, Friedman J, Qunaibi E, Baameur F, Moore RH, Clark RB. Characterization of agonist stimulation of cAMP-dependent protein kinase and G protein-coupled receptor kinase phosphorylation of the beta2-adrenergic receptor using phosphoserine-specific antibodies. Mol. Pharmacol. 2004;65:196–206. doi: 10.1124/mol.65.1.196. [DOI] [PubMed] [Google Scholar]
- 29.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 30.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 31.Aaronson DW. The “black box” warning and allergy drugs. J. Allergy Clin. Immunol. 2006;117:40–44. doi: 10.1016/j.jaci.2005.08.060. [DOI] [PubMed] [Google Scholar]
- 32.Jackson CM, Lipworth B. Benefit-risk assessment of long-acting beta2-agonists in asthma. Drug Saf. 2004;27:243–270. doi: 10.2165/00002018-200427040-00003. [DOI] [PubMed] [Google Scholar]
- 33.Sears MR, Taylor DR, C.G. P, Lake DC, Li Q, Flannery EM, Yates DM, Lucas MK, Herbison GP. Regular inhaled beta-agonist treatment in bronchial asthma. Lancet. 1990;336:1391–1396. doi: 10.1016/0140-6736(90)93098-a. [DOI] [PubMed] [Google Scholar]
- 34.Spitzer WO, Suissa S, Ernst P, Horwitz RI, Habbick B, Cockroft D, Boivin J-F, McNutt M, Buist AS, Rebuck AS. The use of β-agonists and the risk of death and near-death from asthma. N. Engl. J. Med. 1992;326:501–506. doi: 10.1056/NEJM199202203260801. [DOI] [PubMed] [Google Scholar]
- 35.Taylor DR, Town GI, Herbison GP, Boothman-Burrell D, Flannery EM, Hancox B, Harre E, Laubscher K, Linscott V, Ramsay CM, Richards G. Asthma control during long-term treatment with regular inhaled salbutamol and salmeterol. Thorax. 1998;53:744–752. doi: 10.1136/thx.53.9.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mak JC, Hisada T, Salmon M, Barnes PJ, Chung KF. Glucocorticoids reverse IL-1beta-induced impairment of beta-adrenoceptor-mediated relaxation and up-regulation of G-protein-coupled receptor kinases. Br. J. Pharmacol. 2002;135:987–996. doi: 10.1038/sj.bjp.0704545. [DOI] [PMC free article] [PubMed] [Google Scholar]