

Abstract
Fibroblast growth factor-21 (FGF21) has therapeutic potential for metabolic syndrome due to positive effects on fatty acid metabolism in liver and white adipose tissue. FGF21 also improves pancreatic islet survival in excess palmitate; however, much less is known about FGF21-induced metabolism in this tissue. We first confirmed FGF21-dependent activity in islets by identifying expression of the cognate coreceptor Klothoβ, and by measuring a ligand-stimulated decrease in acetyl-CoA carboxylase expression. To further reveal the effect of FGF21 on metabolism, we employed a unique combination of two-photon and confocal autofluorescence imaging of the NAD(P)H and mitochondrial NADH responses while holding living islets stationary in a microfluidic device. These responses were further correlated to mitochondrial membrane potential and insulin secretion. Glucose-stimulated responses were relatively unchanged by FGF21. In contrast, responses to glucose in the presence of palmitate were significantly reduced compared to controls showing diminished NAD(P)H, mitochondrial NADH, mitochondrial membrane potential, and insulin secretion. Consistent with the glucose-stimulated responses being smaller due to continued fatty acid oxidation, mitochondrial membrane potential was increased in FGF21-treated islets by using the fatty acid transport inhibitor etomoxir. Citrate-stimulated NADPH responses were also significantly larger in FGF21-treated islets suggesting preference for citrate cycling rather than acetyl-CoA carboxylase-dependent fatty acid synthesis. Overall, these data show a reduction in palmitate-induced potentiation of glucose-stimulated metabolism and insulin secretion in FGF21-treated islets, and establish the use of autofluorescence imaging and microfluidic devices to investigate cell metabolism in a limited amount of living tissue.
Introduction
The fibroblast growth factor (FGF) family consists of 22 secreted polypeptides divided into seven subfamilies based on phylogeny, sequence identity, and function (1,2). Although the majority of FGF-ligands function as paracrine/autocrine factors, the FGF19 subfamily (FGF15/19, FGF21, and FGF23) has emerged as a unique class of potent endocrine factors. Recently, FGF21 has been shown to exhibit pleiotropic function in regulating obesity, as well as whole body glucose and lipid metabolism (3). Consistently, FGF21 is transcriptionally regulated by PPARα (4,5) and appears functionally independent of insulin (6). Due to these positive effects on metabolic syndrome in combination with apparently few side-effects, recombinant FGF21 is being actively explored as a therapeutic (7).
FGF21 signaling occurs through activation of fibroblast growth factor receptor-1 (FGFR1c) or FGFR2c isoforms, but uniquely requires coexpression of Klothoβ (KLB), a member of the Klotho receptor family (8,9). We have recently confirmed that FGF21 induces dimerization of FGFR1c on the plasma membrane of living cells when coexpressed with KLB (10). Due to the relative ubiquitous expression of FGFR1c and FGFR2c, FGF21 tissue-specificity is dictated by a more limited expression of KLB (11). Accordingly, KLB is predominantly expressed in metabolic tissues including white adipose tissue (WAT), liver, and pancreas (12). We recently demonstrated FGFR1 activity in mouse pancreatic islets and are presently unaware of any studies specifically showing expression and activity of KLB in endocrine pancreas (13). Therefore, our initial goal was to confirm KLB expression and activity in pancreatic islets, and consequently measure the effect of FGF21 activity on islet metabolic responses.
Pancreatic islet β-cells metabolically sense changes in blood glucose and secrete insulin (14). In β-cells, glycolysis-derived pyruvate enters the tricarboxylic acid (TCA) cycle equally through pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC). PDH-dependent metabolism generates NADH to increase mitochondrial membrane potential and ATP production. A rise in the ATP/ADP ratio closes ATP-sensitive potassium (KATP) channels inducing membrane depolarization, Ca2+-influx through voltage-gated channels, and insulin secretion. However, NADH via the electron transport chain also generates reactive oxygen species, which is believed to be a root cause of β-cell failure. In contrast, NADPH is generated by PC-dependent metabolism while supporting anaplerosis (refilling) of TCA cycle intermediates and citrate/isocitrate cycling (15). NADPH provides reducing equivalents to long-chain fatty acid synthesis and is postulated to modulate NADH-driven (KATP-dependent) insulin secretion (14). NADPH also provides electrons to the glutathione redox system, which protects β-cells by scavenging reactive oxygen species. Therefore, metabolism through PDH (NADH-dependent) and PC (NADPH-dependent) differentially impacts glucose-stimulated insulin secretion and β-cell survival. In this study, we use autofluorescence imaging of NAD(P)H and mitochondrial NADH in living islets to infer FGF21-induced changes in PDH- and PC-dependent metabolism (16–18).
β-cells initially show significantly greater glucose-stimulated insulin secretion in the presence of fatty acids due to large metabolic flux and glucose-stimulated reduction in fatty acid oxidation. Glucose stimulates efflux of citrate from mitochondria, which is converted to malonyl CoA to both inhibit fatty acid transport through carnitine palmitoyltransferase-1 (CPT1) and act as a substrate for long-chain fatty acid synthesis. However, chronic inhibition of fatty acid oxidation in the presence of fatty acids is not handled well by β-cells and eventually results in dissipation of the glucose-stimulated response. FGF21 has been shown to protect ex vivo pancreatic islets from palmitate-induced toxicity through ERK1/2 and Akt-dependent signaling mechanisms (19). Due to the link between β-cell metabolism and toxicity, we hypothesized that FGF21 reduces the toxic effects of excess palmitate by modulating glucose-stimulated PDH metabolism. We therefore confirmed KLB expression in islets and further examined the effect of FGF21 on glucose- and palmitate-induced metabolism. Furthermore, these studies demonstrate the use of autofluorescence imaging and microfluidic device trapping to explore islet metabolism in a limited amount of live tissue with high spatial and temporal resolution.
Materials and Methods
Pancreatic islet isolation and tissue culture
Animal procedures were approved by the Animal Care Committee of the University Health Network, Toronto, Ontario, Canada in accordance with the policies and guidelines of the Canadian Council on Animal Care (Animal Use Protocol #1531). Pancreatic islets were isolated from 8- to 12-week-old C57BL6 male mice by using collagenase digestion (Roche, Mississauga, Canada) (20,21). Islets were subsequently cultured in full RPMI 1640 medium supplemented with 11 mM glucose, 10% fetal bovine serum, and 5 U/ml penicillin-streptomycin.
Palmitate preparation
Palmitic acid (Sigma-Aldrich Canada Ltd., Oakville, Canada) was dissolved to palmitate in 0.1 M NaOH at 70°C. Fatty acid free-bovine serum albumin (BSA) (BioShop, Burlington, Canada) was dissolved in ddH2O by shaking at 4°C. Palmitate was subsequently conjugated to fatty acid free-BSA by mixing at a ratio of 1 mM palmitate:1% BSA at 60°C. The final BSA-conjugated palmitate solution was diluted to 0.4 mM in subsequent preparations of either standard islet culture media or imaging media (125 mM NaCl, 5.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, and the indicated glucose concentration, pH 7.4).
Polymerase chain reaction analysis
Oligonucleotide primers were designed to amplify the KLB cDNA sequence (N-terminal sequence from aa119 using 5′-ACCTGTGCCAAACCCATG-3′ and 5′-GCTGTCTGCGTTTGTTCTG-3′, or the C-terminal sequence from aa339 using 5′-AGATGAGTGGTTTCTTCTC-3′ and 5′-TCCATCGGTTATGAAGGAAT-3′). The master mix (total volume 25 μl) was composed of 1 μl mouse islet cDNA (10 ng/μl), 12.5 μl GoTaq Green Mixture, 9.5 μl nuclease free water, and 1 μl of each primer (10 μM final concentration). An MJ Research PTC-200 Peltier Thermal Cycler was used with the following cycling parameters: 94°C for 30 s, 52°C for 1 min, and 72°C for 1 min (30 cycles). The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GADPH) was amplified as a positive control using identical cycling parameters (5′-ATCGAGCTCATCCCATCACCATCTTCCAGG-3′ and 5′-ACATCTAGAGCCATCACGCCACAGTTTCCC-3′), and all reactions included nuclease-free water as a negative control.
Immunofluorescent detection of KLB
Freshly isolated islets were dissociated to single cells using Accutase as described previously (13). Single cell populations were allowed to adhere to poly-L-lysine-coated Labtech dishes for 2 h. Dispersed cells and intact islets were subsequently fixed with 2% paraformaldehyde and nonspecifically blocked in phosphate buffered saline containing 5% normal donkey serum and 0.1% Triton X-100 (2 h; room temperature (RT)). The dispersed cells were incubated with goat antimouse KLB antibody (AF2619, R&D Systems, Minneapolis, MN, 1:500, 4°C overnight) and donkey antigoat Alexa568 antibody (Invitrogen, Grand Island, NY, 1:1000, RT for 1 h), and subsequently with guinea pig antiinsulin antibody (Dako, Glostrup, Denmark, 1:500, RT for 1 h) and goat antiguinea pig Alexa 488 antibody (Invitrogen, 1:1000, RT for 1h). Islets were incubated with goat antimouse KLB antibody (4°C overnight) and donkey antigoat Alexa 568 antibody (RT for 1 h). Whole islets were imaged as 3–5 z-sections using the 20× 0.95 NA lens and 543-nm excitation of a Zeiss LSM710 confocal to create extended focus maximum projection images.
Western immunoblotting
Purified islets in culture (P35 dishes) were stimulated with FGF21 (100 ng/ml) for 48 h. Islets were subsequently handpicked into microfuge tubes containing islet media and collected by centrifugation (3000 rpm; 3 min). Islets were resuspended in lysis buffer (1% Triton X-100, 100 mM NaCl, 50m M HEPES, 5% glycerol, 1 mM sodium vanadate, and protease inhibitor mix (Roche); 1 μl/islet) and lysed on ice for 45 min. Whole-islet protein lysates (20–80 islets/lane) were separated by 8% SDS-PAGE and transferred to Trans-Blot nitrocellulose membranes (Bio-Rad, Hercules, CA). Membranes were blocked by incubating with 5% nonfat dry milk powder in Tris-buffered saline-Tween 20 (TBS-T) for 1 h at RT. Proteins of interest were detected by overnight incubation at 4°C in 5% BSA/TBS-T containing one of the following primary antibodies: acetyl-CoA carboxylase (ACC) (Cell Signaling Technology (CST), Danvers, MA, 1:1000) or β-actin ((CST); 1:1000). Blots were subsequently incubated with antirabbit peroxidase-conjugated antibody ((CST), 1:2000) diluted in 5% milk/TBS-T (45min RT), and proteins of interest were detected by enhanced chemiluminescence (Pierce, Thermo Fisher, Rockford, IL).
Redox autofluorescence imaging
Pancreatic islets were loaded into microfluidic devices before autofluorescence imaging as described previously (16,17,22). Briefly, NAD(P)H imaging was performed using a 705 nm Ti:Saph laser attenuated to ∼3 mW (Coherent, Santa Clara, CA), and the 40× 1.3 NA oil immersion objective lens of a Zeiss LSM710 microscope. Epifluorescence was directed through a custom-built infrared-blocked band pass filter (385–550 nm, Chroma, Bellows Falls, VT) to an external nondescanned detector (16,17). All autofluorescence images were collected ∼20 μm into the tissue to focus on the β-cells inside the core of the islet. NAD(P)H autofluorescence is from the combined signal of NADH and NADPH, with the majority of the signal from the mitochondrial pools. Lipoamide dehydrogenase (LipDH) autofluorescence imaging was performed on the same sample using sequential 458 and 488 nm excitation, a 505 nm long pass filter, a pinhole size of 3.09 AU, and pixel dwell time of 12.6 μs (17). Changes in LipDH redox state were measured using the 458:488 nm ratio due to red shifting compared to other cellular flavoproteins and are directly proportional to mitochondrial NADH (mNADH) levels (23,24). The 458:488 nm ratio does not isolate the absolute LipDH redox state, but rather can be used to measure relative changes in the redox state. We have reported here the LipDH(mNADH) responses (458:488 nm ratio) indexed to values achieved using pharmacological treatments that maximize and minimize mitochondrial NADH. Islets were routinely imaged 3 min posttreatment with 2 μM FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) and the resulting 458:488 nm ratio was defined as the minimum (0%) LipDH(mNADH) redox index. Islets were also routinely imaged 5 min posttreatment with 3 mM NaCN + 20 mM glucose and the resulting 458:488 nm ratio was defined as the maximum (100%) LipDH(mNADH) redox index.
Image analysis
The mean intensity of islets in each image (NAD(P)H (705 nm), LipDH (458 and 588 nm) was obtained from 20 small circular regions of interest that were randomly selected while avoiding nuclear regions, saturated pixels, and nonresponsive lipofuscin deposits. Five regions distant to the islet were also measured to obtain the average background intensity in each image.
Insulin secretion
Islets (20–40) were cultured as indicated and then handpicked into microfuge tubes for equilibration in 2 mM glucose-imaging buffer at 37°C. The islets were subsequently stimulated as indicated for 40 min intervals. Supernatant was collected from microtubes before addition of the succeeding stimulation. After collection of the final set of supernatants, total islet insulin content was released by 1% Triton X-100. Fractional total insulin was quantified using a sandwich insulin ELISA assay (Millipore, Billerica, MA).
Rhodamine123 imaging
Islets were suspended in 2 mM glucose-imaging buffer and incubated in the presence of Rh123 (10 μg/ml; 30 min at 37°C). Islets were subsequently imaged using a Zeiss LSM710 confocal using the 514-nm laser line, 525–655-nm bandpass filter, pinhole size of 1.73 AU, and a pixel dwell time of 12.6 μs.
Data analysis
Data are expressed as mean ± S.E.M. All statistical calculations were performed with two-tailed ANOVA and the Tukey post hoc test.
Results
Pancreatic islet KLB expression and FG21-responses
We have previously shown that murine β-cells express active fibroblast growth factor receptor-1 (FGFR1), part of the required FGF21 signaling complex (13). To determine whether murine islets express KLB, the recognized coreceptor for FGF21, we examined islet mRNA transcript expression using oligonucleotide primers specific to the N- and C-terminal ends of KLB (Fig. 1 A). We further examined KLB protein expression by immunofluorescence in isolated islets and islets dispersed to single cells (Fig. 1 B). These data show significant KLB expression in islets and more specifically in insulin-positive β-cells. We also observed a fraction of KLB-positive cells that were insulin negative (red cells that were not green) suggesting either the existence of another population of KLB-positive islet cells or contamination from exocrine cells. Combined, these data reveal that murine islet β-cells express detectable levels of KLB mRNA and protein. Previous studies have shown that FGF21-activity regulates expression of ACC in the liver and WAT. We therefore examined ACC expression in FGF21-treated islets (100 ng/ml) by Western immunoblot and observed a significant decrease (∼50%) in islet ACC protein expression levels compared to controls (Fig. 1, C and D). Overall, these data show that murine islets express KLB coreceptor and are responsive to FGF21. The reduction in islet ACC expression is consistent with responses observed in other tissues, and suggests that FGF21 modulates fatty acid metabolism in islets (8,25–27).
Figure 1.

KLB expression and FGF21-dependent responses in pancreatic islets. (A) The cDNA from two separate mouse islet preparations (Islets 1 and 2) were amplified using oligonucleotide primers designed to recognize the N- (KLB-front) and C-terminal (KLB-end) ends of KLB. GAPDH cDNA was amplified to ensure sample integrity, and water was included as a no-DNA negative control. (B) Pancreatic islets were fixed and immunofluorescently labeled using an antibody specific for the extracellular domain of KLB (left panels). Dispersed inlet cells were also immunofluorescently labeled with both KLB (red) and insulin (green) (right panels). Immunostaining in the absence of primary antibody (No Primary) was included as a negative control. Scale bar represents 50 μm. (C) A representative Western immunoblot reveals a reduction in mouse islet ACC protein level when incubated in the presence of FGF21 (48 h; 100 ng/ml). Membranes were stripped and reprobed for β-actin as a loading control. (D) The summarized fold-ACC response normalized to β-actin for control and FGF21-treated islets. Data shown represent the mean ± S.E.M. for islets from five independently assayed and treated mice. (∗P < 0.05).
Autofluorescence imaging of microfluidic device-trapped pancreatic islets
Pancreatic islets isolated from animals are of limited size and quantity, which increases the complexity of traditional biochemical assays. To quantitatively measure the cellular redox state in a limited amount of tissue with high temporal resolution, we used autofluorescence imaging of living pancreatic islets trapped in a microfluidic device (Fig. 2). A custom-built microfluidic device allowed the introduction of islets via inlet tubing (Fig. 2 A). Islets brought into the 100 μm tall × 600 μm wide main channel were stopped from moving completely through the device by a dam wall. In this setup, the islets were held stationary against the glass coverslip for optimal subcellular resolution imaging. The islets were subsequently treated with nutrients and/or pharmacological agents by simply changing the reservoir well, and the responses imaged in real time on an inverted confocal microscope. In these studies, we routinely imaged the autofluorescence of living pancreatic islets using two-photon 710-nm excitation of NAD(P)H and confocal 458- and 488-nm excitation of flavoproteins (Fig. 2 B). Islets were initially treated with 2 mM glucose (Fig. 2 B; top panels) to establish a low redox potential, followed by exposure to sodium cyanide to increase cellular redox potential (Fig. 2 B; bottom panels). A significant rise in NAD(P)H intensity (two-photon 710 nm) and drop in flavoprotein intensity (458- and 488-nm images) by sodium cyanide was distinctly evident by imaging. We have previously shown that the ratio of the 458- and 488-nm images (458:488 nm ratio) quantitatively isolates the redox responses of LipDH, a flavoprotein in equilibrium with the mitochondrial NADH (mNADH) pool (17). Treating islets with sodium cyanide or FCCP maximally reduces or oxidizes the cellular redox state, respectively (Fig. 2 C). The data shown in Fig. 2 C reveal consistent changes in the fold NAD(P)H responses (Fig. 2 C; left graph) and 458:488 nm image ratio (Fig. 2 C; right graph). These pharmacological treatments were used to subsequently index the nutrient-induced responses of LipDH(mNADH) (Fig. 2 C; right axis). These data set the dynamic range for the LipDH(mNADH) index of our microscope, and further show the redox imaging and microfluidic methods used to tease apart metabolic responses of islets.
Figure 2.
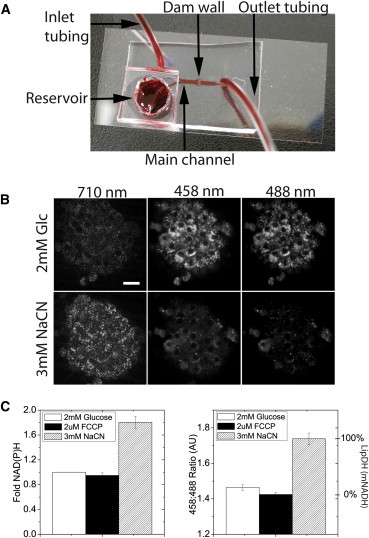
Autofluorescence imaging of living pancreatic islets in a microfluidic device. Pancreatic islets were routinely brought into microfluidic devices before imaging the two-photon NAD(P)H and confocal LipDH(mNADH) responses. (A) A representative microfluidic device is shown flooded with food coloring to highlight the inlet and outlet tubing, main channel, dam wall, and reservoir. The device is composed of polydimethylsiloxane bonded to a glass coverslip, which provides an optimized optical window. Inlet- and outlet-tubing were used to bring islets into the microfluidic device and for removing effluent, respectively. Once 3 to 10 islets were loaded into the device, the inlet tubing was capped and flow was initiated from the on-chip reservoir to the outlet tubing. Islets captured in the device were held against the coverslip while media was allowed to flow past. (B) A representative islet stimulated with 2 mM glucose was imaged using two-photon 710 nm excitation and confocal 488 and 488 nm excitation (top row). This same islet was subsequently imaged after treatment with 3 mM sodium cyanide for 5 min (bottom row). Scale bar represents 20 μm. (C) The summarized NAD(P)H and 458:488 nm image ratio responses to 2 mM glucose, 3 mM cyanide, and 2 μM FCCP. The NAD(P)H data is plotted as the fold-response to 2 mM glucose. The 458:488 nm ratio plotted as arbitrary units (left axis) is a readout of the LipDH(mNADH) redox state. These pharmacological treatments induce maximal and minimal cellular redox state and were subsequently used to index the LipDH or mitochondrial NADH response (LipDH(mNADH)) (right axis). Data represent the pooled response from 13 and 30 islets harvested from 2 and 3 mice on separate days for the sodium cyanide and FCCP data, respectively (∗P < 0.01).
Glucose-stimulated NAD(P)H responses
A previous study showed FGF21 protects islets from palmitate-induced toxicity (19). We postulated that FGF21 protects islets by modulating glucose-stimulated metabolism. To quantitatively measure the effect of FGF21 on islet metabolism, we imaged the glucose-stimulated NAD(P)H response using two-photon microscopy (16,28) (Fig. 3). Islets showed a significant glucose-stimulated increase in NAD(P)H intensity with exposure to increasing glucose levels (Fig. 3 B). By quantifying NAD(P)H intensity in a number of islets, we observed similar responses from all islets examined over most of the glucose dose-response examined. However, in the presence of excess glucose (20 mM), FGF21-treated islets showed a statistically smaller NAD(P)H response compared to controls (Fig. 3 B). These data show that FGF21 does not dramatically modify islet glucose-stimulated metabolism with only moderate differences revealed at supraphysiological levels of glucose. Chronic fatty acid stimulation induces β-cell stress. To measure the effect of chronic fatty acid-induced stress on islet function, we further explored maintenance of glucose-stimulated NAD(P)H responses after culture for an additional 24 h in media containing palmitate (0.4 mM) in the presence and absence of FGF21 (Fig. 3 C). A more dynamic NAD(P)H response was observed in FGF21-treated islets compared to controls at all concentrations of glucose examined, consistent with a previous study (19) and indicative of improved islet survival.
Figure 3.
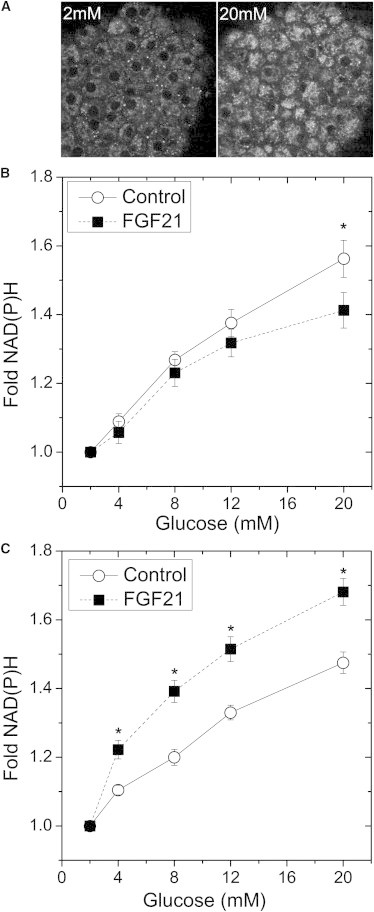
Glucose-stimulated NAD(P)H responses. Pancreatic islets were cultured for 48 h in full RPMI media 1640 at 11 mM glucose in the absence (control) and presence (FGF21) of FGF21. Palmitate (0.4 mM) was added to a separate batch of islets after 24 h to induce palmitate toxicity. All islets were subsequently incubated in imaging media containing 2 mM glucose at 37°C (minimum 30 min), and loaded into microfluidic devices on the microscope stage to image the two-photon NAD(P)H response to varied glucose levels (as indicated). (A) Representative images of NAD(P)H autofluorescence for a device-immobilized islet at 2 and 20 mM glucose. (B) The summarized mitochondrial NAD(P)H intensities throughout the glucose dose-response from islets cultured in the absence of palmitate. Data represent the pooled response from 20 to 30 islets harvested from 3 mice on separate days. (C) The summarized mitochondrial NAD(P)H intensities throughout the glucose dose-response from islets cultured for 24 h in the presence of palmitate (0.4 mM). Data represent the pooled response from 15 to 20 islets harvested from 3 mice on separate days. (∗P < 0.05).
Glucose-stimulated NAD(P)H and LipDH(mNADH) responses
Metabolism of glycolysis-derived pyruvate can lead to PDH-stimulated production of NADH and PC-stimulated production of NADPH. To determine whether FGF21 modulates glucose-stimulated PDH-dependent (NADH) and PC-dependent (NADPH) metabolism, we compared the two-photon NAD(P)H and mitochondrial NADH responses (Fig. 4). LipDH is in direct equilibrium with mitochondrial NADH, therefore the LipDH(mNADH) redox index measures changes in the mitochondrial NADH signal for comparison to the two-photon NAD(P)H response (17,23,24). Islets were cultured in FGF21 (100 ng/ml) for 48 h before imaging responses to glucose, glucose in the presence of palmitate, or citrate. No difference was observed in the two-photon NAD(P)H response of control and FGF21-treated islets at 10 mM glucose. However, we again observed a slightly smaller NAD(P)H response in FGF21-treated islets exposed to 20 mM glucose compared to controls (Fig. 4 A). In contrast, the LipDH(mNADH) redox index was no different at all concentrations of glucose examined (Fig. 4 B). These imaging data are therefore consistent with no difference in the glucose-stimulated mitochondrial NADH (PDH-dependent) response of FGF21-treated islets, and only a small decrease in the NADPH (PC-dependent) response observed in the presence of excess glucose. Furthermore, FGF21 does not induce changes in the mRNA expression of proteins related to mitochondrial uncoupling or citrate cycling suggesting that the autofluorescence responses are due to metabolic output rather than mitochondrial energetics (see the Supporting Material, Fig. S1).
Figure 4.
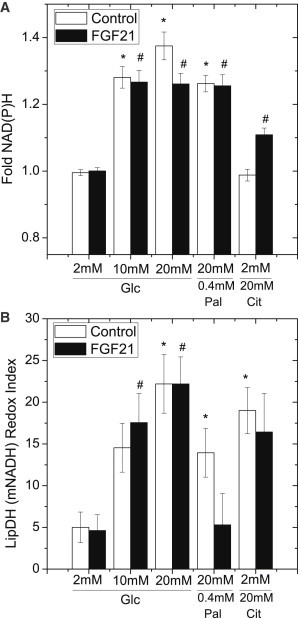
Glucose-stimulated NAD(P)H and LipDH(mNADH) responses. Pancreatic islets were cultured for 48 h in full RPMI media 1640 at 11 mM glucose in the absence (control) and presence (FGF21) of FGF21. Islets were subsequently incubated in imaging media containing 2 mM glucose or 2 mM glucose + 0.4 mM palmitate (37°C; minimum of 30 min) followed by loading into microfluidic devices on the microscope stage to image the glucose-stimulated NAD(P)H and LipDH (mNADH) redox index responses. All responses were measured at least 20 min after subsequent addition of the indicated substrates. (A) The summarized fold NAD(P)H response of mitochondrial regions from islets exposed to glucose (2, 10, and 20 mM), high glucose (20 mM) in the presence of palmitate, and low glucose (2 mM) in the presence of citrate. (B) The summarized LipDH(mNADH) redox index for the same islets shown in (A). The LipDH response (458:488 nm intensity ratio) was indexed to pharmacological treatments that minimize and maximize mitochondrial NADH reduction. Data were collected from control islets (n = 21 (glucose), 14 (palmitate), and 21 (citrate)) and FGF21-treated islets (n = 28 (glucose), 14 (palmitate) 22 (citrate)) harvested from mice on 3 separate days. (∗ and # indicate P < 0.05 compared to 2 mM and 2mM + FGF21, respectively).
In a reaction rate limited by ACC, citrate from the TCA cycle is converted to malonyl-CoA to act as an inhibitor of fatty acid oxidation. Reduced expression of ACC protein in FGF21-treated islets therefore suggested modulation of glucose-stimulated CPT1-closure and fatty acid oxidation. To measure the effect of fatty acids on glucose-induced metabolism, we measured glucose-stimulated responses in the presence of palmitate (Fig. 4). Glucose stimulated a significant increase in aggregate NAD(P)H in both control and FGF21-treated islet islets in the presence of palmitate (Fig. 4 A). This response was accompanied by a small rise in the LipDH(mNADH) redox index in control islets that was unexpectedly absent in FGF21-treated islets (Fig. 4 B). These data are consistent with FGF21-treated islets having a limited mitochondrial NADH response to glucose in the presence of fatty acids, consistent with reduced PDH activity in FGF21-treated islets.
Independent of ACC, citrate is also metabolized to α-ketoglutarate in the cytoplasm producing NADPH. To assess changes in islet metabolism upstream of ACC, we further examined the NAD(P)H and LipDH(mNADH) responses of islets in response to 20 mM citrate (Fig. 4). Citrate caused no change in NAD(P)H intensity in control islets, but a significant increase in the intensity of FGF21-treated islets (Fig. 4 A). By comparing the two-photon NAD(P)H and LipDH(mNADH) responses (Fig. 4 B), citrate induced greater production of NADPH in FGF21-treated islets than in control islets (a rise in NAD(P)H not observed in control islets that is again accompanied by similar mitochondrial NADH responses). These data are consistent with FGF21 altering islet metabolism to favor NADPH production from citrate, and suggest citrate cycling rather than metabolism to malonyl-CoA. These data also suggest that changes in islet metabolism induced by FGF21 go beyond simply regulating ACC expression and will need to be explored further to reveal the entire metabolic outcome and mechanism.
Mitochondrial membrane potential and insulin secretion
Redox imaging suggested that palmitate did not potentiate glucose-induced mitochondrial NADH response in FGF21-treated islets. However, autofluorescence imaging provides an indirect measure of the metabolite-stimulated responses. We therefore aimed to see whether these changes in redox response translated to similar downstream responses in mitochondrial energetics and insulin secretion (Fig. 5). To determine whether the LipDH(mNADH) responses translated to mitochondrial energetics, Rhodamine 123 (Rh123) intensity was imaged and quantified as a measure of islet mitochondrial membrane potential (Fig. 5 A). Increased levels of glucose stimulated a decrease in Rh123 intensity that was similar in control and FGF21-treated islets and reflects a comparable increase in mitochondrial membrane potential in all islets (Fig. 5, A and B). However, FGF21-treated islets showed a significantly smaller glucose-stimulated decrease in Rh123 intensity compared to controls in the presence of palmitate (Fig. 5 B) that was consequently ablated by the coaddition of etomoxir (an inhibitor of CPT1-dependent fatty acid transport). These data suggest that the reduced mitochondrial NADH and membrane potential responses of FGF21-treated islets are due to fatty acid transport and oxidation. To determine whether the LipDH(mNADH) and mitochondrial membrane responses translated to downstream function, the fractional insulin secretion of islets in response to glucose in the presence and absence of palmitate was examined (Fig. 5 C). Consistent with observed LipDH(mNADH) and mitochondrial membrane potential responses, no difference in glucose-stimulated insulin secretion was evident between control and FGF21-treated islets. Similarly, no difference was observed in the palmitate-induced insulin response between control and FGF21-treated islets. However, consistent with previous studies, glucose-stimulated insulin secretion of control islets was significantly greater in the presence of palmitate as compared to glucose-alone. In contrast, FGF21-treated islets stimulated with glucose secreted similar levels of insulin regardless of the presence of palmitate, and significantly less than control islets in palmitate. Overall, these data reveal that FGF21-treated islets exhibit glucose-stimulated responses similar to control islets; however, FGF21-treated islets do not exhibit palmitate-induced potentiation of insulin secretion further confirming the NAD(P)H and LipDH(mNADH) imaging data.
Figure 5.
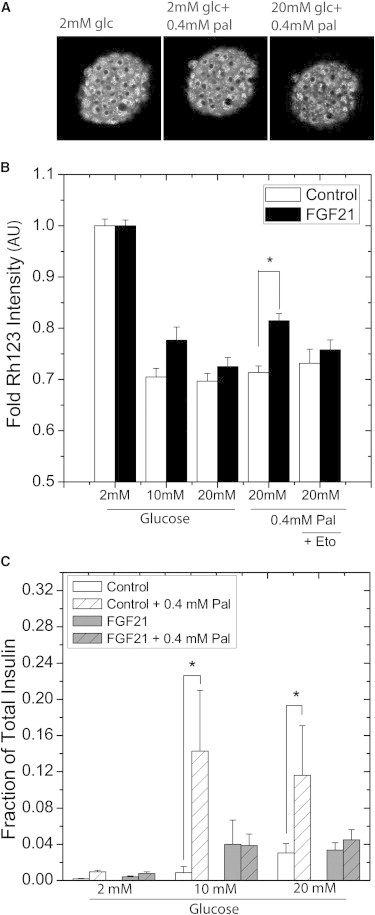
Glucose-stimulated changes in mitochondrial membrane potential and insulin secretion. Pancreatic islets were cultured for 48 h in full RPMI media 1640 supplemented with 11 mM glucose in the absence (control) and presence (FGF21) of FGF21. Islets were subsequently used for measurement of mitochondrial membrane potential or measuring glucose stimulated insulin secretion. (A) Islets were incubated in imaging media containing 2 mM glucose (37°C for 1 h) followed by addition of Rh123 (10 μg/ml). Labeled islets were loaded into microfluidic devices on the microscope stage for imaging. Representative images are shown of the same Rh123-labeled control islet subsequently stimulated with 2 mM glucose (5 min), 2 mM glucose + 0.4 mM palmitate (25 min), and 20 mM glucose + 0.4 mM palmitate (5 min). (B) The summarized data from islets in glucose alone (2, 10, and 20 mM), and glucose in the presence of 0.4 mM palmitate (20 mM glucose, and 20 mM glucose + 10μM etomoxir) is reported as fold Rh123 intensity (arbitrary units, AU) relative to 2 mM glucose. Data was collected from control (n = 20–25) and FGF21-treated (n = 20–25) islets harvested from mice on 3 separate days. (∗P < 0.05). (C) Islets were further incubated in imaging media containing 2 mM glucose (37°C for 30 min) and effluent was sequentially collected following 1 h incubations with the indicated nutrients. Sample supernatant (effluent) was carefully collected and assayed. The fractional insulin response from control and FGF21-treated islets in response to glucose alone (2, 10, and 20 mM glucose) and glucose in the presence of 0.4 mM palmitate (Control + 0.4 mM palmitate and FGF21 + 0.4 mM palmitate) are shown. Data were normalized to total insulin content collected post islet permeabilization with 1% triton X-100. Data shown are summarized from the islets harvested from 4 to 8 mice on independent days. (∗P < 0.05).
Discussion
Recombinant FGF21 is actively being explored as a therapeutic for type 2 diabetes due to potent effects in lowering the blood glucose and lipids in mice. The majority of studies to date explore mechanisms of action in liver and WAT using classical biochemical methods. However, the effect of FGF21 on pancreatic islets has been given significantly less attention particularly considering a previous report showing increased islet survival of palmitate-induced toxicity (19). This may in part be due to the inherent difficulty of working with pancreatic islets due to limited size and availability. The current study explored the basic mechanisms of FGF21-activity by using a combination of microfluidic device capture and quantitative autofluorescence imaging. Our goal was to determine whether FGF21 treatment of islets modulated islet metabolism, or more specifically, reduced PDH-dependent metabolism due to a direct link between NADH production and cell stress. Our unique combination of methods allowed us to gain mechanistic insight into the interaction of glucose and fatty acid mitochondrial metabolism in a limited amount of living tissue.
FGFR1c and KLB form the cognate receptor complex for FGF21 (11). We previously demonstrated FGFR1 expression and activity in mouse islets (13). Our data now confirms coexpression of KLB in islets consistent with the endocrine pancreas having the capacity to be FGF21-responsive. FGF21-treated islets also showed a decrease in ACC expression similar to responses observed in liver and WAT (25,27). ACC is the rate limiting enzyme for converting citrate into malonyl-CoA, a substrate for long-chain fatty acid synthesis and potent inhibitor of mitochondrial transport and oxidation of fatty acids. These data were collected to confirm a response to FGF21 in pancreatic islets. Glucolipotoxicity models suggest that long-chain fatty acid synthesis induces β-cell stress and that enhancing fatty acid oxidation is a mechanism to reduce this stress (29,30). Consistent with a role for FGF21 in stimulating islet survival, we also observed a more dynamic glucose-stimulated NAD(P)H response in treated islets cultured for 24 h in palmitate. We therefore postulate that FGF21 signaling decreases islet ACC expression as one compensatory mechanism to reduce palmitate-induced toxicity. Further studies will be required to obtain a more global picture of the metabolic changes induced by FGF21 in pancreatic islet β-cells.
Using autofluorescence imaging of living islets, we subsequently examined the effect of FGF21 on the glucose-stimulated NAD(P)H and mitochondrial NADH responses. FGF21-treated islets showed only moderate differences in glucose-stimulated NAD(P)H and mitochondrial NADH responses compared to controls. In contrast, more significant differences were revealed in the presence of palmitate where FGF21-treated islets showed reduced glucose-stimulated mitochondrial NADH. Of importance, we correlated this autofluorescence data with complementary methods including measuring decreased mitochondrial membrane potential and insulin secretion. These readouts are all consistent with reduced PDH-dependent production of NADH in FGF21-treated islets when in the presence of palmitate. Critically, mitochondrial membrane potential was restored in FGF21-treated islets by the CPT1 inhibitor etomoxir suggesting PDH-dependent NADH is somehow limited by fatty acid oxidation. A more direct measurement of fatty acid oxidation will be needed to confirm these indirect data.
Similar to the Randle model, our data suggest that continued fatty acid oxidation in FGF21-treated islets inhibits glucose oxidation (31). An essential component of the Randle model is that a fatty acid-induced accumulation of acetyl CoA in the fasted state inhibits PDH activity through phosphorylation by PDH kinase (31). Our data are consistent with similar regulation of PDH-activity; however, in contrast to Randle’s work, our observations were made in the presence of excess glucose (the fed state). Excess glucose stimulates mitochondrial efflux of citrate to subsequently decrease fatty acid oxidation by closure of CPT1 by ACC-dependent production of malonyl CoA. We postulate that continued transport and oxidation of fatty acids in FGF21-treated islets, even in the presence of excess glucose, limits PDH-dependent metabolism of glycolysis-derived pyruvate. In effect, FGF21-treated islets maintain PDH activity in a fasted state despite the presence of glucose and palmitate. We further postulate that continued fatty acid oxidation in the presence of glucose lessens toxicity associated with glucose oxidation through PDH. Future studies are required to explore the regulation of PDH activity by citrate/malonyl CoA production and CPT1 transport, and will critically depend on the temporal resolution provided by autofluorescence imaging of islets in a microfluidic device.
The reduction in insulin secretion from FGF21-treated islets is a phenotype generally not considered beneficial in type 2 diabetes. However, reducing glucose-stimulated mitochondrial NADH and membrane potential would also contribute to the long-term survival of islets (19). Mitochondrial NADH induces reactive oxygen species (ROS) due to leakage of electrons at complex I and III of the electron transport chain. This leakage is particularly prevalent during state IV respiration, defined as a state of excess metabolite (high NADH) and minimal ADP (32). Limiting mitochondrial NADH would therefore be expected to decrease ROS production. We observed a significant glucose-stimulated NADPH response in FGF21-treated islets. NADPH provides reducing potential to glutathione and thereby benefits ROS scavenging. Mitochondrial ROS has recently been implicated as an amplifying mechanism for insulin secretion and may also in part account for the reduction in glucose-stimulated insulin secretion observed in FGF21-treated islets (33,34). Overall, a reduction in mitochondrial energetics would limit ROS production and insulin secretion, whereas an increase in NADPH would reduce the effect of ROS and ultimately improve long-term survival.
Due to the reduced mitochondrial membrane potential and insulin secretion found in FGF21-treated islets, we also explored UCP2 and NNT expression as an alternative mechanism of uncoupling mitochondrial energetics rather than reduced PDH-dependent production of NADH (see the Supporting Material). UCP2 has been shown to have a protective role in islets, although this role is still debated (35,36). NNT converts NADH to NADPH resulting in mild uncoupling and has also been shown to play a protective role in β-cells (37–39). Our data showed no change in mRNA expression of UCP2 and NNT, inconsistent with increasing mitochondrial uncoupling in the presence of palmitate. However, our data do not discount posttranslational regulation of the activity of these proteins. A number of alternative mechanisms also exist for uncoupling mitochondria and reducing mitochondrial NADH including the mitochondrial permeability transition pore (MPTP). The MPTP increases permeability to ions such as Ca2+ resulting in depletion of mitochondrial membrane potential. The MPTP is generally considered to have a role in pathophysiology (40); however, MPTP has also been shown to act in a low conductance state triggered by stress (41). A varied number of mechanisms and techniques will need to be considered to determine the effective role of uncoupling rather than PDH-dependent metabolism in controlling mitochondrial energetics in FGF21-treated islets.
In summary, we have used a unique strategy to measure FGF21-stimulated changes in islet metabolism by combining autofluorescence imaging with microfluidic capture. These studies revealed changes in glucose-stimulated NAD(P)H and mitochondrial NADH when concurrently exposed to palmitate. Our data show pancreatic islets treated with FGF21 modify glucose-stimulated responses only at supraphysiological levels or in the presence of excess fatty acids. Using multiple methods to follow up on our initial discovery, we further revealed that FGF21-treated islets do not show palmitate-induced potentiation of glucose-stimulated mitochondrial energetics and insulin secretion. All changes induced by FGF21 to islet metabolism were consistent with reduced PDH-dependent metabolism stimulated by fatty acid stimulation; however, further studies are required to validate this conclusion. Nevertheless, to our knowledge, these studies suggest a novel paradigm that enhancing fatty acid oxidation may be protective against glucolipotoxicity due to inhibition of PDH-dependent metabolism.
Acknowledgments
Funding was provided by the Canadian Institutes of Health Research (CIHR) (NMD106997) and the Heart and Stroke Foundation of Ontario (HSFO) (NA6591). The microscope used in these studies was purchased through a Canada Foundation Innovation (CFI) Leaders Opportunity Fund (18301). These funding sources had no role in the study design or other aspects of the manuscript.
Supporting Material
References
- 1.Itoh N., Ornitz D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Ornitz D.M., Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:3005. doi: 10.1186/gb-2001-2-3-reviews3005. REVIEWS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kharitonenkov A. FGFs and metabolism. Curr. Opin. Pharmacol. 2009;9:805–810. doi: 10.1016/j.coph.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Badman M.K., Pissios P., Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Inagaki T., Dutchak P., Kliewer S.A. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Kharitonenkov A., Shiyanova T.L., Shanafelt A.B. FGF-21 as a novel metabolic regulator. J. Clin. Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kharitonenkov A., Shanafelt A.B. Fibroblast growth factor-21 as a therapeutic agent for metabolic diseases. BioDrugs. 2008;22:37–44. doi: 10.2165/00063030-200822010-00004. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa Y., Kurosu H., Kuro-o M. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl. Acad. Sci. USA. 2007;104:7432–7437. doi: 10.1073/pnas.0701600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki M., Uehara Y., Imamura T. betaKlotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol. Endocrinol. 2008;22:1006–1014. doi: 10.1210/me.2007-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ming A.Y., Yoo E., Rocheleau J.V. Dynamics and Distribution of Klothoβ (KLB) and fibroblast growth factor receptor-1 (FGFR1) in living cells reveal the fibroblast growth factor-21 (FGF21)-induced receptor complex. J. Biol. Chem. 2012;287:19997–20006. doi: 10.1074/jbc.M111.325670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurosu H., Choi M., Kuro-o M. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 2007;282:26687–26695. doi: 10.1074/jbc.M704165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito S., Kinoshita S., Nabeshima Y.I. Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein. Mech. Dev. 2000;98:115–119. doi: 10.1016/s0925-4773(00)00439-1. [DOI] [PubMed] [Google Scholar]
- 13.Kilkenny D.M., Rocheleau J.V. Fibroblast growth factor receptor-1 signaling in pancreatic islet beta-cells is modulated by the extracellular matrix. Mol. Endocrinol. 2008;22:196–205. doi: 10.1210/me.2007-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jitrapakdee S., Wutthisathapornchai A., MacDonald M.J. Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia. 2010;53:1019–1032. doi: 10.1007/s00125-010-1685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 16.Rocheleau J.V., Head W.S., Piston D.W. Pancreatic islet beta-cells transiently metabolize pyruvate. J. Biol. Chem. 2002;277:30914–30920. doi: 10.1074/jbc.M202314200. [DOI] [PubMed] [Google Scholar]
- 17.Rocheleau J.V., Head W.S., Piston D.W. Quantitative NAD(P)H/flavoprotein autofluorescence imaging reveals metabolic mechanisms of pancreatic islet pyruvate response. J. Biol. Chem. 2004;279:31780–31787. doi: 10.1074/jbc.M314005200. [DOI] [PubMed] [Google Scholar]
- 18.Sankar K.S., Green B.J., Rocheleau J.V. Culturing pancreatic islets in microfluidic flow enhances morphology of the associated endothelial cells. PLoS ONE. 2011;6:e24904. doi: 10.1371/journal.pone.0024904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wente W., Efanov A.M., Gromada J. Fibroblast growth factor-21 improves pancreatic beta-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes. 2006;55:2470–2478. doi: 10.2337/db05-1435. [DOI] [PubMed] [Google Scholar]
- 20.Scharp D.W., Kemp C.B., Lacy P.E. The use of ficoll in the preparation of viable islets of langerhans from the rat pancreas. Transplantation. 1973;16:686–689. doi: 10.1097/00007890-197312000-00028. [DOI] [PubMed] [Google Scholar]
- 21.Stefan Y., Meda P., Orci L. Stimulation of insulin secretion reveals heterogeneity of pancreatic B cells in vivo. J. Clin. Invest. 1987;80:175–183. doi: 10.1172/JCI113045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam A.K., Silva P.N., Rocheleau J.V. Quantitative imaging of electron transfer flavoprotein autofluorescence reveals the dynamics of lipid partitioning in living pancreatic islets. Integr. Biol. (Camb) 2012;4:838–846. doi: 10.1039/c2ib20075a. [DOI] [PubMed] [Google Scholar]
- 23.Kunz W.S. Evaluation of electron-transfer flavoprotein and alpha-lipoamide dehydrogenase redox states by two-channel fluorimetry and its application to the investigation of beta-oxidation. Biochim. Biophys. Acta. 1988;932:8–16. doi: 10.1016/0005-2728(88)90134-x. [DOI] [PubMed] [Google Scholar]
- 24.Kunz W.S., Gellerich F.N. Quantification of the content of fluorescent flavoproteins in mitochondria from liver, kidney cortex, skeletal muscle, and brain. Biochem. Med. Metab. Biol. 1993;50:103–110. doi: 10.1006/bmmb.1993.1051. [DOI] [PubMed] [Google Scholar]
- 25.Coskun T., Bina H.A., Kharitonenkov A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 26.Palou M., Priego T., Picó C. Sequential changes in the expression of genes involved in lipid metabolism in adipose tissue and liver in response to fasting. Pflugers Arch. 2008;456:825–836. doi: 10.1007/s00424-008-0461-1. [DOI] [PubMed] [Google Scholar]
- 27.Xu J., Lloyd D.J., Véniant M.M. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blinova K., Levine R.L., Balaban R.S. Mitochondrial NADH fluorescence is enhanced by complex I binding. Biochemistry. 2008;47:9636–9645. doi: 10.1021/bi800307y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prentki M., Joly E., Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51(Suppl 3):S405–S413. doi: 10.2337/diabetes.51.2007.s405. [DOI] [PubMed] [Google Scholar]
- 30.Prentki M., Nolan C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Invest. 2006;116:1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Randle P.J. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 1998;14:263–283. doi: 10.1002/(sici)1099-0895(199812)14:4<263::aid-dmr233>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 32.Chance B., Williams G.R. Respiratory enzymes in oxidative phosphorylation. IV. The respiratory chain. J. Biol. Chem. 1955;217:429–438. [PubMed] [Google Scholar]
- 33.Pi J., Bai Y., Collins S. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 34.Rebelato E., Abdulkader F., Carpinelli A.R. Control of the intracellular redox state by glucose participates in the insulin secretion mechanism. PLoS ONE. 2011;6:e24507. doi: 10.1371/journal.pone.0024507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan C.B., Saleh M.C., Wheeler M.B. Uncoupling protein 2 and islet function. Diabetes. 2004;53(Suppl 1):S136–S142. doi: 10.2337/diabetes.53.2007.s136. [DOI] [PubMed] [Google Scholar]
- 36.Zhang C.Y., Baffy G., Lowell B.B. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 37.Freeman H., Shimomura K., Ashcroft F.M. Nicotinamide nucleotide transhydrogenase: a link between insulin secretion, glucose metabolism and oxidative stress. Biochem. Soc. Trans. 2006;34:806–810. doi: 10.1042/BST0340806. [DOI] [PubMed] [Google Scholar]
- 38.Freeman H., Shimomura K., Ashcroft F.M. Nicotinamide nucleotide transhydrogenase: a key role in insulin secretion. Cell Metab. 2006;3:35–45. doi: 10.1016/j.cmet.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Freeman H.C., Hugill A., Cox R.D. Deletion of nicotinamide nucleotide transhydrogenase: a new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes. 2006;55:2153–2156. doi: 10.2337/db06-0358. [DOI] [PubMed] [Google Scholar]
- 40.Lemasters J.J., Theruvath T.P., Nieminen A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zoratti M., Szabò I. The mitochondrial permeability transition. Biochim. Biophys. Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.