

Abstract
Voltage-gated sodium channels (NaVs) underlie the upstroke of the action potential in the excitable tissues of nerve and muscle. After opening, NaVs rapidly undergo inactivation, a crucial process through which sodium conductance is negatively regulated. Disruption of inactivation by inherited mutations is an established cause of lethal cardiac arrhythmia, epilepsy, or painful syndromes. Intracellular calcium ions (Ca2+) modulate sodium channel inactivation, and multiple players have been suggested in this process, including the cytoplasmic NaV C-terminal region including two EF-hands and an IQ motif, the NaV domain III-IV linker, and calmodulin. Calmodulin can bind to the IQ domain in both Ca2+-bound and Ca2+-free conditions, but only to the DIII-IV linker in a Ca2+-loaded state. The mechanism of Ca2+ regulation, and its composite effect(s) on channel gating, has been shrouded in much controversy owing to numerous apparent experimental inconsistencies. Herein, we attempt to summarize these disparate data and propose a novel, to our knowledge, physiological mechanism whereby calcium ions promote sodium current facilitation due to Ca2+ memory at high-action-potential frequencies where Ca2+ levels may accumulate. The available data suggest that this phenomenon may be disrupted in diseases where cytoplasmic calcium ion levels are chronically high and where targeted phosphorylation may decouple the Ca2+ regulatory machinery. Many NaV disease mutations associated with electrical dysfunction are located in the Ca2+-sensing machinery and misregulation of Ca2+-dependent channel modulation is likely to contribute to disease phenotypes.
Sodium Channel Architecture and Inactivation
NaVs are large ∼250-kDa membrane proteins comprised of four homologous domains (DI, DII, DIII, and DIV), each housing six α-helical transmembrane segments that form the voltage-sensing (Fig. 1 A: S1–S4, shown in gray and red) and the pore-forming (S5–S6, in blue) modules. The conductance of sodium ions through these channels is exquisitely sensitive to changes in voltage, where, upon opening, they allow the rapid inward flow of Na+ ions which act to depolarize the cellular membrane. In mammals, at least nine different NaV isoforms have been isolated (NaV1.1–1.9). Within milliseconds of channel opening, the influx of Na+ is reduced through a process known as “inactivation” (Fig. 1 B). This process is distinct from the normal “closing”, and presents a way for the channel to minimize and regulate the depolarizing signal. Channels recover rapidly from inactivation under normal conditions within milliseconds, thus allowing for Na+ influx during the next action potential. The inherent speed and efficiency of this opening/inactivating cycle is imperative for the electrical stability of the cell because small perturbations to the equilibrium among open, closed, and inactivated channel states can have devastating effects on electrical function.
Figure 1.

Sodium channel architecture, gating, and modulation by calcium. (A) Cartoon of a simplified sodium channel highlighting the four homologous domains (DI–DIV), with six transmembrane segments each (S1–S6). Voltage-sensing domain (gray, S1–S3 and red, S4); pore-forming domain (blue, S5–S6). Cytoplasmic regions implicated in Ca2+ modulation are indicated with inherited mutations (Asterisks). (B) Representative example of a rapid inward sodium current under patch-clamp conditions in response to a 15-ms depolarizing pulse from −100 mV to −20 mV, with the fast activation and inactivation indicated (arrow). (C) Steady-state inactivation curve for NaV1.5 in the presence and absence of 10 μM free Ca2+ in the patch pipette. The data were generated with the protocol shown (inset) where a cell under voltage-clamp is held at −120 mV, stepped to a variable prepulse for 500 ms (to reach a new steady-state equilibrium) before a test pulse to −20 mV is employed to assay how many are available to open.
Interestingly, NaVs can also inactivate directly from the closed conformation, as visible in the steady-state inactivation (SSI) curve (Fig. 1 C). The amount of SSI effectively controls the number of channels that are available for opening, and is directly dependent on the membrane voltage (1). In basal conditions near the resting membrane potential of excitable cells (∼−90 mV), a considerable fraction of sodium channels are closed but ready to open upon depolarization, and the other half are inactivated and therefore unavailable to contribute to action potential firing. If the SSI curve is shifted only 10 mV to the right (a depolarizing shift), the number of available channels almost doubles. Thus, seemingly small changes in the steady-state inactivation properties can have significant effects on channel availability, and therefore on the rhythm and stability of the action potential.
The precise mechanism of NaV inactivation is not known, but one possibility is that inactivation proceeds through a hinged-lid mechanism (2) whereby the ∼50 amino-acid cytoplasmic linker between domains III and IV (DIII-IV) acts as the lid to rapidly occlude the permeation pathway (3). This scheme, albeit overly simplified, is likely accompanied by complementary motions within the pore region. In addition to the DIII-IV linker and its putative binding sites in the S4–S5 linker of domains III (4) and IV (5), the inactivation complex includes the C-terminus of the channel (6,7).
Functional Effect of Ca2+ on Sodium Channel Gating
The consensus of published data supports the observation that increased local cytoplasmic Ca2+ levels into the low micromolar range results in a ∼10-mV depolarizing shift in the steady-state availability relationship for NaV1.5 (6,8–12) (Fig. 1 C). Because the resting membrane potential is midway along the falling phase of this relationship, seemingly minor shifts in the equilibrium value can substantially increase the amount of available channels to open for the next action potential. However, a variety of effects mediated by cytoplasmic calcium has been reported, including effects on inactivation rate (6) and slow inactivation (12,13). There also seem to be isoform-specific differences, where shifts in the steady-state availability curve can also be observed upon coexpressing calmodulin or addition of short peptides, but there is no consensus on these effects (13). How is it possible that so many groups have reported conflicting results?
In addition to variable effects of EGTA and BAPTA, one potential culprit is the composition of the patch pipette solutions where CsF (typically >100 mM) is often used to produce very stable whole-cell patch-clamp recording conditions. However, fluoride avidly binds Ca2+ (Ksp ∼ 3.45 × 10−11) (http://pubchem.ncbi.nlm.nih.gov), thus effectively decreasing the free Ca2+ concentrations, especially in low buffering—suggesting that Ca2+ ion regulation measured under such conditions occurs at lower levels of free [Ca2+]. Although Deschênes et al. (14) have reported no difference in modulation between CsF for CsCl, this outcome could differ among isoforms, and the precise amount of CsF used. Moreover, the regions known to support CaM binding and Ca2+ regulation, namely the DIII-IV linker and the IQ motif, are also home to phosphorylation sites (15,16), providing an intriguing form of additional regulation, but yet another confounding variable in the laboratory setting. Lastly, sodium channels expressed in different mammalian cell systems have yielded divergent results in terms of Ca2+ regulation (17).
Ca2+ regulation of sodium channels could act through a combination of mechanisms, including the direct interaction of ions with EF-hand motifs in the C-terminus of the channel, in addition to calmodulin-mediated effects. Calmodulin (CaM) is a ∼17-kDa protein that is able to bind four Ca2+ ions through EF-hand motifs and the available data support a clear role for CaM in the Ca2+-dependent modulation of sodium channel inactivation; however, considerable experimental variability exists. Coexpression of CaM has been shown to produce a Ca2+ dependent hyperpolarizing shift in the steady-state availability of skeletal muscle NaV 1.4, but not of cardiac NaV 1.5 (14). Chimeric ligation of CaM to the C-terminus of NaV1.4 suggests that a single CaM is sufficient to shift the steady-state inactivation curve (18). The addition of a CaM inhibitory peptide via the patch pipette does not affect the Ca2+-dependent shift in steady-state inactivation (10), leading some investigators to conclude that CaM is not essential to Ca2+ regulation. However, it is not known how effectively the inhibitory peptide competes with intact channels for binding to CaM, complicating the extrapolation of such results.
Calmodulin and the IQ Domain
Isoleucine-glutamine (IQ) domains are well-described motifs that were first reported for myosins as an interaction motif for CaM-like essential light chains (19) and neuromodulin (20). It is now clear that these motifs can form binding sites for CaM or CaM-like proteins, with Ca2+ dependencies and affinities differing in individual cases (21), and with a generalized IQ motif consisting of [I,L,V]QXXXRXXXX[R,K]. The C-terminal domain (CTD) of all NaV isoforms encodes an IQ-like domain, first identified in the NaV1.1–1.3 isoforms (22) (Fig. 2), and in some isoforms differs from the canonical IQ motif because it contains an extra fifth residue between the two positively charged residues of the consensus sequence. IQ motifs also occur in many other ion channels, including the closely related voltage-gated calcium channel, where several Ca2+/CaM-IQ structures have been described in the literature (23–29).
Figure 2.

Conservation of calcium regulatory domains and the location of disease mutations. The DIII-DIV linker (A) and proximal CTD (B) sequences of NaV1.1–1.9 are aligned against NaV1.5. (Top) Human NaV1.5 numbering. (Right-hand side, down) Respective NaV1.1–1.9 numbering. From published NMR and crystal structures, the available secondary structure elements are shown above the sequences: (arrows) β-strands; (coils) α-helices. Sites for disease mutations are highlighted: LQT3 (black), BrS (red), or both (brown). The R1902C mutation in NaV1.2 is causative of GEFS+ (blue). The IQ consensus motif is indicated (black box) for NaV1.5.
Early studies identified that mutants of CaM in Paramecium tetraurelia could impact voltage gated sodium currents in a lobe dependent manner (30,31). Numerous studies have since reported that CaM interacts with the NaV IQ domain (6,8,12–14,18,32–34). Using intrinsic tyrosine fluorescence of CaM, the IQ motif peptide of NaV1.5 was shown to bind apoCaM with ∼160 nM Kd, and Ca2+/CaM with ∼2 μM Kd (35) with similar values obtained using isothermal titration calorimetry (8), suggesting that the NaV IQ domain is primarily an apoCaM binding site. The interaction has been confirmed via FRET (12,18) yet co-immunoprecipitation experiments using GST fusion proteins of the CTD of some isoforms have failed to report an interaction with CaM (13). However, the folding and aggregation behavior for these constructs have not been sufficiently established.
NMR structures have been solved for apoC-lobe bound to the IQ domain of NaV1.2 (36) and for apoCaM bound to the NaV1.5 IQ domain (37) suggesting that the apoC-lobe is the primary binding partner for the IQ domain (NaV1.5 residues 1901–1916), with major hydrophobic contacts made by residues I1908 and F1912. Some studies have shown that mutation of NaV1.5 residues I1908A/Q1909A (IQ/AA), as well as the analogous mutations in NaV1.2 and NaV1.4, disrupt IQ domain-CaM interactions (13,18,32). However, more quantitative biochemical approaches have shown that the IQ/AA mutation only weakly affects binding, with only a twofold lower affinity for both apoCaM and Ca2+/CaM (35). In addition, the IQ/AA mutation alone causes a hyperpolarizing shift in the steady-state availability curve, and abolishes the calcium-dependent shift (35), in addition to an increase in sustained, noninactivating current (32). The IQ/AA mutation can also reduce current amplitudes (13), which is apparently the result of impaired channel trafficking (18).
Outright deletion of the IQ domain (stop-codon at amino acid (aa) 1885 in NaV1.5) also reduces current amplitude, produces a −11 mV shift in steady-state availability (38) and increases the amount of noninactivating current (39) Interestingly, the IQ deletion mutant has been reported to retain a Ca2+-dependent shift in steady-state inactivation, suggesting that other players participate in the regulation (12). However, overexpression of CaM or CaM1234 (a mutant that is no longer able to bind Ca2+ in a physiological range), in the absence of Ca2+, can result in a depolarizing shift of the steady-state inactivation curve, and the deletion of the IQ domain abolishes this effect. This somewhat surprising set of results has been explained by a model whereby apoCaM/IQ domain association affects inactivation, independent of Ca2+, and that the Ca2+-dependence of inactivation does not require this interaction.
Thus, as with the variable macroscopic effects of Ca2+/CaM on channel gating, the effects observed by different laboratories seem in direct conflict, with some suggesting the IQ motif is essential, whereas others concluding that it is peripheral. One potential explanation is that the IQ domain simply serves as a sink for a resident CaM molecule, where it serves to enrich CaM in the vicinity of the channel but is not directly involved in the mechanism of Ca2+-dependent regulation. In this scenario, removing the sink can be overcome by overexpression of CaM, and thus different expression levels in the individual experiments, as well as different inherent affinities of NaV isoforms for CaM, could result in functional discrepancies.
EF-hands
The function and role of the CTD proximal EF-hand domain has also been the subject of controversy. These two EF-hand motifs, initially thought to be formed by NaV1.5 residues 1773–1852 (10), are actually encoded by NaV1.5 residues 1788–1862 as shown by NMR (40) (Fig. 3). A similar structure has been reported simultaneously and independently for the NaV1.2 EF-hand domain (41) which extends further toward the C-terminus (NaV 1.2 residues 1777–1882 equivalent to NaV1.5 residues 1773–1878), and includes a partially ordered fifth helix. The EF-hand domain is linked to the last transmembrane segment (IVS6; domain IV S6 segment) through a 13–15 aa flexible linker. As EF-hands typically form Ca2+-binding motifs, it is possible that the Ca2+-dependent effects on NaV inactivation are due, in part, by direct Ca2+ ion binding to the EF-hands, absent of a CaM contribution.
Figure 3.
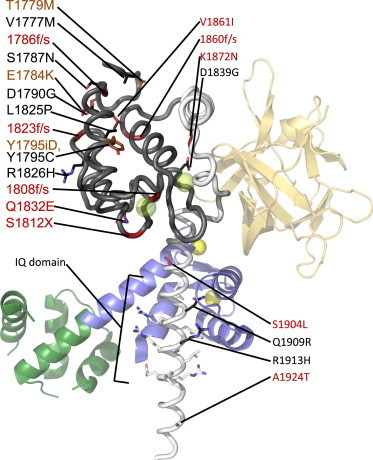
Structure of the human NaV1.5 C-terminal domain. Crystal structure of CaM bound to the NaV1.5 CTD (PDB:4DCK). Mg2+-Calmodulin (dark green N-terminus, blue C-terminus) and fibroblast growth factor 13 (FGF13) (orange) are shown as part of a ternary complex from PDB:4DCK. (Yellow spheres) Magnesium ions. Mutations that cause LQT3 (black), BrS (red), or both (brown), are labelled. (f/s) Mutations that cause frame shifts. (X) Nonsense mutations. (Prefix i) Insertions. The NaV1.5 EF-hand domain, determined separately by NMR (PDB:2KBI), is highlighted (dark gray). It is clear that the portion C-terminal to this region forms interactions with the EF-hand domain. (Transparent green spheres) Calcium ions in the EF-hands, showing where they would bind based on structural superposition with Ca2+-calmodulin. For clarity, only the side chains of the IQ domain as well as missense mutation sites are shown.
However, based on the NMR structure, the EF-hands are predicted to bind, at most, one Ca2+ ion, as the second EF-hand lacks prerequisite acidic residues for Ca2+ ion coordination, thus it is has not been determined if the sodium channel EF-hands are functional or vestigial. However, using either 2 mM BAPTA to simulate Ca2+-free conditions, or 20 mM CaCl2, Shah et al. (35) showed distinct differences in 15N- 1H HSQC NMR spectra for a construct spanning Nav1.5 (1773–1865), and titrations yielded a Kd ∼ 7.5 mM, suggesting that Ca2+ ions are capable of binding the EF-hands. Such a high Kd value would be physiologically irrelevant, but larger constructs (1773–1920) yield a Kd between 1 and 6 μM, suggesting the affinity is strongly affected by the C-terminal region including the IQ domain binding to the EF-hands (10,35). Mg2+ was not found to have an effect. In contrast, Kim et al. (32) initially failed to detect any changes in Trp fluorescence of the EF-hands at varying Ca2+ concentrations, but then using 1H,15N chemical shifts during Ca2+ titrations of the NaV1.2 and NaV1.5 EF-hands, they also found low affinities for Ca2+ (Kd values between 1.6 and 3.3 mM) (41) However, the largest chemical shifts were found outside of the canonical EF-hand loop motifs, suggesting that Ca2+ binds weakly near the N-terminus of helix I, the linker between helices II and III, the C-terminus of helix IV, and the partially structured helix V.
In terms of functionality of expressed channels, a quadruple knock-out of Ca2+ binding to the EF-hands (E1788A, D1790A, D1792A, E1799A) effectively removes the Ca2+-dependent shift in steady-state availability (10,12). Of note, the locations of these mutations were based on an early model of the EF-hands, and subsequent identification in the NMR structure shows, surprisingly, that none of these sites could be involved in coordinating Ca2+. It is therefore puzzling that these mutations abolish Ca2+ binding, especially when both CD and NMR did not detect any structural changes (10). Further, the NaV1.5 double-mutant D1802A/E1804A, which should knock out Ca2+ binding to the EF-hands based upon the NMR structure, disrupted Ca2+-dependent shifts in the steady-state availability curve.
An additional level of complexity in Ca2+ regulation emerges from several reports that the EF-hands and the IQ domain can physically interact. NMR experiments indicate binding of NaV1.5 residues 1897–1925 to an EF-hand construct (residues 1773–1865), thus helping to explain the difference in Ca2+ affinity between isolated EF-hands and longer constructs (35). Consistent with this, mutation of IQ/AA, which disrupts the EF-IQ interaction, lowers the affinity (Kd ∼ 4 mM). Isothermal titration calorimetry (ITC) experiments between the EF-hands and IQ domain suggest a Kd ∼ 27–38 μM, and using NMR, the IQ domain was mapped to bind between helices I and IV of the EF-hand domain (40) However, these experiments were performed on EF-hands as a distinct polypeptide from the IQ motif, and it is not clear whether the same interactions also occur in a physiological setting where both domains are within the same polypeptide.
Transition-metal FRET studies have been used to measure distances at three positions to a Trp residue within the EF-hand region, and these were used as restraints to build a model of the IQ domain bound to the EF-hands (42) Thus, one step of the mechanism is that Ca2+/CaM could displace the EF-hands from the IQ domain, but as Ca2+/CaM-IQ binding is not affected by the presence of the EF-hands (8), they could have the simple role of properly positioning the IQ domain relative to the rest of the channel.
These elements have been recently captured in a crystal structure of a near-full-length C-terminus, containing both the EF-hands and IQ domain of the NaV1.5 isoform (residues 1776–1928) in complex with apo-CaM and the fibroblast growth factor homologous factor, FGF13 (43) (Fig. 3). This structure further confirms the preferred nature of the apo-C-lobe with the NaV IQ motif and highlights the residues that support the interaction. Although considered an apo-CaM complex, Mg2+ is bound to the C-lobe. However, the C-lobe interactions with the IQ domain are very similar to the ones observed in the apoCaM-IQ domain NMR structures (36,37) In addition, no direct interaction between the EF-hands and the IQ motif was observed.
Coupling Inactivation to Ca2+: Interaction between Calmodulin and the DIII-IV Linker
Several studies have shown that CaM can interact with the isolated NaV1.5 DIII-IV linker. Using both NMR and ITC, Potet et al. (11) showed binding of Ca2+-loaded CaM to residues 1471–1523 of NaV1.5, with a Kd of 0.6 μM and a 1:1 stoichiometry. In addition, a peptide corresponding to NaV 1.5 1510–1530 binds Ca2+/CaM with a Kd of ∼8 μM and mutation of distal loop residues 1520–1522 FIF to AAA decreases the affinity of the latter peptide for Ca2+/CaM.
In contrast, it has also been shown that Ca2+/CaM can bind to the N-terminal portion of the DIII-IV linker (8,9). Here, ITC experiments yield a Kd of ∼3 μM, and demonstrate that the interaction is driven by the Ca2+/C-lobe, and importantly, no binding is measured in the absence of Ca2+. Alanine scanning of several amino-acid residues suggests that the Ca2+/C-lobe forms major contacts with a double Tyrosine motif (NaV 1.5 residues 1494 and 1495), an observation that has been confirmed with a crystal structure that highlights intricate contacts between both tyrosines and the Ca2+/C-lobe (8) (Fig. 4). There are additional energetically important contacts with M1498. Notably, no portion of the DIII-IV linker was found to interact with the Ca2+/N-lobe, and whereas the isolated Ca2+/N-lobe is able to interact with the DIII-IV linker in vitro, the affinity is extremely weak (Kd > 500 μM) and the binding site overlaps with the Ca2+/C-lobe site. Thus, in the physiological setting the N-lobe is more likely to bind to a different site, such as the C-terminal IQ domain.
Figure 4.
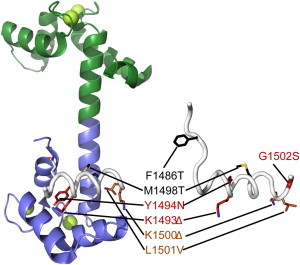
Structures of the sodium channel DIII-DIV linker alone and in complex with Ca2+/CaM. Comparison between a Ca2+/CaM-bound human NaV1.5 DIII-DIV linker (PDB:4DJC) with the rat NaV1.2 DIII-DIV linker (PDB:1BYY). The human NaV1.5 numbering is used. Mutations that cause LQT3 (black), BrS (red), or both (brown), respectively. (Δ) Deletion mutations. (Green) N-lobe of calmodulin, (blue) C-lobe, and (green spheres) Ca2+ ions.
The two reports on Ca2+/CaM binding to the DIII-IV linker are thus in conflict. However, full-length expressed NaV1.5 channels containing the FIF/AAA or FIF/AIA mutations, that have been proposed to eliminate CaM N-lobe binding, still display a Ca2+-dependent shift in steady-state inactivation, minimizing the role, if any, for the CaM N-lobe binding to the DIII-IV linker in Ca2+ regulation (8,11). This suggests that the 1510–1530 peptide is not the functional binding site for Ca2+/CaM within intact channels. Indeed, as the DIII-IV linker is likely to end around residue 1522 and become the transmembrane helix of DIV S1, a substantial portion of this site may not be available to cytoplasmic binding partners. Moreover, mutation of M1498 (highlighted in the Ca2+/C-lobe interaction in the crystal structure) to alanine decreases the affinity as measured by ITC, and abolishes the Ca2+-dependent shift in steady-state inactivation. In addition, introduction of a mutant that enhances the affinity of the DIII-IV linker for Ca2+/CaM (E1489A/E1490A double mutant) also increases the Ca2+ sensitivity for shifting the steady-state availability curve. The correlation between loss of binding with loss of modulation, and gain of binding with gain of modulation, suggests that the Y1494/Y1495 motif as observed in the crystal structure is physiologically relevant, and although the distal DIII-IV linker FIF motif can bind CaM, these side chains are clearly not necessary for Ca2+ regulation.
A Simplified Mechanism for Ca2+ Regulation of Sodium Channels
In total, the in vitro and in vivo data using NMR, ITC, and crystallographic approaches suggest a simple yet dynamic lobe-switching mechanism for Ca2+/CaM binding to intracellular channel domains, and provides a testable model biasing SSI inactivation in NaVs. In low Ca2+ levels, apoCaM is bound to the C-terminal IQ domain via the apoC-lobe, and as Ca2+ levels rise, Ca2+/C-lobe gains affinity for the DIII-IV linker, opening access to the Ca2+/N-lobe which now freely binds to the C-terminus IQ motif. The constrained configuration of the Ca2+/C-lobe bound to the DIII-IV linker inherently destabilizes the interactions between the DIII-IV linker and the inactivation gate receptor. As the kinetics of inactivation of WT channels are unaffected by Ca2+, CaM dissociation from the DIII-IV linker is not limiting. Indeed, the IFM motif known to be essential for inactivation lies well outside of the Ca2+/C-lobe binding site. However, whether CaM does indeed bridge the DIII-IV linker and IQ domain within intact channels to form a tripartite complex remains to be fully visualized.
Even though the above model may be attractive, several additional complications remain. For one, mutations in both the DIII-IV linker and the CTD can affect inactivation in the absence of coexpressed CaM (44,45) For example, both the ΔKPQ in the NaV1.5 DIII-IV linker, and a truncation of the CTD after the EF-hands (stop codon at aa 1885) result in an increased sustained current (39) suggesting that the DIII-IV linker and CTD may form a complex in the absence of CaM (32,39,46). Indeed, ITC experiments between isolated CTD and the DIII-IV linkers indicate a Kd of ∼5 μM (46) and the interaction is only visible with longer CTD constructs containing both the EF-hands and IQ domain (39) However, other studies have failed to see a direct interaction between the DIII-IV linker and the CTD via ITC (8), a discrepancy that may be due to a different choice of constructs, as the positive ITC experiment made use of a longer CTD (ending at NaV1.5 residue 1937 instead of 1924) and DIII-IV linker (ending at 1523 versus 1522).
A Physiological Role for Calcium ION Regulation of Voltage-Gated Sodium Channels: Use-Dependent Facilitation of Channel Availability
While the details of the mechanism of Ca2+ regulation continue to unfold, it is worth considering the role such a modulation might play in the physiological context. The consensus effect on Ca2+ regulation of NaVs is a destabilization of inactivation, which is manifest as an ∼10-mV depolarizing shift in the steady-state availability relationship (8,10,11,18). Because this relationship is at equilibrium at physiological resting membrane potentials, roughly half of the channels are inactivated at resting conditions, and such a shift would significantly increase the number of channels that are available to fire for the next action potential.
How might this mechanism become relevant in an electrically excitable cell such as a cardiac myocyte? One might simply conclude that it would have little impact because the upstroke of the cardiac action potential, where NaV channels are open, precedes the ensuing Ca2+ transient. However, a comprehensive synthesis of the available data points toward a different type of regulation that could, in theory, take place on a rate-dependent basis to encode for a type of use-dependent facilitation. At low firing rates, Na+ channels would sense diastolic, low Ca2+ levels and the DIII-IV linker would not engage significantly with the CaM-C-lobe. In addition, during the Ca2+ transient, interactions between calcium ions and CaM, an effector of Ca2+ regulation, would be functionally irrelevant as most channels are inactivated, and Ca2+ would have time to dissociate from CaM by the next upstroke of the action potential. However, as the firing rate increases, and in particular, when the rate of Ca2+ transient impinges upon the Ca2+/C-lobe dissociation kinetics, then a type of Ca2+ memory would take place whereby Ca2+ ions would still be bound to the CaM C-lobe at the beginning of the next action potential. This would bias the steady-state availability relationship to more depolarized potentials and bring more NaV channels to drive the upstroke of the action potential. Such a mechanism might not necessarily destabilize electrical signaling because it would only be employed transiently when having more channels would be efficacious. Moreover, the domains and specific residues that support the interaction between NaVs and Ca2+/CaM are highly conserved among all nine sodium channel isoforms, and this mechanism would be expected to be widespread throughout the excitable cells of the cardiovascular and nervous systems.
Similarities with Voltage-Gated Calcium Channels
Voltage-gated calcium channels (CaVs) share a fair amount of sequence identity with NaVs. Members of the CaV1 and CaV2 families are also modulated by CaM binding to the pore-forming α1 subunit. CaM can mediate at least two Ca2+-dependent feedback processes: Ca2+-dependent inactivation and Ca2+-dependent facilitation (for a limited set of examples, see Peterson et al. (48), Zülhke et al. (49), and Lee et al. (50)). Like NaVs, CaV1 and CaV2 family members encode EF-hands in the CTD, followed by an IQ domain that is able to bind both Ca2+/CaM and apoCaM, suggesting that some of the mechanisms by which CaM alters inactivation may be conserved. However, there are some intrinsic differences. The CaV IQ domain favors Ca2+/CaM binding, and both lobes contribute to binding of apoCaM and Ca2+/CaM (23,24,26,28). In addition, a second CaM binding site is present between the EF-hands and the IQ domain (25,29). Although only a single CaM is required to mediate Ca2+-dependent inactivation (51), this raises the possibility that multiple CaMs can associate with CaVs. No binding has been detected between CaM and the CaV DIII-IV linker, although some CaV isoforms encode a CaM binding site in the N-terminal region (52). Thus, whereas some of the machinery seems conserved between NaVs and CaVs, there are substantial discrepancies that mediate the different functional effects in both channels.
Disease Mutations in the Ca2+-Sensing Machinery
Sodium channel mutations have been linked to a wide range of debilitating diseases, ranging from epilepsy (53) to myotonia (54), erythermalgia (55), cardiac arrhythmias (56) and migraine (57). Much attention has been given to mutations in the NaV1.5 isoform, where gain-of-function mutations which impair the inactivation process result in the Long QT syndrome variant 3 (LQT3), a rare inherited disorder that is associated with an increased propensity to arrhythmogenic syncope, polymorphous ventricular tachycardia, and sudden cardiac death (58,59). The first reported LQT3 mutation, a deletion of three amino acids (KPQ) in the inactivation gate, results in a small, but significant, sustained sodium current over the duration of the action potential (60), a phenotype displayed by many LQT3 mutations. An additional seven LQT3 mutations have since been found in the inactivation gate, with 10 in the EF-hand region, and two between the EF-hands and the end of the IQ domain (mutations available on www.fsm.it/cardmoc/). Thus, the domains known to be essential to the NaV Ca2+ regulatory apparatus (DIII-IV linker, EF-hand, and IQ-motif) form hot-spot regions for LQT3 syndrome (61) (Figs. 2–4). Thus, it is possible that disruption of Ca2+ sensing may serve to compound the direct effects of mutations on channel gating.
Loss of function mutations that result in reduced sodium current of NaV1.5, mostly due to trafficking defects, underlie Brugada syndrome (BrS) (62). As such, many of the reported mutations provide truncated proteins that likely do not fold. However, a number of point mutations not resulting in truncations are present in the Ca2+-sensing machinery: eight within the inactivation gate, five in the EF-hand region, and three following the EF-hands until the end of the IQ domain (Figs. 2–4). It is likely that several of these simply result in decreased folding stability of the channels, and hence a decreased functional expression.
The NaV1.5 A1924T Brugada syndrome mutation, located immediately C-terminal to the IQ domain, has been shown to affect gating and Ca2+ regulation (6,35). Additionally, the NaV1.5 D1790G LQT3 mutation in the EF-hands, at the N-terminus of the first helix (Fig. 4) reduces the Ca2+-dependent shift in steady-state inactivation, and Trp fluorescence experiments indicate a ∼20-fold reduction in Ca2+ affinity (10,12). Compared to wild-type channels, this mutation also significantly speeds up entry into inactivation in high Ca2+ (12). The LQT3 NaV1.5 S1904L mutation near the IQ motif may affect the interaction between the IQ domain and the EF-hands (42,46) and reduces the affinity of the CTD for the DIII-IV linker to a level that can no longer be quantified via ITC.
Inherited mutations in neuronal channels may also impact Ca2+ sensing. The NaV1.2 R1902C mutation has been linked to the inherited seizure disorder Generalized Epilepsy with Febrile Seizures Plus (GEFS+). Located near the IQ motif (Fig. 2), this mutation causes a threefold reduction in the affinity for Ca2+/CaM (63). In the NMR structure of the NaV1.2 IQ motif complexed to apoC-lobe, R1902 was not found to interact with CaM (36) However, chemical-shift analysis indicates that Ca2+/CaM binding differs from apoCaM binding to the IQ domain, and R1902 may therefore contribute to Ca2+/CaM interactions.
Concluding Comments
The mechanistic basis for Ca2+/CaM regulation of NaVs has thus far been a difficult biological problem to solve. Seemingly contradictory observations may be due to variable experimental conditions and inherent differences between NaV isoforms, in addition to overlapping, redundant signaling pathways within NaV channels themselves. In addition, while a growing number of experimental approaches promise to bring clarity to a historically murky topic, much mystery remains regarding the molecular mechanism and physiological role of this potentially important regulatory process. NaV mutations associated with inheritable excitability disorders are likely to also disrupt Ca2+ sensing, and alternatively, given that many pathological disorders have chronically high Ca2+ levels, such as in the failing heart or catecholaminergic polymorphic ventricular tachycardia, it is feasible that targeted modulation Ca2+/CaM regulation of NaVs could be a novel therapeutic strategy for these largely untreatable diseases.
Acknowledgments
This work is funded by grant No. 259572 from the Canadian Institutes of Health Research (to C.A.A. and F.V.P.). C.A.A. is a Heart and Stroke Foundation of Canada New Investigator and a Michael Smith Foundation for Health Research Career Investigator. F.V.P. is a Canadian Institutes of Health Research New Investigator and a Michael Smith Foundation for Health Research Career Investigator.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Contributor Information
Filip Van Petegem, Email: filip.vanpetegem@gmail.com.
Christopher A. Ahern, Email: chrisahern@gmail.com.
References
- 1.Horn R., Patlak J., Stevens C.F. Sodium channels need not open before they inactivate. Nature. 1981;291:426–427. doi: 10.1038/291426a0. [DOI] [PubMed] [Google Scholar]
- 2.Joseph D., Petsko G.A., Karplus M. Anatomy of a conformational change: hinged “lid” motion of the triosephosphate isomerase loop. Science. 1990;249:1425–1428. doi: 10.1126/science.2402636. [DOI] [PubMed] [Google Scholar]
- 3.Stühmer W., Conti F., Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 4.Smith M.R., Goldin A.L. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophys. J. 1997;73:1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McPhee J.C., Ragsdale D.S., Catterall W.A. A critical role for the S4-S5 intracellular loop in domain IV of the sodium channel α-subunit in fast inactivation. J. Biol. Chem. 1998;273:1121–1129. doi: 10.1074/jbc.273.2.1121. [DOI] [PubMed] [Google Scholar]
- 6.Tan H.L., Kupershmidt S., Balser J.R. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–447. doi: 10.1038/415442a. [DOI] [PubMed] [Google Scholar]
- 7.Kass R.S. Sodium channel inactivation in heart: a novel role of the carboxy-terminal domain. J. Cardiovasc. Electrophysiol. 2006;17(Suppl 1):S21–S25. doi: 10.1111/j.1540-8167.2006.00381.x. [DOI] [PubMed] [Google Scholar]
- 8.Sarhan M.F., Tung C.C., Ahern C.A. Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. USA. 2012;109:3558–3563. doi: 10.1073/pnas.1114748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarhan M.F., Van Petegem F., Ahern C.A. A double tyrosine motif in the cardiac sodium channel domain III-IV linker couples calcium-dependent calmodulin binding to inactivation gating. J. Biol. Chem. 2009;284:33265–33274. doi: 10.1074/jbc.M109.052910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wingo T.L., Shah V.N., Balser J.R. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat. Struct. Mol. Biol. 2004;11:219–225. doi: 10.1038/nsmb737. [DOI] [PubMed] [Google Scholar]
- 11.Potet F., Chagot B., Balser J.R. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J. Biol. Chem. 2009;284:8846–8854. doi: 10.1074/jbc.M806871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas S., DiSilvestre D., Tomaselli G.F. Calcium-mediated dual-mode regulation of cardiac sodium channel gating. Circ. Res. 2009;104:870–878. doi: 10.1161/CIRCRESAHA.108.193565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herzog R.I., Liu C., Cummins T.R. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J. Neurosci. 2003;23:8261–8270. doi: 10.1523/JNEUROSCI.23-23-08261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deschênes I., Neyroud N., Tomaselli G.F. Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ. Res. 2002;90:E49–E57. doi: 10.1161/01.res.0000012502.92751.e6. [DOI] [PubMed] [Google Scholar]
- 15.Ahern C.A., Zhang J.F., Horn R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ. Res. 2005;96:991–998. doi: 10.1161/01.RES.0000166324.00524.dd. [DOI] [PubMed] [Google Scholar]
- 16.Huttlin E.L., Jedrychowski M.P., Gygi S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young K.A., Caldwell J.H. Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J. Physiol. 2005;565:349–370. doi: 10.1113/jphysiol.2004.081422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biswas S., Deschênes I., Tomaselli G.F. Calmodulin regulation of Nav1.4 current: role of binding to the carboxyl terminus. J. Gen. Physiol. 2008;131:197–209. doi: 10.1085/jgp.200709863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheney R.E., Mooseker M.S. Unconventional myosins. Curr. Opin. Cell Biol. 1992;4:27–35. doi: 10.1016/0955-0674(92)90055-h. [DOI] [PubMed] [Google Scholar]
- 20.Alexander K.A., Wakim B.T., Storm D.R. Identification and characterization of the calmodulin-binding domain of neuromodulin, a neurospecific calmodulin-binding protein. J. Biol. Chem. 1988;263:7544–7549. [PubMed] [Google Scholar]
- 21.Bähler M., Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002;513:107–113. doi: 10.1016/s0014-5793(01)03239-2. [DOI] [PubMed] [Google Scholar]
- 22.Rhoads A.R., Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 23.Van Petegem F., Chatelain F.C., Minor D.L., Jr. Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat. Struct. Mol. Biol. 2005;12:1108–1115. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim E.Y., Rumpf C.H., Minor D.L., Jr. Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation. Structure. 2008;16:1455–1467. doi: 10.1016/j.str.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim E.Y., Rumpf C.H., Minor D.L., Jr. Multiple C-terminal tail Ca2+/CaMs regulate CaV1.2 function but do not mediate channel dimerization. EMBO J. 2010;29:3924–3938. doi: 10.1038/emboj.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fallon J.L., Halling D.B., Quiocho F.A. Structure of calmodulin bound to the hydrophobic IQ domain of the cardiac Cav1.2 calcium channel. Structure. 2005;13:1881–1886. doi: 10.1016/j.str.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 27.Halling D.B., Georgiou D.K., Hamilton S.L. Determinants in CaV1 channels that regulate the Ca2+ sensitivity of bound calmodulin. J. Biol. Chem. 2009;284:20041–20051. doi: 10.1074/jbc.M109.013326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mori M.X., van der Kooi C.W., Yue D.T. Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high-resolution mechanistic implications for channel regulation by Ca2+ Structure. 2008;16:607–620. doi: 10.1016/j.str.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fallon J.L., Baker M.R., Quiocho F.A. Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+∗ calmodulins. Proc. Natl. Acad. Sci. USA. 2009;106:5135–5140. doi: 10.1073/pnas.0807487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kink J.A., Maley M.E., Kung C. Mutations in paramecium calmodulin indicate functional differences between the C-terminal and N-terminal lobes in vivo. Cell. 1990;62:165–174. doi: 10.1016/0092-8674(90)90250-i. [DOI] [PubMed] [Google Scholar]
- 31.Saimi Y., Ling K.Y. Calmodulin activation of calcium-dependent sodium channels in excised membrane patches of Paramecium. Science. 1990;249:1441–1444. doi: 10.1126/science.2169650. [DOI] [PubMed] [Google Scholar]
- 32.Kim J., Ghosh S., Pitt G.S. Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 2004;279:45004–45012. doi: 10.1074/jbc.M407286200. [DOI] [PubMed] [Google Scholar]
- 33.Mori M., Konno T., Nagayama K. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+-sensitivity? Biochemistry. 2000;39:1316–1323. doi: 10.1021/bi9912600. [DOI] [PubMed] [Google Scholar]
- 34.Theoharis N.T., Sorensen B.R., Shea M.A. The neuronal voltage-dependent sodium channel type II IQ motif lowers the calcium affinity of the C-domain of calmodulin. Biochemistry. 2008;47:112–123. doi: 10.1021/bi7013129. [DOI] [PubMed] [Google Scholar]
- 35.Shah V.N., Wingo T.L., Chazin W.J. Calcium-dependent regulation of the voltage-gated sodium channel hH1: intrinsic and extrinsic sensors use a common molecular switch. Proc. Natl. Acad. Sci. USA. 2006;103:3592–3597. doi: 10.1073/pnas.0507397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feldkamp M.D., Yu L., Shea M.A. Structural and energetic determinants of apo calmodulin binding to the IQ motif of the NaV1.2 voltage-dependent sodium channel. Structure. 2011;19:733–747. doi: 10.1016/j.str.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chagot B., Chazin W.J. Solution NMR structure of Apo-calmodulin in complex with the IQ motif of human cardiac sodium channel NaV1.5. J. Mol. Biol. 2011;406:106–119. doi: 10.1016/j.jmb.2010.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cormier J.W., Rivolta I., Kass R.S. Secondary structure of the human cardiac Na+ channel C terminus: evidence for a role of helical structures in modulation of channel inactivation. J. Biol. Chem. 2002;277:9233–9241. doi: 10.1074/jbc.M110204200. [DOI] [PubMed] [Google Scholar]
- 39.Motoike H.K., Liu H., Kass R.S. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J. Gen. Physiol. 2004;123:155–165. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chagot B., Potet F., Chazin W.J. Solution NMR structure of the C-terminal EF-hand domain of human cardiac sodium channel NaV1.5. J. Biol. Chem. 2009;284:6436–6445. doi: 10.1074/jbc.M807747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miloushev V.Z., Levine J.A., Palmer A.G., 3rd Solution structure of the NaV1.2 C-terminal EF-hand domain. J. Biol. Chem. 2009;284:6446–6454. doi: 10.1074/jbc.M807401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glaaser I.W., Osteen J.D., Kass R.S. Perturbation of sodium channel structure by an inherited long QT syndrome mutation. Nat. Commun. 2012;3:706. doi: 10.1038/ncomms1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C., Chung B.C., Pitt G.S. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure. 2012;20:1167–1176. doi: 10.1016/j.str.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viswanathan P.C., Bezzina C.R., Balser J.R. Gating-dependent mechanisms for flecainide action in SCN5A-linked arrhythmia syndromes. Circulation. 2001;104:1200–1205. doi: 10.1161/hc3501.093797. [DOI] [PubMed] [Google Scholar]
- 45.Mantegazza M., Yu F.H., Scheuer T. Role of the C-terminal domain in inactivation of brain and cardiac sodium channels. Proc. Natl. Acad. Sci. USA. 2001;98:15348–15353. doi: 10.1073/pnas.211563298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bankston J.R., Sampson K.J., Kass R.S. A novel LQT-3 mutation disrupts an inactivation gate complex with distinct rate-dependent phenotypic consequences. Channels (Austin) 2007;1:273–280. doi: 10.4161/chan.4956. [DOI] [PubMed] [Google Scholar]
- 47.Reference deleted in proof.
- 48.Peterson B.Z., DeMaria C.D., Yue D.T. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 49.Zühlke R.D., Pitt G.S., Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 50.Lee A., Wong S.T., Catterall W.A. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 51.Mori M.X., Erickson M.G., Yue D.T. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 2004;304:432–435. doi: 10.1126/science.1093490. [DOI] [PubMed] [Google Scholar]
- 52.Dick I.E., Tadross M.R., Yue D.T. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kearney J.A., Plummer N.W., Meisler M.H. A gain-of-function mutation in the sodium channel gene SCN2A results in seizures and behavioral abnormalities. Neuroscience. 2001;102:307–317. doi: 10.1016/s0306-4522(00)00479-6. [DOI] [PubMed] [Google Scholar]
- 54.Yang N., Ji S., George A.L., Jr. Sodium channel mutations in Paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc. Natl. Acad. Sci. USA. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Y., Wang Y., Shen Y. Mutations in SCN9A, encoding a sodium channel α-subunit, in patients with primary erythermalgia. J. Med. Genet. 2004;41:171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keating M.T., Sanguinetti M.C. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 57.Kahlig K.M., Rhodes T.H., George A.L., Jr. Divergent sodium channel defects in familial hemiplegic migraine. Proc. Natl. Acad. Sci. USA. 2008;105:9799–9804. doi: 10.1073/pnas.0711717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Splawski I., Shen J., Keating M.T. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 59.Napolitano C., Priori S.G., Leonardi S. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294:2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 60.Wang Q., Shen J., Keating M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 61.Ruan Y., Liu N., Priori S.G. Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol. 2009;6:337–348. doi: 10.1038/nrcardio.2009.44. [DOI] [PubMed] [Google Scholar]
- 62.Chen Q., Kirsch G.E., Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 63.Weiss L.A., Escayg A., Meisler M.H. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry. 2003;8:186–194. doi: 10.1038/sj.mp.4001241. [DOI] [PubMed] [Google Scholar]