

Abstract
Inositol phosphatases are important regulators of cell signaling and membrane trafficking. Mutations in inositol polyphosphate 5-phosphatase, INPP5E, have been identified in Joubert syndrome, a rare congenital disorder characterized by midbrain malformation, retinitis pigmentosa, renal cysts, and polydactyly. Previous studies have implicated primary cilia abnormalities in Joubert Syndrome, yet the role of INPP5E in cilia formation is not well understood. In this study, we examined the function of INPP5E in cilia development in zebrafish. Using specific antisense morpholino oligonucleotides to knockdown Inpp5e expression, we observed phenotypes of microphthalmia, pronephros cysts, pericardial effusion, and left-right body axis asymmetry. The Inpp5e morphant zebrafish exhibited shortened and decreased cilia formation in the Kupffer’s vesicle and pronephric ducts as compared to controls. Epinephrine-stimulated melanosome trafficking was delayed in the Inpp5e zebrafish morphants. Expression of human INPP5E expression rescued the phenotypic defects in the Inpp5e morphants. Taken together, we showed that INPP5E is critical for the cilia development in zebrafish.
Keywords: Inositol phosphatase, primary cilia, Kupffer’s vesicle, INPP5E, zebrafish
1. Introduction
The primary cilium is an evolutionarily conserved subcellular structure that protrudes from nearly all post-mitotic eukaryotic cells (Rohatgi & Snell, 2010). By sensing changes in the extracellular environment, the primary cilium can coordinate signaling cascades that subsequently become amplified throughout the cell (Fisch & Dupuis-Williams, 2011). A highly specialized extension of the plasma membrane, the ciliary membrane is enriched with many signaling precursors, such as Patched1 (Ptc1) (Corbit, et al., 2005, Rohatgi, et al., 2007). Upon ligand binding of Sonic Hedgehog (Hg), Ptc1 is removed from the cilium and Smoothened is then accumulated within the ciliary membrane, allowing initiation of downstream signaling cascades. The ciliary membrane covers a microtubule-based axoneme, which is anchored by a basal body (Pearson, et al., 2007, Pearson & Winey, 2009). Within the ciliary membrane are phospholipids, including phosphoinositides that may serve as second messengers in signal transduction. Defects in cilia formation or maintenance have been found to underlie a wide range of human diseases, including retinitis pigmentosa, renal cysts, polydactyly, and developmental delays, which are collectively called ciliopathies (Jacoby, et al., 2009, Novarino, et al., 2011, Schurman & Scheinman, 2009).
Joubert syndrome, a rare form of autosomal recessive ciliopathy, is characterized by an underdevelopment of cerebellar vermis, with a distinctive “molar tooth sign” of cerebellar vermis hypoplasia on MRI (Bielas, et al., 2009). The most common features of Joubert syndrome include retinitis pigmentosa, hypotonia, severe psychomotor delay, and ataxia (Lee & Gleeson, 2011). Other physical deformities may include polydactyly, cleft palate, renal cysts, and liver disease. A rapidly expanding number of genes have been implicated in Joubert syndrome, including NPHP1, NPHP6/CEP290, NPHP8, ARL13B and INPP5E (Arts, et al., 2007, Bielas et al., 2009, Cantagrel, et al., 2008, Kim, et al., 2008, McEwen, et al., 2007, Travaglini, et al., 2009, Valente, et al., 2010).
INPP5E belongs to a family of inositol polyphosphate 5-phosphatases, which dephosphorylate the D5 position of the inositol ring (Asano, et al., 1999, Kisseleva, et al., 2000, Kong, et al., 2000). There are ten mammalian members of the 5-phosphatase family, which play critical yet distinct roles in a number of biological processes, such as the regulation of insulin signaling, vesicular trafficking, synaptic vesicle formation, and hematopoietic cell proliferation (Ooms, et al., 2009, Pirruccello & De Camilli, 2012). The members of inositol polyphosphate 5-phosphatase family share a common inositol phosphatase domain, but these individual 5-phosphatases have different protein-protein interaction domains that regulate their subcellular localization and function (Dyson, et al., 2012). For instance, in response to growth factor stimulation, INPP5E regulates the intracellular levels of PI(4,5)P2 and PI(3,4,5)P3 by controlling downstream AKT activation (Kisseleva, et al., 2002). Overexpression of INPP5E also results in the hydrolysis of PI(3,5)P2 to PI(3)P at the plasma membrane, and translocation of GLUT4 glucose transporter into the plasma membrane (Kong, et al., 2006). Mutations in the INPP5E phosphatase domain have been identified in a series of patients with Joubert syndrome, thus highlighting the role of inositol phosphatases in cilia development (Bielas et al., 2009). In addition, a C-terminal deletion mutant of INPP5E has been reported in a family with MORM syndrome, a variant of the Bardet-Biedel group of syndromic ciliopathies (Jacoby et al., 2009). In vitro studies revealed that the INPP5E deletion mutant failed to localize to the cilia while retaining its inositol phosphatase activity, thus suggesting that spatial localization of INPP5E, as well as enzymatic activity, is critical to its function in the cilia (Jacoby et al., 2009).
Although INPP5E has been implicated in ciliogenesis, the functional role of inositol phosphatase in the cilia is not understood. The murine model of MORM syndrome yielded knockout animals that died shortly after birth (Jacoby et al., 2009). Thus, to seek a viable INPP5E model system, we examined the function of INPP5E in zebrafish cilia development by transient knockdown with morpholino anti-sense oligonucleotides specific for Inpp5e.
2. Materials and Methods
2.1 Reagents and DNA constructs
Anti-acetylated α-tubulin and anti-Myc monoclonal antibodies were purchased from Sigma (St. Louis, MO). Mouse antibody against INPP5E antibody was obtained from Abcam (Cambridge, MA). Secondary antibodies were AlexaFluor 488 and 546-conjugated donkey anti-mouse IgG, Cy3-conjugated donkey anti-mouse IgG, and horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse IgG (Jackson ImmunoResearch Laboratories, Inc. West Grove, PA). IRDye goat anti-mouse and anti-rabbit (680 and 800) were obtained from Li-cor Bioscience (Lincoln, NB). Myc-tagged INPP5E was previously described (Kisseleva et al., 2002). Site-directed mutagenesis for INPP5E R378C and R435Q mutants was performed using QuikChange II (Aglient, Santa Clara, CA).
2.2 Immunoblot analysis
Cell lysates were subjected to SDS-PAGE followed by immunoblot analysis with the indicated antibodies. Equal amounts of protein were resolved on 10–12% polyacrylamide gels, and protein bands were transferred to nitrocellulose membranes (BioRad, Hercules, CA), which were blocked with 5% non-fat dried milk in PBST; and incubated with the primary and then secondary antibodies as indicated. Odyssey imaging system (Li-Cor Bioscience, Lincoln, NE) was used to analyze the immunoblots.
2.3 Zebrafish immunohistochemistry and cilia measurements
Zebrafish (wildtype strain: AB tevbigan) (gift of Dr. Ryan Anderson, Indiana University, Indianapolis, IN) were raised and maintained at the Laboratory Animal Resource Center of Indiana University. All animal procedures were subject to the Institutional Animal Care and Use Committee of Indiana University approved protocols. The phenotypes of morphants were photographed with Leica DFC310 FX. Eye size was determined by the longest diameter of eye area. The diameter was measured using Leica Appication Suite V4.1 and NIH Image J v1.46.
Embryos were fixed overnight at 4°C in 4% PFA and 1% sucrose in PBS. Embryos were dechorionated and washed with PBST for 6–8 times 10 min each. Following one hour of blocking with 10% NGS and 0.5% 0BSA, immunostaining was performed with 1:200 anti-acetylated α-tubulin monoclonal antibody and later with 1:500 Alexa Fluor 546 goat anti-mouse conjugated IgG at 4°C overnight. KV cilia measurements were performed as described (Luo et al., 2012). Cresyl violet staining was performed as described (Luo et al., 2012).
2.4 Morpholino (MO) antisense oligonucleotides knockdown and mRNA rescue in zebrafish
Antisense MOs were designed and purchased from Gene Tools, Inc (Gene Tools, Philomath, OR). The Inpp5e ATG initiation codon sequence is GCTCACTCATCCTATTGGCGGGCTT. A mismatch morpholino MO: sequence ATGCGAAATCAAGGTTCGATCATCA served as a negative control. We also used a p53 ATG morpholino: GCGCCATTGCTTTGCAAGAATTG to test for off-target effects. Morpholino stocks were dissolved at 1 mM in water and 2 or 4 nl of injection solution (0.25% phenol red) containing 125–500 μM morpholino was injected into fertilized eggs at the one- to two-cell stage using a pressure injector, Pressure System IIe (Toohey Company, Fairfield, NJ). Synthetic mRNA was prepared from linearized human INPP5E-pcDNA3.1 DNA with Ambion mMessage mMachine® high-yield Capped RNA transcription kit, and purified with phenol-chloroform; mRNA was co-injected for rescue experiments.
2.5 Retrograde melanosome transport assay
The melanosome transport assay was performed as described (Yen, et al., 2006). Briefly, zebrafish 5 dpf larvae were exposed to epinephrine (50 mg/ml, Sigma) in the final concentration of 2 mg/ml in a dark room, and melanosome retractions were observed under the brightfield microscope Leica DFC310 FX. The end of melanosome transport was marked when all melanosomes in the head were perinuclear.
2.6 Statistical analysis
Statistical analysis was performed using SPSS software (SPSS, Chicago, IL) and the p value less than 0.05 was considered significant. An unpaired t-test was carried out to analyze if there was a significant difference between the cilia length or percentage of cilia formation as described above. ANOVA test was performed to analyze the difference observed in different groups of hTERT-RPE1 cells transfected with INPP5E constructs.
3. Results
3.1 Inpp5e knockdown results in cilia-dependent phenotypes
Inositol metabolism has been explored in a number of model organisms, including zebrafish (Danio rerio) (Sarmah, et al., 2007). We have examined zebrafish orthologs of known inositol polyphosphate 5-phosphatases and identified the orthologous zebrafish Inpp5e. Human INPP5E contains an N-terminal proline-rich domain (PRD), an inositol polyphosphate 5-phosphatase domain, and a lipid modification domain (ie, CAAX) in the C-terminus (Figure 1A). Similarly, the zebrafish Inpp5e also contains a PRD, an inositol polyphosphate 5-phosphatase domain, and a CAAX domain. Known mutations in patients with Joubert syndrome are found within the 5-phosphatase domain, which is conserved between the human and zebrafish. Comparative studies of the protein sequence show 65% identity and 82% similarity between the human and zebrafish INPP5E. In the 5-phosphatase domain, there is 71% identity and 86% similarity between these two species (Figure 1B). The conserved amino acid sequences in the 5-phosphatase domains, FWFGDFNFR and KQRTPSYTDRVLY, are nearly identical in both human and zebrafish Inpp5e genes. Interestingly, the disease-causing mutations in humans are found in basic residues of arginine and lysine (R378, R435, R512, R515, R563, and K580), which are conserved in both species. However, an important difference is in the PRD, which contains 23% proline in the N-terminus of the human gene but only 11% in the zebrafish gene. In addition, the C-terminal prenylation signal, CAAX, is conserved in both species; CSVS is found in human INPP5E and CSIS is found in zebrafish Inpp5e, suggesting a conserved role of lipid modification in both zebrafish and humans.
Figure 1. INPP5E in human and zebrafish.

(A) Domain structure of human INPP5E. Known mutations in patients with Joubert syndrome (red) and MORM syndrome (yellow) are marked within the 5-phosphatase domain.
(B) Comparison of 5-phosphatase domain between human and zebrafish INPP5E. Conserved 5-phosphatase domain (boxed), and Joubert syndrome mutations (X) are indicated (summary based on (Jacoby et al., 2009) (Bielas et al., 2009)). Sequence alignment generated by Sequence Manipulation Suite (www.bioinformatics.org/sms2).
To determine the functional significance of Inpp5e in cilia development, we established a zebrafish model by using antisense morpholino oligonucleotides to knockdown Inpp5e expression. To assess the in vivo effects of Inpp5e knockdown, we injected an Inpp5e translation-blocking morpholino and a mismatched control MO. As shown in Figure 2A, injection of Inpp5e morpholino specifically decreased Inpp5e protein expression as compared to β-actin; we verified Inpp5e expression by immunoblot analysis and demonstrated the specificity of INPP5E antibody. At 48 hpf stage, Inpp5e morphants exhibited phenotypes of microphthalmia, pericardial edema, body axis asymmetry, kinked tail, and pronephric cyst formation (Fig. 2B and Fig. 3A). Morphant embryos injected with increasing concentrations of Inpp5e MO demonstrated dose-dependent phenotypes in response to Inpp5e knockdown (Fig. 2C). Approximately 60% of morphants exhibited microphthalmia, which was observed over three independent sets of experiments (Fig. 2E). Many morphants also developed hypopigmentation and hydrocephalus (Fig. 2B and Fig. 3A).
Figure 2. Phenotype of Inpp5e morphants.

(A) Immunoblot analysis of 30 μg of total lysates of zebrafish embryo injected with control MO (4 ng) or Inpp5e MO (4 ng) at 48 hpf with anti-INPP5E and anti-β-actin antibodies.
(B) Zebrafish embryos were injected with p53 MO (2 ng) or p53 MO (2 ng) and Inpp5e MO (4 ng). Representative phenotypes of microphthalmia (black arrowhead), pericardial edema (small arrow), body axis asymmetry, kinked tail (white arrow), pronephric cyst formation (red arrow), and hypopigmentation were observed at 48 hpf (Top panel, scale bar 250 μm). The ventral sides of morphants are shown (Bottom, scale bar 100 μm).
(C) Dose-dependent effect of morpholinos in zebrafish. Control and Inpp5e MO at indicated doses were injected into zebrafish embryos, and phenotypes of microphthalmia, kinked tail, and body asymmetry were quantified at 48 hpf (ANOVA, F = 92, p = 3.6E-10), kinked tail (ANOVA, F = 3.6, p = 0.08), and body asymmetry (ANOVA, F = 5.2, p=0.04). (n = the number of injected embryos).
(D) Quantification of eye size of morphants at 24 hpf and 48 hpf. The eye size was determined by the longest diameters of eye balls in dorsal view. (n = 20 embryos, three independent experiments, unpaired t-test, * p = 1.2E-08, ns means not statistically significant).
(E) Inpp5e MO1 (4 ng) and control MO (4 ng) were injected into 1-cell zebrafish embryos. At 48 hpf, all the phenotypes were assessed and the total numbers of defective morphants in four independent experiments were quantified (n, the number of injected embryos, unpaired t-test, p=0.02).
(F) Cresyl violet staining of ocular sections of zebrafish larvae (5 dpf) injected with control MO (4 ng) or Inpp5e MO (4 ng). Scale bar 30 μm.
(G) Immunostaining of 3 dpf zebrafish larvae injected with control MO (4 ng) or Inpp5e MO (4 ng), followed by antibody staining against INPP5E (green) (DAPI, blue). Scale bar 10 μm.
Figure 3. Human INPP5E rescue phenotypes of Inpp5e morphants.
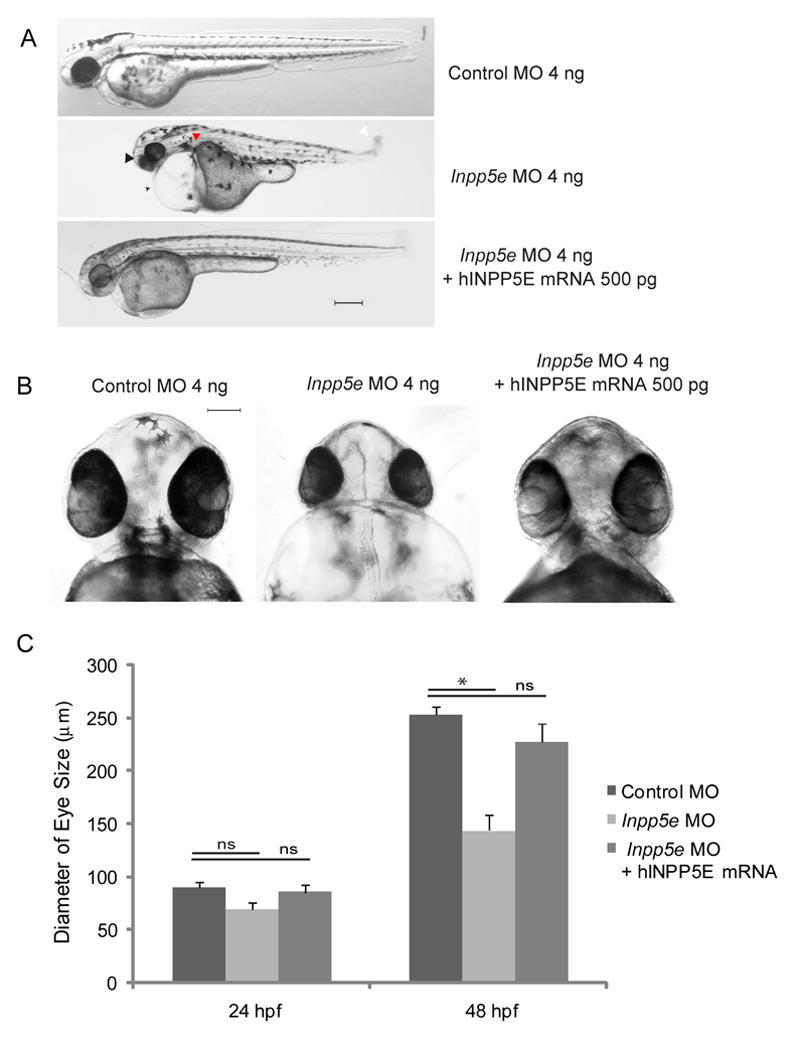
(A) Zebrafish embryos were injected with control MO (4 ng), Inpp5e MO (4 ng) or Inpp5e MO (4 ng) and human INPP5E mRNA (hINPP5E mRNA, 500 pg). Representative phenotypes of microphthalmia (arrowhead), pericardial edema (arrow), body axis asymmetry, kinked tail (white arrow), pronephric cyst formation (red arrow), and hypopigmentation were observed at 48 hpf (scale bar 250 μm).
(B) The ventral sides of embryos were injected with control MO (4 ng), Inpp5e MO (4 ng) or Inpp5e MO (4 ng) and human INPP5E mRNA (hINPP5E mRNA, 500 pg), scale bar 100 μm.
(C) Quantification of eye size of zebrafish morphants at 24 hpf and 48 hpf. (N=20 embryos, three independent experiments, unpaired t-test, * p = 1.61E-12).
To verify the phenotypes are specific to Inpp5e knockdown, we injected p53 MO into zebrafish embryos and showed that p53 MO alone did not result in either decreased eye size, generalized edema, or body axis asymmetry in zebrafish, but these phenotypes appeared in the embryos when co-injected with p53 MO and Inpp5e MO (Fig. 2B). After measuring the longest diameter of the eyes, we found a statistically significant decrease in eye size (unpaired t-test, p = 1.2E-08) at 48 hpf between p53 morphants (247 μm ± 3.5 μm) and p53 plus Inpp5e morphants (152 μm ± 11 μm) (Fig. 2D).
We examined the ocular phenotypes in detail and found the Inpp5e morphants exhibited abnormal retinal development compared to control zebrafish (Fig. 2F). The zebrafish larvae injected with control MO developed normal retinal structures with a well-defined nerve-fiber layer, inner nuclear and photoreceptor layers, but the Inpp5e MO-treated morphants exhibited disorganized retina with a lack of separation between the retinal cell layers. To examine the expression pattern of Inpp5e in the zebrafish retina, we performed immunofluorescence studies using anti-INPP5E antibodies. We confirmed the immunoreactivity of inositol phosphatase in the outer segment of photoreceptors; however, in zebrafish treated with Inpp5e MO, resulted in a loss of immunoreactivity in the eyes when compared to the controls (Fig. 2G).
To show that the body asymmetry, microphthalmia, and kinked tail are dependent on the loss-of-function of Inpp5e, we co-injected human INPP5E (hINPP5E) mRNA with Inpp5e MO to potentially rescue the observed phenotypes. Indeed, the expression of wildtype human INPP5E was able to partially rescue the loss-of-function of Inpp5e. The phenotypes of edema, body asymmetry and kinked tail reduced dramatically (Fig. 3A–B). The eye size of morphants co-injected with hINPP5E mRNA recovered to 215 μm ± 13 μm, compared with the morphants injected with Inpp5e MO (144 μm ± 14 μm) and control MO (253 μm ± 7.8 μm) (Fig 3C), thus supporting our hypothesis that the phenotypes observed are caused by the loss of Inpp5e.
3.2 Cilia defects in Kupffer’s vesicle of Inpp5e morphants
To evaluate whether INPP5E is necessary for cilia development, we examined the Kupffer’s vesicle (KV) of young larvae. The KV is a monociliated, fluid-containing structure in the zebrafish, orthologous to the mouse embryonic node (Hirokawa, et al., 2006). The KV is a spherical vesicle originating from dorsal forerunner cells and is readily visible in the tail-bud region. During early embryogenesis, the primary cilia within the KV generate directional flow just prior to the expression of asymmetric genes in the lateral cells. Previous studies have demonstrated that the ciliated KV is required for early somitogenesis for left-right patterning in the heart, gut, and the brain (Essner, et al., 2005, Kawakami, et al., 2005, Oteiza, et al., 2008). The cilia within the KV regulate left-right body axis and organ development, thus, we examined the KV by immunohistochemistry with anti-acetylated α-tubulin to stain the cilia at the 6-somite stage (Fig. 4A). Inpp5e morphants showed an approximate 50% decrease in the number of cilia within the KV (59 ± 3 in control embryos vs 35 ± 7 in Inpp5e morphants, unpaired t-test, t = 14.1, p = 0.02) when compared to control MO-injected embryos (Fig. 4B). In addition, the average length of cilia within the KV was found to be shorter in the Inpp5e morphants (2.9 μm ± 0.6 μm) as compared to controls (4.9 μm ± 0.4 μm) (unpaired t-test, t=12.4, p =1.56E-08) (Fig. 4C). Cilia formation in the KV can be rescued to a greater number (51 ± 5) and longer cilia length (4.3 μm ± 0.5 μm) after co-injection with hINPP5E mRNA. Thus, our results support that zebrafish Inpp5e function is required for KV cilia development.
Figure 4. Kupffer’s vesicle cilia defect in Inpp5e morphants.
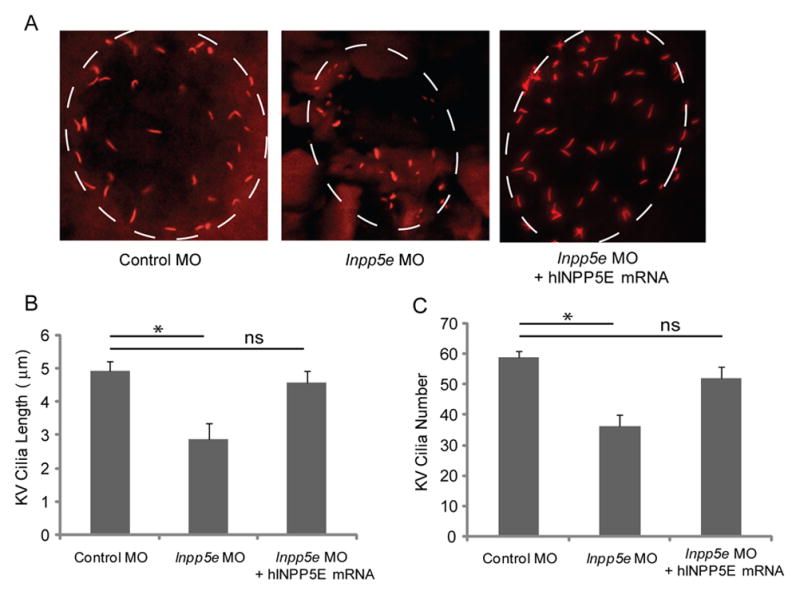
(A) Human INPP5E mRNA could rescue the loss of Inpp5e. KV cilia of zebrafish embryos injected with control MO (4 ng), Inpp5e MO (4 ng) or Inpp5e MO (4 ng) and hINPP5E mRNA (500 ng) at 6-somite stage were immunostained with acetylated α-tubulin (red), representative images are shown (dash line indicates border of KV).
(B–C) Quantification of number (B) and length (C) of KV cilia in zebrafish embryos injected with control MO (4 ng), Inpp5e MO (4 ng) or Inpp5e MO (4 ng) and hINPP5E mRNA (500 ng). (N=20 embryos, three independent experiments, unpaired t-test, * p = 0.02 in B and * p = 1.56E-08 in C).
3.3 Pronephric duct cilia defects of Inpp5e morphants
The pronephric kidney is the first stage of kidney development in vertebrate embryos (Otto, et al., 2003). Arising from the intermediate mesoderm, the pronephros has a single nephron that is attached to the nephric duct, which forms the Wolffian duct and ureter of the adult human kidney (Paces-Fessy, et al., 2012). In zebrafish, the primitive pronephros contains motile cilia that may generate flow by unidirectional beating (Essner, et al., 2002). It has been shown that obstruction of fluid flow may result from the loss of ciliary function, leading to cystic kidney disease (Kramer-Zucker, et al., 2005). As cystic kidney has been implicated in human Joubert syndrome, we examined the zebrafish cilia in the pronephric ducts by immunofluorescence with anti-acetylated α-tubulin at the 24 hpf stage (Fig. 5A). As compared to control MO injected embryos, there was an approximate 50% shortening in the length of cilia of Inpp5e morphants (22 μm ± 2.1 μm) vs control embryos (10 μm ± 1.4 μm) (unpaired t-test, t = 21.3, p = 8.57E-08), and cilia could be rescued (18 μm ± 1.8 μm) by co-injection with hINPP5E mRNA (Fig. 5B). This finding may explain the phenotypes of renal cysts and generalized edema in the Inpp5e morphants. Our data supports that zebrafish Inpp5e function is involved in the cilia development in pronephric kidney.
Figure 5. Pronephric cilia defect in Inpp5e morphants.
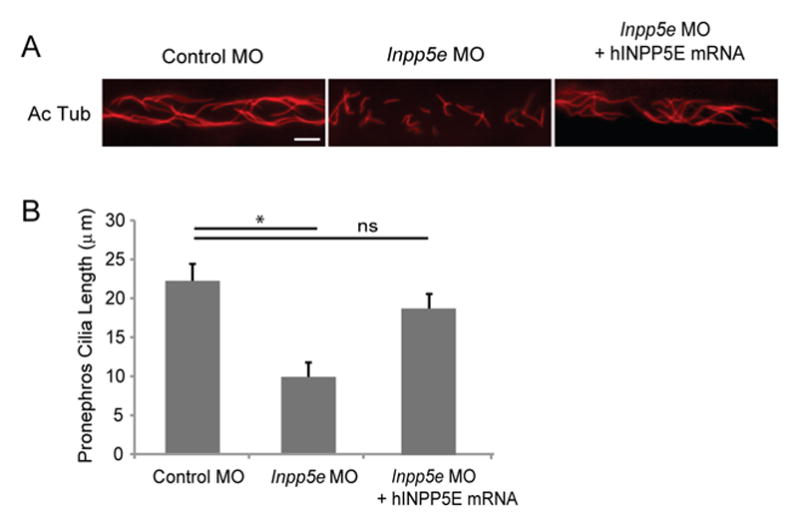
(A) INPP5E mRNA rescue of Inpp5e pronephric cilia formation. Representative image of pronephric cilia of zebrafish embryos at 24 hpf stage, injected with control MO (4 ng), Inpp5e MO (4 ng) or Inpp5e MO (4 ng) and hINPP5E mRNA (500 ng), immunostaining with acetylated α-tubulin (red). Scale bar 10 μm.
(B) Pronephric cilia length of control and Inpp5e MO. Pronephric cilia of zebrafish embryos injected with control MO (4 ng), Inpp5e MO (4 ng) or Inpp5e MO (4 ng) and hINPP5E mRNA (500 ng) at 24 hpf stage were analyzed by immunostaining with acetylated α-tubulin and cilia length was measured. (N=20 embryos, three independent experiments, unpaired t-test, * p = 8.57E-08).
3.4 Melanosome transport defects in Inpp5e morphants
In addition to KV and pronephros cilia, melanosome transport has been shown to be dependent on ciliary motor proteins (Yen et al., 2006). Melanosome shuttling between the cell periphery and the perinuclear region is mediated by kinesin II and dynein. Anterograde transport of the melanosomes to the cell periphery is caused by kinesin II motors while retrograde transport to the perinuclear region is performed by dyneins. Many ciliary proteins, such as BBS family members, are involved in retrograde melanosome transport in the zebrafish (Yen et al., 2006). Since we noted a pigmentary defect in the Inpp5e morphants (Fig. 2B and Fig. 3A), we hypothesized that melanosome transport may also be affected in the Inpp5e-deficient zebrafish. Retrograde or anterograde trafficking of melanosomes is known to be stimulated by epinephrine or caffeine, respectively (Nascimento, et al., 2003); therefore, we measured the rates of epinephrine-induced melanosome retraction in zebrafish morphants. In 5 dpf zebrafish larvae treated with epinephrine, the melanosomes contracted rapidly and perinuclear accumulation of melanosomes were observed (Fig. 6A). The pigment accumulation of Inpp5e morphants takes three times longer to complete than the control MO injected (6.0 ± 0.4 minutes in control vs 1.9 ± 0.2 minute in Inpp5e zebrafish, unpaired t-test, t = 28.9, p = 1.17E-18) (Fig. 6B). Moreover, the delayed melanosome retraction was partially rescued by co-injecting human INPP5E mRNA (Fig. 6B).
Figure 6. Impaired melanosome transport in Inpp5e morphants.
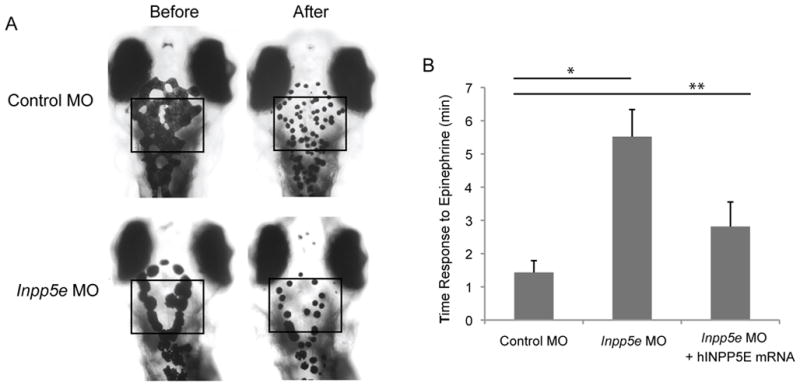
(A) Inpp5e morphants showed slowed retrograde melanosome transport. Five day-old larvas injected with morpholinos were treated with epinephrine, and the time required for all melanosomes in the head and trunk to retract was determined. Representative photos are shown.
(B) Quantification of the response time for epinephrine treatments in the control MO (2 ng), Inpp5e MO (2 ng), and Inpp5e MO (2 ng) and hINPP5E mRNA (400 pg) embryos. (N=20 embryos, three independent experiments, unpaired t-test, * p = 1.17E-18, ** p = 1.5E-12).
3.5 INPP5E mutants affect eye development in zebrafish
Recently INPP5E was identified in the connecting cilium in the photoreceptors of mice (Jacoby et al., 2009). In their INPP5E transgenic mice, short cilia development caused the absence of the photoreceptor cell layer. Almost all identified INPP5E mutations in Joubert syndrome are localized in the 5-phosphatase domain (Bielas et al., 2009). To examine the effect of mutant INPP5E on cilia formation during eye development, we first examined the localization of INPP5E mutants in the cilia of hTERT-RPE1 cells. In the hTERT-RPE1 cells expressing control vector, WT INPP5E, or mutant INPP5E R378C, and R435Q, we assessed the cilia length and percent of ciliated cells at 48 hours post serum-starvation. The cells transfected with mutant INPP5E R378C and R435Q have shortened cilia length and decreased cilia formation, thus suggesting these mutants may act as dominant negatives in cilia regulation (data not shown). We generated the mRNA of R378C and R435Q INPP5E mutants, which were co-injected with Inpp5e MO zebrafish embryos at the 1-cell stage. In zebrafish embryos injected with R378C and R435Q mRNA resulted in the phenotypes of generalized edema and body asymmetry (Fig. 7A). As shown in Fig. 7A and Fig. 7B, the zebrafish morphants injected with mutant INPP5E R378C and R435Q have smaller eyes (132 μm ± 20 μm in INPP5E R378C and 152 μm ± 18 μm in INPP5E R435Q) as compared to the zebrafish injected with wild type INPP5E (215 μm ± 13 μm) (unpaired t-test, p=3.6E-08). Taken together, we have showed that the disease-causing mutations in INPP5E affected cilia associated eye development.
Figure 7. INPP5E mutants in zebrafish eye development.
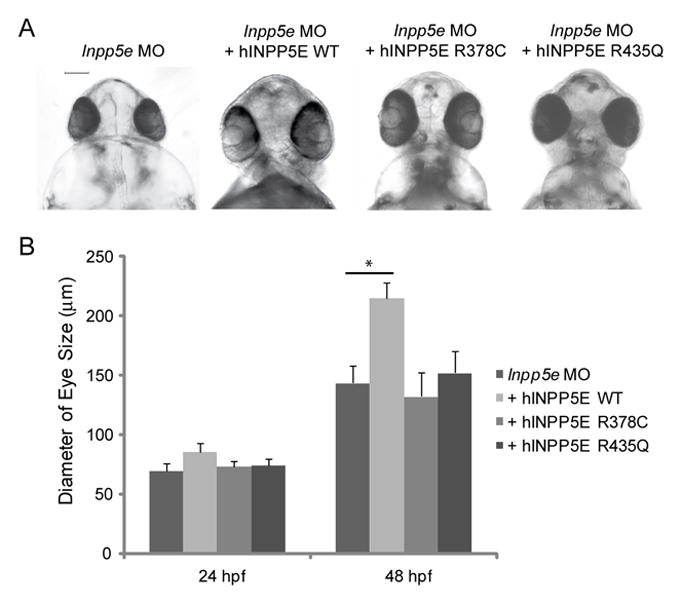
(A) Zebrafish embryos were injected with Inpp5e MO (4 ng), Inpp5e MO (4 ng) and hINPP5E WT mRNA (500 pg), Inpp5e MO (4 ng) and hINPP5E R378C mRNA (500 pg), or Inpp5e MO (4 ng) and hINPP5E R435Q mRNA (500 pg). The ventral sides of morphants are shown, scale bar 100 μm.
(B) Quantification of eye size of morphants at 24 hpf and 48 hpf. (N=20 embryos, three independent experiments, unpaired t-test, * p = 3.6E-08).
4. Discussion
Lipid composition of the ciliary membrane is highly regulated. Phosphoinositide dysregulation has been implicated in ciliopathies in several recent studies (Bielas et al., 2009, Jacoby et al., 2009). INPP5E is an inositol 5-phosphatase involved in a cilia phenotype that underlies two rare human syndromic ciliopathies, Joubert syndrome and MORM syndrome, both of which present with retinal degeneration, kidney cysts, and mental developmental delays (Bielas et al., 2009, Jacoby et al., 2009). Studies in mice and humans have demonstrated that INPP5E plays a critical role in cilia function. In this study, we utilized zebrafish to examine the role of Inpp5e in the development of the cilium. The Inpp5e morphants develop ciliopathy-like phenotypes of hydrocephalus, retinal dysplasia, and body asymmetry, which is consistent with the murine knockout models. Since Inpp5eΔ/Δ mouse died soon after birth (Jacoby et al., 2009), the availability of a zebrafish model can provide a novel animal model to examine the role of inositol phosphatases in cilia development.
As previously shown in other ciliopathy studies, the zebrafish is a robust embryological model system to examine cilia function in early organogenesis (Chakarova, et al., 2011, Ghosh, et al., 2010, Hurd, et al., 2010, Khanna, et al., 2005, Murga-Zamalloa et al., 2010) because it offers optical clarity, rapid development, ease of genetic manipulation and high resolution imaging for in vivo studies that would not be achieved in larger animal models. Cilia function is required for maintaining left-right asymmetry and are conserved in nearly all vertebrates (Capdevila, et al., 2000). During early organogenesis, the left-right axis is maintained by asymmetric expression of LR-specific genes, such as pitx2, nodal, lefty1 and lefty2 (Hamada, et al., 2002). KV cilia are responsible for generating a transient left-biased calcium influx. The calcium flux is required for left-right determination (Schneider, et al., 2008). We show here that Inpp5e morphants exhibited left-right asymmetry as well as abnormal cilia formation in the KV, which support that INPP5E is a ciliary protein. Since PI(4,5)P2 is the precursor for IP3, which was shown to play a role in left-right body axis determination in Xenopus (Hatayama, et al., 2011), it is likely that the soluble inositol molecules may be the effectors for the body axis regulation.
The finding of pronephric duct cilia defects in Inpp5e morphants support the clinical observation of renal cyst development in human Joubert patients. The cilia within the pronephros in Inpp5e morphants were shorter and less organized than the control zebrafish, suggesting the motility of the cilia maybe severely affected. Cilia beating has been shown to be important in flow regulation in the pronephric ducts (Kramer-Zucker et al., 2005); the loss of cilia organization may reduce fluid movement resulting in cystic kidney formation. It will be important to visualize the ciliary beating in both the KV and pronephric ducts to determine if Inpp5e loss-of-function directly affects fluid transport in these embryonic structures.
Previously, Bielas et al. has shown that the fibroblasts from patients with mutations in the INPP5E 5-phosphatase domain exhibited ciliary instability (Bielas et al., 2009). Our results with the two disease-causing INPP5E mutants (R378C and R435Q) support this observation. In both mutants, we found that the cilia localization was unaffected by the loss of the inositol phosphatase activity. However, we did observe increased swelling of ciliary membrane at the distal tip of the cilia (unpublished results), suggesting the 5-phosphatase activity may play a role in retrograde transport of ciliary proteins. In addition to the enzymatic activity of INPP5E, the conservation of the C-terminal CAAX domain in both human and zebrafish INPP5E suggests that the lipid modification may play a vital role in the function of INPP5E in cilia. It will be important to identify the enzyme involved in this post-translational modification; and the zebrafish will be an excellent model organism to test the functional importance of the CAAX domain.
Recently, three independent groups have identified another inositol polyphosphate phosphatase, OCRL, to also be involved in cilia function (Coon, et al., 2012, Luo et al., 2012, Rbaibi, et al., 2012). Mutations in OCRL are found in Lowe syndrome and Dent syndrome (Attree, et al., 1992, Lewis, 1993–2001, Reilly, et al., 1990). Lowe syndrome, also known as Oculocerebrorenal syndrome of Lowe, is a rare congenital X-linked recessive disorder characterized by congenital cataracts, glaucoma, renal tubular dysfunction, and developmental delays (Attree et al., 1992). Similar to Lowe syndrome, Type II Dent syndrome presents with congenital renal tubular dysfunction but lacks the ocular and cerebral phenotypes (Hoopes, et al., 2005, Schurman & Scheinman, 2009). OCRL was found to be trafficked to the cilia by Rab8 and Ses1/Ses2 and that the inositol phosphatase activity may be important in this ciliary recruitment (Coon et al., 2012). Another evidence for inositol regulation of cilia comes from studies of the tubby-like proteins. The tubby mouse, which develops adult-onset obesity, has mutations in the Tub gene; the tubby-like proteins are known to interact with phosphoinositides. Recently, Mukhopadhyay et al. has found Tubby-like protein 3 (TULP3) strongly binds to PI(4,5)P2, followed by PI(3,4)P2, and PI(3,4,5)P3. Mutants of TULP3 that do not bind to phosphoinositides failed to traffic GPCR into the cilium (Mukhopadhyay, et al., 2010). Therefore, it was proposed that PIP2 might regulate the specificity of trafficking of ciliary proteins.
In addition to lipid phosphoinositides, soluble inositol phosphates have been implicated in ciliogenesis and cilia-mediated signaling in zebrafish. Phosphoinositides Kinase 1 (PK1) is a 2-kinase that phosphorylates I(1,3,4,5,6)P5 to generate IP6 (Sarmah et al., 2007). Knockout of IPK1 in zebrafish resulted in loss of cilia maintenance as well as cessation of ciliary beating in the KV (Sarmah et al., 2007). Although the ultrastructure of the cilia does not appear disrupted, the functional transport of melanosomes implicated the loss of ciliary function. Immunofluorescence studies showed the distribution of IPK1 to the basal body in the cilia, suggesting a role of phosphorylation of IP5 to IP6 may be required in cilia function (Sarmah & Wente, 2010b). In addition to the direct cilia function, Sarmah et al. showed that higher order inositol phosphates are important in regulating Hg signaling, which is known to occur through the primary cilium. By knocking down IP6K2 expression in zebrafish, which prevents the formation of diphosphoryl inositol phosphates (PP-IPs), Sarmah et al. showed IP6K2 is a positive regulator of Hg signaling by acting upstream of transcription factor Gli1 (Sarmah & Wente, 2010a).
Although zebrafish offers a robust model system to examine the function of inositol metabolism in cilia formation, there are limitations to this approach. One of the drawbacks is the duration of Hg signaling the effectiveness of morpholinos; the degradation of the oligonucleotides over the first 48–72 hour period limits long-term studies of eye development and potential delayed phenotypes. Also, similar to the mouse model, another drawback is that the pleiotropic effect of inpp5e knockdown may result in early lethality of the zebrafish morphants. In the future, this may be circumvented by transplant technology, removing only the eyes into wild type zebrafish, which can allow specific examinations of ocular phenotypic rather than a global effect.
5. Conclusion
In this study, we present a knockdown zebrafish model using antisense morpholinos against Inpp5e that resemble human Joubert syndrome. This phenotype can be rescued by concomitant injection of normal human INPP5E mRNA. Following the knockdown of Inpp5e, we demonstrated a progressive loss of cilia at Kupffer’s vesicle, pronephric kidney, and subsequent alterations to organ laterality, such as bent body axes and cardiac left-right asymmetry. The high degree of homology between human and zebrafish INPP5E, and the rescue of human INPP5E shows that the zebrafish is a good model organism to study inositol phosphatase function in the cilia. The data presented supports a role for INPP5E in ciliary formation to explain the phenotypes in Joubert syndrome, and the knockdown zebrafish provides an animal model to test potential treatments in the future.
Highlights.
We present a knockdown zebrafish model for inositol 5-phosphatase INPP5E
Morphants defects of cilia in Kupffer’s vesicle and pronephros ducts
Wildtype human INPP5E can rescue the defect in zebrafish morphants
Acknowledgments
We thank Drs. Philip Majerus, Jeffrey Travers, Timothy Corson, Akhilesh Kumar, and Michael Conwell for thoughtful comments during the preparation of the manuscript. This work was funded by NIH K08 EY022058, a Pediatric Research Grant from the Knights Templar Eye Foundation, and a Clinician-Scientist award from American Glaucoma Society (Y.S.).
Abbreviations
- RPE
retinal pigmented epithelium
- NHF
normal human fibroblasts
- MORM syndrome
mental retardation, truncal obesity, retinal dystrophy, and micropenis
- RBD
RAB binding domain
- ONL
outer nuclear layer
- INL
inner nuclear layer
- GCL
Ganglion cell layer
- IFT
intraflagellar transport
- hTERT-RPE1
human telomerase transformed RPE cells
- PFA
paraformaldehyde
- BSA
bovine serum albumin
- NGS
Normal goat serum
- PBS
phosphate buffered saline
- FCS
fetal calf serum
- PRD
proline rich domain
- dpf
days post-fertilization
- hpf
hours post-fertilization
- KV
Kupffer’s vesicle
Footnotes
Conflict of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39(7):882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- Asano T, Mochizuki Y, Matsumoto K, Takenawa T, Endo T. Pharbin, a novel inositol polyphosphate 5-phosphatase, induces dendritic appearances in fibroblasts. Biochemical and biophysical research communications. 1999;261(1):188–195. doi: 10.1006/bbrc.1999.0998. [DOI] [PubMed] [Google Scholar]
- Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, et al. The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature. 1992;358(6383):239–242. doi: 10.1038/358239a0. [DOI] [PubMed] [Google Scholar]
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41(9):1032–1036. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83(2):170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila J, Vogan KJ, Tabin CJ, Izpisua Belmonte JC. Mechanisms of left-right determination in vertebrates. Cell. 2000;101(1):9–21. doi: 10.1016/S0092-8674(00)80619-4. [DOI] [PubMed] [Google Scholar]
- Chakarova CF, Khanna H, Shah AZ, Patil SB, Sedmak T, Murga-Zamalloa CA, et al. TOPORS, implicated in retinal degeneration, is a cilia-centrosomal protein. Hum Mol Genet. 2011;20(5):975–987. doi: 10.1093/hmg/ddq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon BG, Hernandez V, Madhivanan K, Mukherjee D, Hanna CB, Barinaga-Rementeria Ramirez I, et al. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Human molecular genetics. 2012;21(8):1835–1847. doi: 10.1093/hmg/ddr615. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Dyson JM, Fedele CG, Davies EM, Becanovic J, Mitchell CA. Phosphoinositide phosphatases: just as important as the kinases. Sub-cellular biochemistry. 2012;58:215–279. doi: 10.1007/978-94-007-3012-0_7. [DOI] [PubMed] [Google Scholar]
- Essner JJ, Amack JD, Nyholm MK, Harris EB, Yost HJ. Kupffer’s vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development. 2005;132(6):1247–1260. doi: 10.1242/dev.01663. [DOI] [PubMed] [Google Scholar]
- Essner JJ, Vogan KJ, Wagner MK, Tabin CJ, Yost HJ, Brueckner M. Conserved function for embryonic nodal cilia. Nature. 2002;418(6893):37–38. doi: 10.1038/418037a. [DOI] [PubMed] [Google Scholar]
- Fisch C, Dupuis-Williams P. Ultrastructure of cilia and flagella - back to the future! Biology of the cell/under the auspices of the European Cell Biology Organization. 2011;103(6):249–270. doi: 10.1042/BC20100139. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Murga-Zamalloa CA, Chan L, Hitchcock PF, Swaroop A, Khanna H. Human retinopathy-associated ciliary protein retinitis pigmentosa GTPase regulator mediates cilia-dependent vertebrate development. Hum Mol Genet. 2010;19(1):90–98. doi: 10.1093/hmg/ddp469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada H, Meno C, Watanabe D, Saijoh Y. Establishment of vertebrate left-right asymmetry. Nature reviews Genetics. 2002;3(2):103–113. doi: 10.1038/nrg732. [DOI] [PubMed] [Google Scholar]
- Hatayama M, Mikoshiba K, Aruga J. IP3 signaling is required for cilia formation and left-right body axis determination in Xenopus embryos. Biochemical and biophysical research communications. 2011;410(3):520–524. doi: 10.1016/j.bbrc.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Tanaka Y, Okada Y, Takeda S. Nodal flow and the generation of left-right asymmetry. Cell. 2006;125(1):33–45. doi: 10.1016/j.cell.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Hoopes RR, Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, et al. Dent Disease with mutations in OCRL1. Am J Hum Genet. 2005;76(2):260–267. doi: 10.1086/427887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd T, Zhou W, Jenkins P, Liu CJ, Swaroop A, Khanna H, et al. The retinitis pigmentosa protein RP2 interacts with polycystin 2 and regulates cilia-mediated vertebrate development. Hum Mol Genet. 2010;19(22):4330–4344. doi: 10.1093/hmg/ddq355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41(9):1027–1031. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Raya A, Raya RM, Rodriguez-Esteban C, Izpisua Belmonte JC. Retinoic acid signalling links left-right asymmetric patterning and bilaterally symmetric somitogenesis in the zebrafish embryo. Nature. 2005;435(7039):165–171. doi: 10.1038/nature03512. [DOI] [PubMed] [Google Scholar]
- Khanna H, Hurd TW, Lillo C, Shu X, Parapuram SK, He S, et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J Biol Chem. 2005;280(39):33580–33587. doi: 10.1074/jbc.M505827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Krishnaswami SR, Gleeson JG. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet. 2008;17(23):3796–3805. doi: 10.1093/hmg/ddn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva MV, Cao L, Majerus PW. Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits Akt/protein kinase B phosphorylation and leads to apoptotic cell death. The Journal of biological chemistry. 2002;277(8):6266–6272. doi: 10.1074/jbc.M105969200. [DOI] [PubMed] [Google Scholar]
- Kisseleva MV, Wilson MP, Majerus PW. The isolation and characterization of a cDNA encoding phospholipid-specific inositol polyphosphate 5-phosphatase. J Biol Chem. 2000;275(26):20110–20116. doi: 10.1074/jbc.M910119199. [DOI] [PubMed] [Google Scholar]
- Kong AM, Horan KA, Sriratana A, Bailey CG, Collyer LJ, Nandurkar HH, et al. Phosphatidylinositol 3-phosphate [PtdIns3P] is generated at the plasma membrane by an inositol polyphosphate 5-phosphatase: endogenous PtdIns3P can promote GLUT4 translocation to the plasma membrane. Molecular and cellular biology. 2006;26(16):6065–6081. doi: 10.1128/MCB.00203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong AM, Speed CJ, O’Malley CJ, Layton MJ, Meehan T, Loveland KL, et al. Cloning and characterization of a 72-kDa inositol-polyphosphate 5-phosphatase localized to the Golgi network. The Journal of biological chemistry. 2000;275(31):24052–24064. doi: 10.1074/jbc.M000874200. [DOI] [PubMed] [Google Scholar]
- Kramer-Zucker AG, Olale F, Haycraft CJ, Yoder BK, Schier AF, Drummond IA. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development. 2005;132(8):1907–1921. doi: 10.1242/dev.01772. [DOI] [PubMed] [Google Scholar]
- Lee JE, Gleeson JG. Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr Opin Neurol. 2011;24(2):98–105. doi: 10.1097/WCO.0b013e3283444d05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RA, Nussbaum RL, Brewer ED. Lowe Syndrome. GeneReviews. 1993–2001 Jul 24; [Google Scholar]
- Luo N, West CC, Murga-Zamalloa CA, Sun L, Anderson RM, Wells CD, et al. OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Human molecular genetics. 2012 doi: 10.1093/hmg/dds163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen DP, Koenekoop RK, Khanna H, Jenkins PM, Lopez I, Swaroop A, et al. Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proc Natl Acad Sci U S A. 2007;104(40):15917–15922. doi: 10.1073/pnas.0704140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, et al. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes & development. 2010;24(19):2180–2193. doi: 10.1101/gad.1966210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga-Zamalloa CA, Atkins SJ, Peranen J, Swaroop A, Khanna H. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum Mol Genet. 2010;19(18):3591–3598. doi: 10.1093/hmg/ddq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- Nascimento AA, Roland JT, Gelfand VI. Pigment cells: a model for the study of organelle transport. Annual review of cell and developmental biology. 2003;19:469–491. doi: 10.1146/annurev.cellbio.19.111401.092937. [DOI] [PubMed] [Google Scholar]
- Novarino G, Akizu N, Gleeson JG. Modeling human disease in humans: the ciliopathies. Cell. 2011;147(1):70–79. doi: 10.1016/j.cell.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms LM, Horan KA, Rahman P, Seaton G, Gurung R, Kethesparan DS, et al. The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem J. 2009;419(1):29–49. doi: 10.1042/BJ20081673. [DOI] [PubMed] [Google Scholar]
- Oteiza P, Koppen M, Concha ML, Heisenberg CP. Origin and shaping of the laterality organ in zebrafish. Development. 2008;135(16):2807–2813. doi: 10.1242/dev.022228. [DOI] [PubMed] [Google Scholar]
- Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nature genetics. 2003;34(4):413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paces-Fessy M, Fabre M, Lesaulnier C, Cereghini S. Hnf1b and Pax2 cooperate to control different pathways in kidney and ureter morphogenesis. Human molecular genetics. 2012 doi: 10.1093/hmg/dds141. [DOI] [PubMed] [Google Scholar]
- Pearson CG, Culver BP, Winey M. Centrioles want to move out and make cilia. Dev Cell. 2007;13(3):319–321. doi: 10.1016/j.devcel.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Pearson CG, Winey M. Basal body assembly in ciliates: the power of numbers. Traffic. 2009;10(5):461–471. doi: 10.1111/j.1600-0854.2009.00885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirruccello M, De Camilli P. Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends in biochemical sciences. 2012;37(4):134–143. doi: 10.1016/j.tibs.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rbaibi Y, Cui S, Mo D, Carattino M, Rohatgi R, Satlin LM, et al. OCRL1 modulates cilia length in renal epithelial cells. Traffic. 2012;9999(999A) doi: 10.1111/j.1600-0854.2012.01387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly DS, Lewis RA, Nussbaum RL. Genetic and physical mapping of Xq24–q26 markers flanking the Lowe oculocerebrorenal syndrome. Genomics. 1990;8(1):62–70. doi: 10.1016/0888-7543(90)90226-k. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Snell WJ. The ciliary membrane. Current opinion in cell biology. 2010;22(4):541–546. doi: 10.1016/j.ceb.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmah B, Wente SR. Inositol hexakisphosphate kinase-2 acts as an effector of the vertebrate Hedgehog pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010a;107(46):19921–19926. doi: 10.1073/pnas.1007256107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmah B, Wente SR. Zebrafish inositol polyphosphate kinases: new effectors of cilia and developmental signaling. Adv Enzyme Regul. 2010b;50(1):309–323. doi: 10.1016/j.advenzreg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmah B, Winfrey VP, Olson GE, Appel B, Wente SR. A role for the inositol kinase Ipk1 in ciliary beating and length maintenance. Proc Natl Acad Sci U S A. 2007;104(50):19843–19848. doi: 10.1073/pnas.0706934104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider I, Houston DW, Rebagliati MR, Slusarski DC. Calcium fluxes in dorsal forerunner cells antagonize beta-catenin and alter left-right patterning. Development. 2008;135(1):75–84. doi: 10.1242/dev.004713. [DOI] [PubMed] [Google Scholar]
- Schurman SJ, Scheinman SJ. Inherited cerebrorenal syndromes. Nat Rev Nephrol. 2009;5(9):529–538. doi: 10.1038/nrneph.2009.124. [DOI] [PubMed] [Google Scholar]
- Travaglini L, Brancati F, Attie-Bitach T, Audollent S, Bertini E, Kaplan J, et al. Expanding CEP290 mutational spectrum in ciliopathies. Am J Med Genet A. 2009;149A(10):2173–2180. doi: 10.1002/ajmg.a.33025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42(7):619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HJ, Tayeh MK, Mullins RF, Stone EM, Sheffield VC, Slusarski DC. Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer’s vesicle cilia function. Hum Mol Genet. 2006;15(5):667–677. doi: 10.1093/hmg/ddi468. [DOI] [PubMed] [Google Scholar]