

Abstract
Elucidating the principles governing anesthetic-protein interactions requires structural determinations at high resolutions not yet achieved with ion channels. Protein kinase C (PKC) activity is modulated by general anesthetics. We solved the structure of the phorbol-binding domain (C1B) of PKCδ complexed with an ether (methoxymethylcycloprane) and with an alcohol (cyclopropylmethanol) at 1.36-Å resolution. The cyclopropane rings of both agents displace a single water molecule in a surface pocket adjacent to the phorbol-binding site, making van der Waals contacts with the backbone and/or side chains of residues Asn-237 to Ser-240. Surprisingly, two water molecules anchored in a hydrogen-bonded chain between Thr-242 and Lys-260 impart elasticity to one side of the binding pocket. The cyclopropane ring takes part in π-acceptor hydrogen bonds with the amide of Met-239. There is a crucial hydrogen bond between the oxygen atoms of the anesthetics and the hydroxyl of Tyr-236. A Tyr-236-Phe mutation results in loss of binding. Thus, both van der Waals interactions and hydrogen-bonding are essential for binding to occur. Ethanol failed to bind because it is too short to benefit from both interactions. Cyclopropylmethanol inhibited phorbol-ester-induced PKCδ activity, but failed to do so in PKCδ containing the Tyr-236-Phe mutation.
Introduction
Gaining a detailed understanding of the intermolecular interactions governing anesthetic-protein interactions is a critical step in elucidating the molecular mechanisms underlying general anesthesia and in developing more selective anesthetic agents (1–5). However, little progress can be made until such intermolecular interactions are characterized at high resolution. The physiological sites of anesthetic action remain difficult to define, and some potential targets, such as the transmembrane domains of the mammalian ligand-gated ion channel superfamily, are poorly characterized. Furthermore, the structures of prokaryotic channels have been solved at low resolution (6–8) relative to that necessary to reveal atomic-level principles governing anesthetic binding (9–11). Consequently, crystallizable soluble proteins have been used as surrogate targets to obtain structural information on protein-anesthetic interactions (12–18). Although these surrogates are not involved in signal transduction, they provide principles of protein-anesthetic interactions that may apply at any site.
Protein kinase C (PKC), a key signal transduction protein, has been proposed as the target of anesthetics such as alcohols, halothane, and enflurane (19,20) (for a review, see Rebecchi and Pentyala (21)). The PKC family of serine-threonine kinases plays a central role in signal transduction mediated by lipid second messengers such as sn-1,2 diacylglycerols, regulating divergent cellular functions by phosphorylation of target proteins such as ion channels (22,23). Members of the PKC family that respond to DGs can be separated into two major categories; the conventional (α, βI, βII, γ) and the novel (δ, ε, θ, η) PKCs, each with four domains, termed C1–C4, which play distinct roles in kinase function. C1 and C2 are regulatory domains, C3 is the ATP-binding domain, and C4 is the catalytic domain. The C1 domain is a tandem repeat of highly conserved cysteine-rich zinc finger subdomains C1A and C1B. These domains differ in their binding affinities for phorbol esters and sn-1,2 diacylglycerols (24–27).
In intact PKCα, volatile anesthetics and long- and short-chain alcohols modulate PKC activation in vitro and alter the binding affinities of activators. The isolated catalytic domain, C4, is unaffected by anesthetics, indicating that the primary anesthetic interaction occurs within the regulatory domains, most likely C1, because these agents interact allosterically with phorbol binding (19,26,28,29). To test this hypothesis, we chose PKCδ. Although it is the least sensitive to anesthetics (30), its phorbol-binding domain (C1B) is the only one for which a crystal structure is available (31). We previously used azialcohols to photolabel Tyr-236 on this domain of PKCδ (32). Encouraged by this finding, we have now solved high-resolution crystal structures of unliganded PKCδ C1B as well as complexes with methoxymethylcycloprane (CPE) and cyclopropylmethanol (CPM). Both compounds bind in the same surface pocket, forming a single hydrogen bond to Tyr-236. Eliminating the hydrogen bond by mutation of Tyr-236 to phenylalanine resulted in no detectable alcohol binding. Furthermore, we show that this binding site is important in anesthetic-induced inhibition of PKCδ activity.
Materials and Methods
Reagents
Glutathione Sepharose 4B was obtained from Amersham Biosciences (Piscataway, NJ). Protein concentration was determined with a BCA protein assay reagent kit (Pierce, Rockford, IL). COS-7 cells were purchased from the American Type Culture Collection. Dulbecco's modified Eagle's medium, L-glutamine, and penicillin-streptomycin were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum, Anti-Flag affinity gel, Histone-IIIS, Phorbol 12-myristate 13-acetate (PMA), CPM, and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. sn-1,2 Dioleoylglycerol (DG) and L-α-phosphatidylserine (PS) were purchased from Avanti Polar Lipids (Alabaster, AL). [32γ-P]ATP was purchased from PerkinElmer Life Sciences (Waltham, MA).
Expression, purification, and characterization of PKCδ C1B
PKCδ C1B and its mutants were expressed in Escherichia coli as glutathione S-transferase fusion products, purified by glutathione-Sepharose affinity chromatography, and characterized by circular dichroism spectra as previously described (32). Before crystallization, the protein was further purified by fast-performance liquid chromatography (Biologic Workstation, BioRad, Hercules, CA) using a Superdex 75 column (GE Healthcare, Uppsala, Sweden), a mobile phase of 50 mM Tris, 100 mM NaCl, pH 7.0, and a flow rate of 0.5 ml/min (32).
Site-directed mutagenesis of PKCδ C1B
Site-directed mutagenesis of the glutathione-S-transferase-PKCδ C1B was performed using the QuikChange kit (Stratagene, La Jolla, CA). One round of polymerase chain reaction (PCR) amplification was carried out using two suitably designed complementary primers, and the variant was amplified by PCR in the pGEX plasmid containing the C1B domain sequence (kindly provided by P. M. Blumberg of the National Institutes of Health National Cancer Institute). DNA sequences were confirmed at the Massachusetts General Hospital DNA sequencing facility.
Crystallization, protein-alcohol complex formation, and data collection
Crystallization was carried out at 293°K using the hanging-drop vapor-diffusion method. Initial screening was with crystallization screens (Hampton Research, Aliso Viejo, CA). The hanging-drop solution contained equal volumes (2 μl) of a solution containing 25 mg/ml protein and the reservoir solution. The reservoir solution consisted of 20% polyethylene glycol (PEG) 3350 in 0.2 M ammonium sulfate and 25 mM HEPES, pH 7.2. The crystals exhibited plate morphology and reached their full dimensions of 0.05 × 0.2 × 0.5 mm within a week. For cocrystallization, anesthetic was included in the reservoir solution. For soaking, preformed crystals were incubated in reservoir solution supplemented with anesthetic.
During cocrystallization, loss of anesthetic was monitored by headspace gas chromatography performed on an HP 5890 (Agilent Technologies, Palo Alto, CA) using a DB-17HT column (30 m × 0.321 mm, 0.15 mm from J&W Scientific, Philadelphia, PA). When 0.5 M CPM in 0.2 M ammonium sulfate and 20% PEG 3350 was present during cocrystallization, 40% of CPM was lost over a 4-week period, which is twice as long as the usual cocrystallization condition time.
Crystals were flash-frozen in liquid nitrogen in 40% PEG 3350 in the buffer ± anesthetic. Synchrotron data were collected at beam lines X6A (National Synchrotron Light Source, Brookhaven National Laboratory, Upton, NY) and 8BM (Advanced Photon Source, Argonne National Laboratory, Argonne, IL). Images were integrated and scaled using DENZO and SCALEPACK as implemented in HKL-2000 (33). Crystals belonged to the space group P21 and contained two molecules in the asymmetric unit. Data collection and refinement statistics are given in Table 1.
Table 1.
Data collection and refinement statistics
| Wild-type C1B | CPM | CPE | C1B Tyr-236-Phe | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P21 | P21 | P21 | P21 |
| Cell dimensions | ||||
| a, b, c (Å) | 43.598, 32.475, 49.965 | 43.715, 32.597, 49.719 | 43.530, 32.459, 49.584 | 43.876, 32.522, 49.816 |
| β (°) | 94.10 | 94.21 | 94.55 | 94.02 |
| Resolution (Å) | 30.0–1.30 (1.35–1.30) | 30.0–1.36 (1.38–1.36) | 30.0–1.36 (1.38–1.36) | 30.0–1.3 (1.35–1.30) |
| ∗Rmerge | 8.2 (36.3) | 5.3 (34.9) | 6.8 (37.0) | 11.2 (46.8) |
| I/σI | 19.7 (2.6) | 19.8 (1.9) | 20.9 (2.0) | 16.1 (1.2) |
| Completeness (%) | 99.5 (99.0) | 97.0 (78.9) | 98.8 (93.8) | 95.7 (79.3) |
| Redundancy | 4.3 (4.0) | 4.3 (3.2) | 4.5 (3.6) | 4.0 (3.2) |
| Refinement | ||||
| Ramachandran plot | ||||
| Most favored (%) | 92.1 | 92.1 | 93.0 | 92.1 |
| Allowed (%) | 7.9 | 7.9 | 7.0 | 7.9 |
| Total number of reflections | 32,058 | 28,244 | 27,793 | 30,558 |
| †Rwork/Rfree | 14.2/15.8 | 14.7/17.0 | 15.3/18.2 | 18.8/22.8 |
| Est. coordinate error (Å) | 0.13 | 0.14 | 0.16 | 0.17 |
| RMS deviation of | ||||
| Bond lengths (Å) | 0.007 | 0.006 | 0.006 | 0.007 |
| Bond angles (°) | 1.21 | 1.09 | 1.09 | 1.13 |
Values in parentheses refer to the last shell. Rfree was computed for 5% randomly selected reflections (61). Est., estimated.
Rmerge = ΣhklΣi|Ii (hkl) − <I(hkl)>|/ΣhklΣiIi(hkl), where the summation is over all symmetry-equivalent reflections, excluding reflections observed only once.
Rwork = Σhkl|Fo(hkl) – kFc(hkl)|/Σhkl|Fo(hkl)|, where Fo and Fc are the observed and calculated structure factors, respectively.
Structure determination and refinement
The Phenix suite (34) was used for the final structure solution and refinement. The 1.95-Å structure of PKCδ (Protein Data Bank code, 1PTQ) was the search model in molecular-replacement calculations. The AutoMR program provided the correct solution with two molecules in the asymmetric unit. After a few cycles of refinement and convergence, the model was manually checked using COOT (35). The refinement and model building were then repeated until Rwork and Rfree converged to constant values. This model was then anisotropically refined until such convergence occurred (Table 1).
The same procedure was followed for the complexes with anesthetics. After the convergence of the refinement of the protein model, if extra electron density was found near Tyr-236, appropriate ligands were fit into that density and refined. Adjustments of models were made at the end of each refinement cycle guided by 2Fo – Fc and Fo – Fc electron density maps.
Using a standard protocol implemented in CNS, we refined the structures and calculated simulated annealing omit maps for each structure (36) to eliminate model bias for molecules occupying the binding site. A box including the ligand was defined and enlarged by 2 Å on each side. A refinement protocol, including simulated annealing from 1000 to 50 K and energy minimization, was then carried out without using any of the atoms inside the box. Electron density for the entire structure was then calculated using the phases derived from this procedure, resulting in maps that are independent of the contribution from the molecule within the box.
Anesthetic potency
General anesthetic potency was assessed with institutional approval in pre-limb-bud Xenopus tadpoles 1.5–2 cm in length (Xenopus 1, Dexter, MI), as previously described (37,38).
Cell culture and plasmids for PKCδ
COS-7 cells were grown in Dulbecco's modified Eagle's medium, supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 μg/mL streptomycin, and 100 units/mL penicillin. The cells were maintained at 37°C, in a humidified atmosphere of 90% air and 10% CO2. Flag-epitope-tagged PKCδ was generated by PCR using pRSV-rat PKCδ (provided by Peter. J. Parker, Imperial Cancer Research Fund, London, UK) as the template with 5′ primer (5′-CGCGGATCCATGGACTATAAGGACGATGATGACAA AGCACCGTTCCTGCGCATCTCCTTC-3′) and 3′ primer (5′-CCGGAATTCCTATTC CAGGAATTGCTCATATTTGG-3′). The PCR product was digested with BamHI and EcoRI, and ligated into pcDNA3 (Invitrogen) to add the epitope tag. The PKCδ Tyr-236-Phe mutant (PKCδ Y236F) was generated using a QuikChange site-directed mutagenesis kit (Stratagene) with 5′ primer (5′-CCTCACCGATTCAAGGTCTTCAACTACATGAGCCCCAC-3′) and 3′ primer (5′-GTGGGGCTCATGTAGTTGAAGACCTTGAATCGGTGAGG-3′).
Kinase expression and purification
FLAG-PKCδ (wild-type or Y236F) plasmids (10 μg) were transfected into COS-7 cells plated in 10-cm plates using Superfect (Qiagen, Hilden, Germany). After 72 h, the cells were washed first in phosphate-buffered saline and then with lysis buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, phosphatase inhibitor cocktail 1 (Roche Applied Science, Penzberg, Germany), and complete protease inhibitor cocktail (Roche Applied Science). After 20 min on a rocker at 4°C, cells were collected and centrifuged (12,000 × g, 10 min). Supernatants were added to anti-FLAG M2-agarose gel (Sigma-Aldrich) and incubated at 4°C for 3 h. The beads were transferred to a column and washed sequentially with 15 ml of lysis buffer, 15 ml of buffer B (150 mM NaCl and 50 mM Tris-HCl, pH 7.4) and 5 ml of PKC storage buffer (20 mM HEPES, pH 7.4, 250 mM NaCl, 2 mM EGTA, 2 mM EDTA, 5 mM DTT, 50% glycerol, and 0.05% Triton X-100). To elute FLAG-tagged proteins, the beads were incubated with PKC storage buffer containing 0.15 mg/ml 3X FLAG peptide (Sigma-Aldrich) for 30 min at 4°C and then centrifuged for 10 s at 10,000 × g. Concentrations of wild-type and Y236F PKCδ were determined by comparison with purchased recombinant PKCδ (Invitrogen) using Sypro Ruby (Molecular Probes, Eugene, OR). The kinases were aliquoted and stored in PKC storage buffer at –80°C.
In vitro PKCδ kinase assay
PKCδ (10 nM) was added to kinase buffer containing 20 mM HEPES, pH 7.4, 10 mM MgCl2, 0.1 mM EGTA, 0.23% TritonX-100, and 1 mM dithiothreitol in 30 μL with 0.28 μg/μL Histone-IIIS, with or without 300 mM CPM, as indicated. The reaction was started by adding either 150 nM PMA and 8% PS, or 4% DG and 8% PS, prepared in 3% TritonX-100 micelles, followed by 20 μM ATP and 0.5 μCi [32γ–P]ATP (39). The PMA stock was in dimethylsulfoxide, whose final concentration in the reaction mixture was 3%. Controls showed that the inhibitory action of CPM was the same at 0.015% and 3% dimethylsulfoxide. After 30 min at 37°C, the reaction was terminated by adding sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and heating (95°C for 5 min). Samples (12 μL) were separated by SDS-PAGE (4–12% Bis-Tris gradient gels; Invitrogen). Radioactivity incorporated into histone III-S was quantified by phosphorimaging (Typhoon 9410, GE Healthcare). Specific PKCδ activity was calculated as the difference between total counts per minute measured with (PMA + PS) or (DG + PS) minus nonspecific activity measured in the absence of activators. Percentage inhibition by CPM was calculated as (1 − (specific PKCδ activity with inhibitor/specific activity without inhibitor)) × 100.
Results
Characterization of proteins and agents
Purified PKCδ C1B and its mutant Tyr-236-Phe both showed a single band ∼7 kDa on 18% SDS-PAGE (data not shown) (32) and migrated as single symmetrical peaks with the expected Stokes radius by fast-performance liquid chromatography on a gel filtration column. The mutant's circular dichroism spectra between 195 and 240 nm were similar to that of the wild-type C1B. Spectra for both proteins had secondary structures very similar to those previously reported (50% β-sheet, 7% α-helix, remainder random) (32).
CPE was synthesized by a routine Williamson ether synthesis with excess CPM as solvent. Sodium (460 mg, 20 mmol) was added in small portions to CPM (2.88 g, 40 mmol; two-fold excess) and stirred at room temperature until evolution of hydrogen gas ceased (a thick slurry of yellow sodium salt in CPM was observed). Iodomethane (2.84 g, 20 mmol) was added, and the mixture was stirred for 2 h (white sodium iodide solid was observed). CPM and CPE were distilled off, and the ether was separated by five to seven aqueous extractions of 5 mL each to afford pure ether (1.52 g, 88%). The analytical data were analogous to those described earlier (40).
CPE-induced loss of righting reflexes was determined in four groups of six tadpoles exposed to 1–15 mM CPE. Quantal analysis yielded an EC50 of 5 ± 1.2 mM and a slope of 3 ± 1.5. The published EC50 value for CPM is 54 ± 3.2 mM (41).
Crystal structures of the wild-type PKCδ C1B domain
Our crystals of the PKCδ C1B domain belonged to space group P21 and contain two molecules of the protein in the asymmetric unit (see Materials and Methods). Both molecules in the asymmetric unit of our crystals are nearly identical in structure, with a root-mean-square deviation (rmsd) of 0.25 Å for Cα. All 65 residues of each chain could be built into the electron density maps, including the 50 residues of the C1B domain and the 15 residues of the construct that were not previously reported (Table 1).
Our wild-type structure, which superimposed well on that previously published (rmsd of 0.27 Å for the Cα atoms of residues His-231 to Cys-280) (31), had all residues within the allowed region of the Ramachandran plot, well-defined side chains, and stereochemical parameters well within the statistical error (Table 1). Our structure has an especially low coordinate error of 0.13 Å, calculated using the Luzzati plot (42) (Table 1), allowing us to discuss interactions with high accuracy. The B-factors (Fig. S1 in the Supporting Material) are lowest (7–11 Å2) at the Cys and His residues coordinating the two zinc ions and tend to be higher (∼17–26 Å2) in the loop regions and around the residues previously shown to be in contact with phorbol esters (Fig. 1 and Fig. S2) (31), an observation consistent with a recent NMR study (43).
Figure 1.
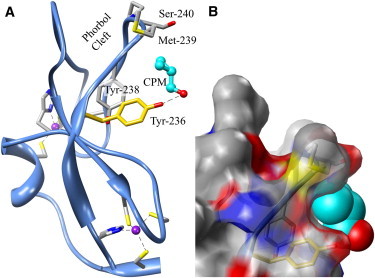
The overall structure of the PKCδ C1B subdomain complexed with CPM. (A) CPM (cyan) binds to the protein in a pocket created by Tyr-236, Asn-237, Tyr-238, Met-239, and Ser-240, and its hydroxyl group is hydrogen-bonded (dashed line) to Tyr-236 (2.8 Å). The site includes both Tyr-236 (gold), photolabeled by azialcohols, and part of the phorbol-binding site (Met-239, Ser-240) discovered by Hurley's group (31). Dashed lines are the coordination bonds to zinc (purple). (B) Surface representation in the same orientation as in A. Tyr-236, Tyr-238, Met-239, and Ser-240 are visible through the translucent surface. Nitrogen and oxygen atoms are blue and red, respectively. Molecular graphics images were produced using the UCSF Chimera package (59) (see Acknowledgments).
In the phorbol-binding site, 11 water molecules form an extensive hydrogen-bond network (Fig. S2 A). Water-71 and Water-84 form a four-sided hydrogen-bonded ring linking the carboxyl oxygens of Tyr-238 and Leu-254. The remaining waters form a hydrogen-bonded network each with two to four hydrogen bonds in the vicinity of Pro-241 and Gly-253.
In the vicinity of Tyr-236, there are three water molecules that play important roles in ligand binding. Water-1 is hydrogen-bonded to the amide nitrogen of Met-239 (3.1 Å). It is tightly held on the axis of that bond but undergoes significantly higher motion parallel to the backbone. Water-2 and Water-3 form a hydrogen-bonded chain between Thr-242 and Lys-260 (2.7–2.9 Å). The anisotropic B-factors indicate that motion along one axis perpendicular to the hydrogen bonds is greater than that in line with the bonds, as shown by the thermal ellipsoids in Fig. 2 A.
Figure 2.
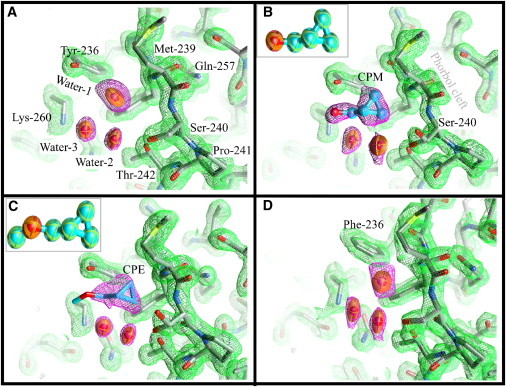
Details of the structure of the anesthetic binding site and the phorbol-binding cleft of the PKCδ C1B subdomain. Carbon atoms are colored gray, oxygen atoms red, nitrogen atoms blue, and sulfur atoms yellow. Thermal ellipsoids at 50% probability represent the water molecules. Waters in the phorbol cleft are omitted for clarity (see Fig. S2 for details). Ligand carbons are cyan. The 2Fo electron density map of the protein (green mesh) is drawn at 1σ cut-off. The equivalent maps around the ligands and water (magenta mesh) are at 1σ, except for CPE where the map is drawn at 0.9σ cut-off. (A) Wild-type C1B containing a water molecule (Water-1) at the anesthetic site. Water-2 and Water-3 form a hydrogen-bond network connecting Thr-242 and Lys-260. (B) CPM bound to C1B. CPM forms a hydrogen bond with Tyr-236. Water-2 and Water-3 form one side of the ligand-binding pocket. The hydroxyl of Ser-240 now adopts two rotamers. (Inset) Thermal ellipsoid representation of CPM. (C) CPM bound to C1B. The hydroxyl of Ser-240 again adopts two rotamers. (Inset) Thermal ellipsoid representation of CPE. (D) Structure of the C1B mutant Tyr-236-Phe. A water molecule occupies the anesthetic binding site in a way very similar to that observed for the wild-type. Neither CPM nor CPE were detected in this mutant (see text). Figures were prepared using Chimera (59).
An interesting observation was that in each molecule, the construct's C- and N-terminal residues form a two-stranded antiparallel β-sheet (Fig. S3). Two such sheets from molecules in adjacent unit cells interact to form a four-stranded antiparallel β-sheet held together by four (2.8–2.9 Å) backbone intermolecular hydrogen bonds, with each pair of Ala-226 and Val-228 residues contributing two of these bonds. This structure propagates to form an array of extended planar sheets of C1B domains sandwiched between similar sheets of solvent.
Crystal structures of PKCδ's C1B domain in the presence of anesthetics
The structure of C1B cocrystallized in the presence of 500 mM CPM was solved to 1.36 Å. A similar structure was obtained by soaking a preformed crystal with 500 mM CPM (data not shown). The unit cell and the structure of C1B were unchanged by CPM; the rmsd for the Cα atoms compared to the unliganded structure was 0.06 Å (His-231 to Cys-280). CPM tended to slightly lower isotropic B-factors except in the loop regions (Fig. S1).
It is important in small ligand studies to establish that any excess density originates from the added ligand and is not an artifact of the crystallization conditions, such as changes in hydration or the presence of PEG (44). We chose CPM because its restricted geometry and characteristic shape would aid in validating the assignment. After refinement of the protein model, extra electron density was found near Tyr-236 (Fig. 2 B and Fig. S5 A). This density consisted of a triangular blob with one side close to the backbone of residues Met-239 and Ser-240. From the apex opposite this side of the triangle there was an extension of the electron density toward Lys-260. The triangular excess density could be fitted with a cyclopropane ring. The remaining positive density could not be satisfactorily fitted by adding water molecules, but an excellent fit was obtained with CPM leaving no residual density at 3.6σ (Fig. 2 B). The oxygen was well placed to form a hydrogen bond with Tyr-236's hydroxyl (2.8–3.0 Å) (Fig. 3).
Figure 3.
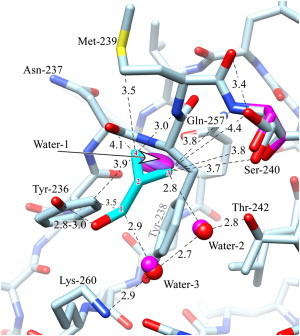
Interactions between CPM and the C1B domain of PKCδ. The carbons of the ligand are in cyan and are numbered for reference by superimposed red numerals. The unliganded structure (magenta) is omitted unless it is different from that of the ligand-bound structure (light blue). Oxygen is red, nitrogen is blue, except that the waters in the unliganded state are magenta. Distances are in Ångstroms. The ligand's hydroxyl forms a hydrogen bond to Tyr-236. Good van der Waals contact is made between the ligand and several residues, as well as Water-2 and Water-3, which are hydrogen-bonded between Thr-242 and Lys-260 (see text). The ligand-bound structure shows two rotamers for Ser-240; one of these superposes with the single wild-type structure. The ligand replaces water-1, whose thermal ellipsoid is shown) (see Discussion). The figure was prepared using Chimera (59).
The structure with bound CPE was determined at a resolution of 1.36 Å. The position of the cyclopropane ring was similar to that of CPM, and the side chain exhibited a similar degree of mobility. The ether oxygen was well positioned to form a hydrogen bond to Tyr-236, whereas the terminal methyl (C5) was ill-defined, suggesting rotation around the C1—O bond (Figs. 2 C).
Once the models were complete, omit maps were calculated, confirming the presence of water or ligand in the electron density (see Fig. S5).
Water-1, adjacent to Met-239 and displaced by CPM and CPE, was not displaced when crystals were soaked in the following agents: ethanol, ≤10 M; propanol, 3 M; butanol, 1 M; octanol, 4 mM; chloropropanol, 0.5 M; bromobutanol 20 mM; and chloroform 0.5 M.
Crystal structure of PKCδ's C1B domain with the mutation Tyr-236-Phe
This mutant also crystallized in the native condition, yielding similar platelike crystals. No significant deviation was observed in the unit-cell parameters, and superposition on the wild-type structure gave an rmsd of 0.16 Å for the Cα carbons of all 128 residues of the two copies present in the asymmetric unit, indicating that the overall structure and crystal packing are very similar in both the wild-type and mutant structures. The only change was the loss of the solvent-exposed tyrosine hydroxyl group (Fig. 2 D). Minor increases in the B-factors of the residues adjacent to Phe-236 were also observed, but not in the aromatic ring itself. The statistics are given in Table 1 and the omit map is shown in Fig. S5 D.
The mutant Tyr-236-Phe C1B was cocrystallized with 0.3 and 0.5 M CPM, and structures were determined for both. In addition, two different data sets were collected for crystals soaked with 1 M CPM. These conditions are similar to those successfully employed with wild-type C1B, but although the resolutions were all comparable to those of the unliganded mutant, in no case was excess electron density found. We therefore conclude that the two ligands do not bind to Tyr-236-Phe C1B.
Pharmacology of protein kinase Cδ activity
DG stimulated Histone-IIIS phosphorylation by three- to fourfold and PMA stimulated it by approximately twofold over the basal activity, independent of the presence of the Tyr-236-Phe mutation. CPM inhibited basal activity by ∼20%, independent of the presence of the mutation. After correction for this effect on basal activity, CPM still inhibited DG-stimulated activity of wild-type rat PKCδ in a concentration-dependent manner, but at a concentration ≥600 mM, inhibition was only partially reversible. Therefore, all further experiments were carried out using 300 mM CPM, which reversibly inhibited DG-stimulated PKCδ activity by 35–40%, independent of the Tyr-236-Phe mutation (Table 2). CPM also inhibited PMA-stimulated wild-type PKCδ to a similar extent. In contrast, it was barely effective in inhibiting PMA-stimulated PKCδ Tyr-236-Phe (Table 2). The PMA-activity concentration-response curve, determined over the range 10–1,000 nM, fit mass-action kinetics (see Fig. 4). In the presence of 300 mM CPM, the EC50 for PMA was unchanged (p = 0.9) for wild-type PKCδ, but the maximal effect was reduced significantly (p = 0.02), consistent with noncompetitive inhibition. In contrast, CPM did not alter the EC50 or the maximal effect of PMA for PKCδ Tyr-236-Phe.
Table 2.
Inhibition of PKCδ activity by CPM
| PKCδ | Percent inhibition of stimulated activity by 300 mM CPM |
|||||
|---|---|---|---|---|---|---|
| DG |
PMA |
|||||
| mean | SE | n | mean | SE | n | |
| Wild-type | 39.4 | ± 7.2 | 3 | 27.5 | ± 3.4 | 7 |
| Tyr-236-Phe | 35.5 | ± 4.8 | 3 | 7.1∗ | ± 1.3 | 7 |
Each data point in each experiment was calculated as the average of three to five replicates. n is the number of independent experiments.
p = 0.0001 compared with wild-type PKCδ activated with PMA (two-tailed, unpaired t-test).
Figure 4.
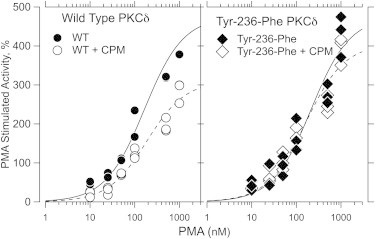
| PKCδ | Agent | EC50 ± SD | Max activity ± SD |
|---|---|---|---|
| Wild-type | — | 165 ± 38 | 475 ± 35 |
| Wild-type | + CPM | 186 ± 37 | 311 ± 20 |
| Tyr-236-Phe | — | 215 ± 58 | 475 ± 43 |
| Tyr-236-Phe | + CPM | 162 ± 40 | 406 ± 31 |
Discussion
We have determined structures at 1.36-Å resolution of the mouse PKCδ C1B phorbol-binding domain complexed with an alcohol, CPM, and its methyl ether, CPE. The anesthetics bind near the side chain of Tyr-236, a residue that has been photolabeled in solution by 3-azioctanol and 3-azibutanol (32), affirming the utility of photoaffinity labeling. The anesthetic binding site is unusual. First, there is no α-helix in contact with the anesthetic, whereas this motif is often present at general anesthetic sites in ion channels, soluble proteins, and model systems (9–11,16,45). Instead, part of the pocket is formed by a loop between two β-sheets. This loop motif is not uncommon, but it usually occurs together with other motifs in anesthetic-binding pockets such as those on albumin, cholesterol oxidase, luciferase, and LUSH (45) (Fig. S4). Second, the site is a shallow surface pocket in which the bound ligand remains partially exposed to the solvent. The sites in LUSH and luciferase are inaccessible to bulk solvent (45–47), although that of halothane in human serum albumin is less buried (16).
We have obtained the highest-resolution anesthetic-bound structures reported to date for signal transduction proteins with the exception of butanol bound to LUSH (1.25 Å, PDB 1OOH) (45). Anesthetics interact with residues from Tyr-236 to Ser-240, as well as with Thr-242 and Lys-260 (Figs. 1 and 2 and Fig. S5), on the loop between the strands of a β-sheet. The backbone amide of Tyr-236 is the only part of this sequence to take part in the preceding β-sheet. The remaining residues in the loop arch over the binding pocket, opening up on the other side to form the phorbol-binding cleft (Fig. 1), in which the phorbol ester interacts with residues Met-239, Pro-241, and Thr-242, and also with Leu-250, Leu-251, Gly-253, and Leu-254 (Fig. S2) (31).
Role of van der Waals interactions
Binding of anesthetics is stabilized by van der Waals interactions with the backbone and side chains of C1B and with water, as well as by hydrogen-bonding interactions with the protein and the solvent. These interactions are considered in order below. We have adopted the numbering convention for the ligand carbons that is given in Fig. 3. C1 is the carbon between the cyclopropane ring and the oxygen, C2 is the adjacent ring carbon, and C3 and C4 follow counterclockwise. The methyl group in CPE is C5. Force fields in proteins are complex. We have empirically judged the strength of van der Waals interactions using Tsai's tabulation of radii (48) and assuming pure dispersion forces. These assumptions place the distance of maximum interaction in the range 3.3–4.1 Å and that of half-maximum interaction in the range 3.8–4.9 Å, This provides a conservative estimate of interaction strength, because static dipolar contributions operate over a longer range.
A major source of van der Waals interactions is between the cyclopropane ring and the backbone atoms running from the carboxyl oxygen of Asn-237 (4.1 Å) through to the amide nitrogen of Ser-240 (3.8 Å) (see Fig. 3). The position of the cyclopropane ring in the ether and alcohol derivatives is nearly identical; distances given are for CPM. The C4 to Met-239 backbone amide nitrogen interaction is particularly short-range (3.0 Å), suggestive of a π-ring…NH interaction (42,49). Side chains also provide significant van der Waals interactions (Fig. 3). Strong interactions are observed with Met-239 (Cγ being 3.5 Å from the cyclopropane ring's C4) and Tyr-236 (Cε being 3.9 Å from C4). Tyr-236 and Tyr-238 provide an aromatic character to the bottom of the binding pocket that may interact favorably with the partially aromatic cyclopropane rings of the ligands. Weaker interactions (∼4.5 Å) are seen with the side chains of Tyr-238, Ser-240, Thr-242, and Lys-260.
An unusual feature of this ligand-binding site is that two water molecules (Water-2 and Water-3), which are anchored in a hydrogen-bonding chain stretching from Thr-242 to Lys-260, provide tight van der Waals interactions (≤3 Å) along one side of CPM and CPE (O, C1, C2, and C3 atoms). These water molecules impart elasticity to the site, being displaced 0.5–0.7 Å upon ligand binding without breaking the hydrogen-bonding chain between Thr-242 and Lys-260 (Fig. 3).
The ligands showed very little B-factor anisotropy (Fig. 2, B and C, insets), but their B-factors were higher than those of the atoms contacting them. Indeed, the B-factors are greater than those for the terminal atoms of the longer amino acids, suggesting that the ligands exhibit considerable motion in the binding pocket, as might be expected for such low-affinity agents.
Role of hydrogen-bonding
Both ligands form a hydrogen bond with Tyr-236 (2.8–3.0 Å in length). The ether can only accept a hydrogen bond from Tyr-236, ruling out a contribution from tyrosinate. These hydrogen-bond lengths compare well to the values of 2.6–3.3 Å reported for propofol bound to human serum albumin (Fig. S4 C) and butanol bound to LUSH (50,51) (Fig. S4, A and B). In the cytoplasmic domain of Kir2.1, 2-methyl-2,4-pentanediol forms a hydrogen bond (2.6–3.0 Å) with Tyr-337 (52).
The cyclopropane ring is a common motif in drugs. It has been hypothesized, based on spectroscopic evidence (53) and on surveys of the structures of small molecules (42,54), that cyclopropane rings take part in X-H donor-to-π-acceptor hydrogen bonds. To test whether this occurs in protein-ligand interactions, we compared the interaction between CPM's ring and the backbone amide protons of Met-239 and Ser-240 and the oxygen of Water-2 to the examples of the 22 small molecules given in the above survey (42) (Fig. S6). The two >N–H interactions occupy positions on the boundaries of that data set (Fig. S6, A and C); the interaction of Met-239 is of unusually short range for the angle of interaction, whereas that of Ser-240 appears weaker. Water-2 is capable of adopting very good O–H…π-acceptor hydrogen-bond geometry while maintaining a second hydrogen bond with Thr-242 or Water-3 (Fig. S6 B). Furthermore, the >C3–H of the cyclopropane ring has excellent geometry for donating a hydrogen bond to Water-2 (Fig. S6 D).
The hydrogen bond between Tyr-236 and the ligands is essential, because after the Tyr-236-Phe mutation, bound CPM was not observed and inhibition of the kinase was attenuated. The requirement for a hydrogen bond imposes a geometric constraint that may account for the ligand selectivity of this binding pocket. Thus, although ethanol might form a hydrogen bond with Tyr-236, it is too short to take part in van der Waals interactions with the backbone of the phorbol-binding loop (e.g., Met-239). Butanol and octanol are long enough for their third carbons to make such interactions, but modeling suggests that this would result in steric clashes with subsequent carbons. Furthermore, Tyr-236 is not hydrogen-bonded to water, whereas other sites are (e.g., the Met-239 amide), so that exchanging the water for an alcohol hydrogen bond is less advantageous. This may account for the remarkable observation that 10 M ethanol in 55 M water cannot displace Water-1. In fact, electron density attributable to ethanol is not seen anywhere in the structure, suggesting that the structural requirements for tight ethanol binding are quite strict and in no way nonspecific.
Role of solvation
Water contributes to anesthetic binding in two ways, one entropic and the other enthalpic. Removal of some of the anesthetic's hydrophobic surface from solvation by water will be entropically favorable (the hydrophobic effect). In addition, in the native state, the anesthetic binding pocket is occupied by a water molecule hydrogen-bonded to the backbone nitrogen of Met-239 (3.1 Å) that is displaced when the ligands bind (Fig. 3), thus compensating for the entropic cost of confining a freely diffusing anesthetic molecule to the binding pocket. An enthalpic contribution of water comes from tight van der Waals interactions with the two water molecules that form one side of the binding pocket between Thr-242 and Lys-260, and from the solvation of the displaced water, which would be expected to compensate for the loss of its single hydrogen bond with the protein.
When the previously published (31) phorbol-bound structure is superimposed on our higher-resolution structure, a dramatic role for the solvent is revealed. Nine of the 11 waters in our structure are isosteric with the phorbol and must be displaced by phorbol binding. One of these (Water-28) is isosteric with phorbol oxygen O20, so that upon binding, the phorbol O20 replaces the hydrogen bond between Water-28 and the backbone nitrogen of Thr-242. Notably, Water-28 has the lowest B-factor (20 Å2, compared to 26–49 Å2 for the others). One of the remaining waters (Water-155; Fig. S2 B, green) loses its hydrogen bond to Water-146, which is displaced by phorbol binding, and forms one with O3 of the phorbol ester.
Site of kinase inhibition
When PKCδ activity was stimulated by using PMA, which binds exclusively to the C1B domain of PKCδ adjacent to the alcohol site we have characterized, 300 mM CPM inhibited activity by ∼30%. However, introduction of the Tyr-236-Phe mutation in C1B reduced the inhibition dramatically, suggesting that the inhibitory site is located on this domain in the same locus observed in the structure. As a control, we examined CPM's action on activity stimulated by DG, which acts exclusively on the C1A domain, finding that the wild-type and mutated kinases were equally inhibited. These findings are consistent with CPM inhibiting phorbol-induced kinase activity by binding to the site near Tyr-236 on the C1B domain of PKCδ, whereas inhibition of DG-induced activity must occur at another site. This conclusion is consistent with the hypothesis of Stubbs and co-workers that alcohols interact at or close to the phorbol-binding site of PKC (28).
PKCδ is one of the isoforms least sensitive to anesthetic action (30), so we were unable to pursue the mechanism in more detail. PKCε is quite sensitive to ethanol and has been photolabeled at His-236 (55), a residue homologous to Tyr-236 in PKCδ. Many of the residues in the putative alcohol site in PKCε differ from those in the δ-isoform, perhaps accounting for the different sensitivities to alcohols. This suggests that our findings may be relevant to other isoforms of PKC, and structural information on these isoforms is eagerly awaited.
The mechanism by which anesthetic binding at the Tyr-236 pocket of the kinase affects PMA-stimulated kinase activity needs to be studied further. We have studied anesthetic interactions with a single conformation of the C1B domain of PKCδ, and that conformation is the same one studied with and without phorbol ester bound (31). In both studies, ligand-induced structural changes were modest, even when the ligands were co-crystallized with the protein. Zhang and Hurley (31) concluded "… that phorbol esters activate PKC by altering the nature of the protein surface and by stabilizing the membrane-inserted state, rather than by inducing a conformational change at the activator-binding site." On the other hand, a solution structure of the C1 domain of Munc13-1 reveals a conformation change upon ligand binding. A conserved tryptophan (Trp-252 in our structure) occluded the binding cleft, which meant that a conformation change would be required before diacylglycerol could bind (56). Thus, the possibility that anesthetics cause allosteric interactions in C1B domains cannot be dismissed.
There are no structures of intact novel PKCs, but the structure of PKCβ, a conventional PKC, has been solved at 4.0 Å (57). The C1B domain clamps the NFD helix of the kinase domain in its phorbol-binding cleft, a situation that is reversed upon activation, indicating that this region is involved in state transitions. In a similar way, the C1 domain of β2-chimaerin interacts with four other domains that also occlude the phorbol-binding site (58). The backbone of the C1 domain in both these structures differs little from that in PKCδ C1B. In each case, the homologous anesthetic binding pocket faces the solvent, allowing unhindered access for anesthetics, so that one cannot rule out allosteric actions on interdomain interactions.
Conclusions
We have characterized at very high resolution a binding site for an ether and an alcohol in a surface pocket of the PKCδ C1B domain. Binding involves displacement of a water molecule and the rearrangement of a single side chain in the binding pocket. An unusual feature of the binding pocket is that one side is bounded by two water molecules held in place by a hydrogen-bonding chain between two residues. These waters move modestly to accommodate the ligand, providing the pocket with some elasticity. Although van der Waals interactions contribute to binding, a hydrogen bond between the ligand and Tyr-236 is essential, because mutation of Tyr-236 to Phe abolishes ligand binding. The requirement to maintain both hydrogen bonding and van der Waals interactions imposes geometric constraints that account for why ethanol does not bind. It is possible that the cyclopropane ring plays a unique role as a π-acceptor for X–H hydrogen bonds. The binding site, whose structure we have characterized, mediates inhibition of phorbol-induced PKCδ activity by anesthetics, as shown by site-directed mutagenesis studies of the kinase itself.
Coordinates
The coordinates and structure factors have been deposited in the Protein Data Bank (accession codes 3UEJ, 3UGL, 3UGI, 3UEY).
Acknowledgments
We thank Elizabeth Kelly for assistance with the anesthetic potency measurements, Dr. Peter M. Blumberg for providing the PKCδ C1B construct and Dr. David Jones for providing us with the structure factor file for his LUSH structure. Molecular graphics images were produced using the University of California, San Francisco, Chimera package (59) from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081) or using Pymol (60). S.S. thanks V. Stojanoff, M. Allaire, J. Jakoncic, and F. Yokaichiya of beam line X6A, and N. Sukumar of BM8, for their support during data collection.
This research was supported by grants from the U.S. Public Health Service to K.W.M. (GM 069726) and R.O.M. (AA018316), by a Foundation for Anesthesia Education and Research award to W.S.S., by the Department of Anesthesia, Critical Care & Pain Medicine, Massachusetts General Hospital, and by funds provided by the State of California for medical research on alcohol and substance abuse through University of California, San Francisco, to R.O.M. This work is based upon research conducted at the Northeastern Collaborative Access Team beam lines (BM8) of the Advanced Photon Source, which is supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Research carried out (in whole or in part) at X6A beam line was funded by the National Institute of General Medical Sciences, National Institutes of Health, under agreement GM-0080. The National Synchrotron Light Source, Brookhaven National Laboratory is supported by the U.S. Department of Energy under contract no. DE-AC02-98CH10886.
Footnotes
Joydip Das's present address is Department of Pharmacological and Pharmaceutical Sciences, University of Houston, Houston, TX 77204.
Warren S. Sandberg's present address is Department of Anesthesiology, Vanderbilt University Medical Center, Nashville, TN 37232.
Thilo Stehle's present address is Interfaculty Institute of Biochemistry, Eberhard-Karls Universität Tübingen, 72076 Tübingen, Germany, and Vanderbilt University School of Medicine, Nashville, TN 37232.
Supporting Material
References
- 1.Hemmings H.C., Jr., Akabas M.H., Harrison N.L. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol. Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Miller K.W. The nature of sites of general anaesthetic action. Br. J. Anaesth. 2002;89:17–31. doi: 10.1093/bja/aef167. [DOI] [PubMed] [Google Scholar]
- 3.Yamakura T., Bertaccini E., Harris R.A. Anesthetics and ion channels: molecular models and sites of action. Annu. Rev. Pharmacol. Toxicol. 2001;41:23–51. doi: 10.1146/annurev.pharmtox.41.1.23. [DOI] [PubMed] [Google Scholar]
- 4.Campagna J.A., Miller K.W., Forman S.A. Mechanisms of actions of inhaled anesthetics. N. Engl. J. Med. 2003;348:2110–2124. doi: 10.1056/NEJMra021261. [DOI] [PubMed] [Google Scholar]
- 5.Eckenhoff R.G., Johansson J.S. Molecular interactions between inhaled anesthetics and proteins. Pharmacol. Rev. 1997;49:343–367. [PubMed] [Google Scholar]
- 6.Bocquet N., Nury H., Corringer P.J. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 7.Hilf R.J., Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 8.Hilf R.J., Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–118. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- 9.Hilf R.J., Bertozzi C., Dutzler R. Structural basis of open channel block in a prokaryotic pentameric ligand-gated ion channel. Nat. Struct. Mol. Biol. 2010;17:1330–1336. doi: 10.1038/nsmb.1933. [DOI] [PubMed] [Google Scholar]
- 10.Nury H., Van Renterghem C., Corringer P.J. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- 11.Baenziger J.E., Corringer P.J. 3D structure and allosteric modulation of the transmembrane domain of pentameric ligand-gated ion channels. Neuropharmacology. 2011;60:116–125. doi: 10.1016/j.neuropharm.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Schoenborn B.P., Watson H.C., Kendrew J.C. Binding of xenon to sperm whale myoglobin. Nature. 1965;207:28–30. doi: 10.1038/207028a0. [DOI] [PubMed] [Google Scholar]
- 13.Schoenborn B.P. Binding of xenon to horse haemoglobin. Nature. 1965;208:760–762. doi: 10.1038/208760a0. [DOI] [PubMed] [Google Scholar]
- 14.Sachsenheimer W., Pai E.F., Schirmer R.H. Halothane binds in the adenine-specific niche of crystalline adenylate kinase. FEBS Lett. 1977;79:310–312. doi: 10.1016/0014-5793(77)80809-0. [DOI] [PubMed] [Google Scholar]
- 15.Franks N.P., Jenkins A., Brick P. Structural basis for the inhibition of firefly luciferase by a general anesthetic. Biophys. J. 1998;75:2205–2211. doi: 10.1016/S0006-3495(98)77664-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharya A.A., Curry S., Franks N.P. Binding of the general anesthetics propofol and halothane to human serum albumin. High resolution crystal structures. J. Biol. Chem. 2000;275:38731–38738. doi: 10.1074/jbc.M005460200. [DOI] [PubMed] [Google Scholar]
- 17.Gursky O., Fontano E., Caspar D.L. Stereospecific dihaloalkane binding in a pH-sensitive cavity in cubic insulin crystals. Proc. Natl. Acad. Sci. USA. 1994;91:12388–12392. doi: 10.1073/pnas.91.26.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu R., Loll P.J., Eckenhoff R.G. Structural basis for high-affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 2005;19:567–576. doi: 10.1096/fj.04-3171com. [DOI] [PubMed] [Google Scholar]
- 19.Slater S.J., Cox K.J., Stubbs C.D. Inhibition of protein kinase C by alcohols and anaesthetics. Nature. 1993;364:82–84. doi: 10.1038/364082a0. [DOI] [PubMed] [Google Scholar]
- 20.Hemmings H.C., Jr., Adamo A.I. Effects of halothane and propofol on purified brain protein kinase C activation. Anesthesiology. 1994;81:147–155. doi: 10.1097/00000542-199407000-00021. [DOI] [PubMed] [Google Scholar]
- 21.Rebecchi M.J., Pentyala S.N. Anaesthetic actions on other targets: protein kinase C and guanine nucleotide-binding proteins. Br. J. Anaesth. 2002;89:62–78. doi: 10.1093/bja/aef160. [DOI] [PubMed] [Google Scholar]
- 22.Newton A.C. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 23.Cho W. Membrane targeting by C1 and C2 domains. J. Biol. Chem. 2001;276:32407–32410. doi: 10.1074/jbc.R100007200. [DOI] [PubMed] [Google Scholar]
- 24.Kazanietz M.G., Barchi J.J., Jr., Blumberg P.M. Low affinity binding of phorbol esters to protein kinase C and its recombinant cysteine-rich region in the absence of phospholipids. J. Biol. Chem. 1995;270:14679–14684. doi: 10.1074/jbc.270.24.14679. [DOI] [PubMed] [Google Scholar]
- 25.Szallasi Z., Bogi K., Blumberg P.M. Non-equivalent roles for the first and second zinc fingers of protein kinase Cδ. Effect of their mutation on phorbol ester-induced translocation in NIH 3T3 cells. J. Biol. Chem. 1996;271:18299–18301. doi: 10.1074/jbc.271.31.18299. [DOI] [PubMed] [Google Scholar]
- 26.Slater S.J., Ho C., Stubbs C.D. Protein kinase Cα contains two activator binding sites that bind phorbol esters and diacylglycerols with opposite affinities. J. Biol. Chem. 1996;271:4627–4631. doi: 10.1074/jbc.271.9.4627. [DOI] [PubMed] [Google Scholar]
- 27.Medkova M., Cho W. Interplay of C1 and C2 domains of protein kinase C-α in its membrane binding and activation. J. Biol. Chem. 1999;274:19852–19861. doi: 10.1074/jbc.274.28.19852. [DOI] [PubMed] [Google Scholar]
- 28.Slater S.J., Kelly M.B., Stubbs C.D. Interaction of alcohols and anesthetics with protein kinase Cα. J. Biol. Chem. 1997;272:6167–6173. doi: 10.1074/jbc.272.10.6167. [DOI] [PubMed] [Google Scholar]
- 29.Slater S.J., Malinowski S.A., Stubbs C.D. The nature of the hydrophobic n-alkanol binding site within the C1 domains of protein kinase Cα. Biochemistry. 2004;43:7601–7609. doi: 10.1021/bi049755z. [DOI] [PubMed] [Google Scholar]
- 30.Slater S.J., Stagliano B.A., Stubbs C.D. Effects of ethanol on protein kinase C activity induced by filamentous actin. Biochim. Biophys. Acta. 2001;1544:207–216. doi: 10.1016/s0167-4838(00)00222-3. [DOI] [PubMed] [Google Scholar]
- 31.Zhang G., Kazanietz M.G., Hurley J.H. Crystal structure of the cys2 activator-binding domain of protein kinase Cδ in complex with phorbol ester. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 32.Das J., Addona G.H., Miller K.W. Identification of a general anesthetic binding site in the diacylglycerol-binding domain of protein kinase Cδ. J. Biol. Chem. 2004;279:37964–37972. doi: 10.1074/jbc.M405137200. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z., Minor R. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Adams P.D., Afonine P.V., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P., Lohkamp B., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brünger A.T., Adams P.D., Warren G.L. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 37.Downes H., Courogen P.M. Contrasting effects of anesthetics in tadpole bioassays. J. Pharmacol. Exp. Ther. 1996;278:284–296. [PubMed] [Google Scholar]
- 38.Husain S.S., Forman S.A., Miller K.W. Synthesis and properties of 3-(2-hydroxyethyl)-3-n-pentyldiazirine, a photoactivable general anesthetic. J. Med. Chem. 1999;42:3300–3307. doi: 10.1021/jm9806300. [DOI] [PubMed] [Google Scholar]
- 39.Hannun Y.A., Loomis C.R., Bell R.M. Activation of protein kinase C by Triton X-100 mixed micelles containing diacylglycerol and phosphatidylserine. J. Biol. Chem. 1985;260:10039–10043. [PubMed] [Google Scholar]
- 40.Dauben W.G., Ellis D.J., Templeton W.H. The preparation of AB-dinor steroids. J. Org. Chem. 1969;34:2297–2301. doi: 10.1021/jo01260a012. [DOI] [PubMed] [Google Scholar]
- 41.Raines D.E., Korten S.E., Miller K.W. Anesthetic cutoff in cycloalkanemethanols. A test of current theories. Anesthesiology. 1993;78:918–927. doi: 10.1097/00000542-199305000-00017. [DOI] [PubMed] [Google Scholar]
- 42.Allen F.H., Lommerse J.P.M., Desiraju G.R. The hydrogen-bond C-H donor and ϕ-acceptor characteristics of three-membered rings. Acta Crystallogr. B. 1996;52:734–735. [Google Scholar]
- 43.Brian P.Z., Jamie C.B., David J.N. 1H, 13C and 15N NMR assignments of the C1A and C1B subdomains of PKC-δ. Biomol. NMR Assign. 2011;5:125–129. doi: 10.1007/s12104-010-9283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kleywegt G.J. Crystallographic refinement of ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2007;63:94–100. doi: 10.1107/S0907444906022657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kruse S.W., Zhao R., Jones D.N. Structure of a specific alcohol-binding site defined by the odorant binding protein LUSH from Drosophila melanogaster. Nat. Struct. Biol. 2003;10:694–700. doi: 10.1038/nsb960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Auld D.S., Lovell S., Inglese J. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc. Natl. Acad. Sci. USA. 2010;107:4878–4883. doi: 10.1073/pnas.0909141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakatsu T., Ichiyama S., Kato H. Structural basis for the spectral difference in luciferase bioluminescence. Nature. 2006;440:372–376. doi: 10.1038/nature04542. [DOI] [PubMed] [Google Scholar]
- 48.Tsai J., Taylor R., Gerstein M. The packing density in proteins: standard radii and volumes. J. Mol. Biol. 1999;290:253–266. doi: 10.1006/jmbi.1999.2829. [DOI] [PubMed] [Google Scholar]
- 49.Baker E.N., Hubbard R.E. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 1984;44:97–179. doi: 10.1016/0079-6107(84)90007-5. [DOI] [PubMed] [Google Scholar]
- 50.Langkilde A., Kristensen S.M., Larsen S. Short strong hydrogen bonds in proteins: a case study of rhamnogalacturonan acetylesterase. Acta Crystallogr. D Biol. Crystallogr. 2008;D64:851–863. doi: 10.1107/S0907444908017083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rajagopal S., Vishveshwara S. Short hydrogen bonds in proteins. FEBS J. 2005;272:1819–1832. doi: 10.1111/j.1742-4658.2005.04604.x. [DOI] [PubMed] [Google Scholar]
- 52.Pegan S., Arrabit C., Choe S. Andersen's syndrome mutation effects on the structure and assembly of the cytoplasmic domains of Kir2.1. Biochemistry. 2006;45:8599–8606. doi: 10.1021/bi060653d. [DOI] [PubMed] [Google Scholar]
- 53.Joris L., Schleyer P.R., Gleiter R. Cyclopropane rings as proton-acceptor groups in hydrogen bonding. J. Am. Chem. Soc. 1968;90:327–336. [Google Scholar]
- 54.Panigrahi S.K., Desiraju G.R. Strong and weak hydrogen bonds in the protein-ligand interface. Proteins. 2007;67:128–141. doi: 10.1002/prot.21253. [DOI] [PubMed] [Google Scholar]
- 55.Das J., Pany S., Slater S.J. PKC epsilon has an alcohol-binding site in its second cysteine-rich regulatory domain. Biochem. J. 2009;421:405–413. doi: 10.1042/BJ20082271. [DOI] [PubMed] [Google Scholar]
- 56.Shen N., Guryev O., Rizo J. Intramolecular occlusion of the diacylglycerol-binding site in the C1 domain of munc13-1. Biochemistry. 2005;44:1089–1096. doi: 10.1021/bi0476127. [DOI] [PubMed] [Google Scholar]
- 57.Leonard T.A., Różycki B., Hurley J.H. Crystal structure and allosteric activation of protein kinase C βII. Cell. 2011;144:55–66. doi: 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Canagarajah B., Leskow F.C., Hurley J.H. Structural mechanism for lipid activation of the Rac-specific GAP, β2-chimaerin. Cell. 2004;119:407–418. doi: 10.1016/j.cell.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 59.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 60.Schrodinger, LLC. 2010. The PyMOL Molecular Graphics System, Version 1.3r1. New York.
- 61.Brünger A.T. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.