

Abstract
Traumatic brain injury (TBI) results in significant inflammation which contributes to the evolving pathology. Previously we have demonstrated that cyclic AMP (cAMP), a molecule involved in inflammation, is downregulated after traumatic brain injury (TBI). To determine the mechanism by which cAMP is downregulated after TBI, we determined whether TBI induces changes in phosphodiesterase (PDE) expression. Adult male Sprague Dawley rats received moderate parasagittal fluid-percussion brain injury (FPI) or sham injury, and the ipsilateral, parietal cortex was analyzed by western blotting. In the ipsilateral parietal cortex, expression of PDE1A, PDE4B2, and PDE4D2, significantly increased from 30 min to 24 hr post-injury. PDE10A significantly increased at 6 and 24 hr after TBI. Phosphorylation of PDE4A significantly increased from 6 hr to 7 days post-injury. In contrast, PDE1B, PD4A5, and PDE4A8 significantly decreased after TBI. No changes were observed with PDE1C, PDE3A, PDE4B1/3, PDE4B4, PDE4D3, PDE4D4, PDE8A, or PDE8B. Colocalization studies showed that PDE1A, PDE4B2, and phospho-PDE4A were neuronally expressed, whereas PDE4D2 was expressed in neither neurons nor glia. These findings suggest that therapies to reduce inflammation after TBI could be facilitated with targeted therapies, in particular for PDE1A, PDE4B2, PDE4D2, or PDE10A.
Keywords: cAMP, fluid-percussion, inflammation, isoform, phosphodiesterase, traumatic brain injury
Traumatic brain injury (TBI) initiates a range of biochemical reactions that have deleterious effects on recovery. Cyclic cAMP (cAMP) is a ubiquitous second messenger involved in many physiological mechanisms including cell survival, inflammation, synaptic plasticity, and learning. After TBI and other CNS injuries, cAMP levels decrease and return to baseline by 1–3 days post-injury (Pearse et al. 2004, Atkins et al. 2007b). cAMP is synthesized from ATP via adenylyl cyclases and hydrolyzed by phosphodiesterases (PDEs) into the inactive 5′AMP. PDEs exert strong control on cAMP levels because the capacity for cAMP hydrolysis in most cells is an order of magnitude greater than the maximum synthesis rate for cAMP.
Several PDE inhibitors are proven anti-inflammatory drugs, and thus PDE inhibitors are potential therapeutics for TBI since TBI has a significant inflammatory component (Dietrich et al. 2004, Morganti-Kossmann et al. 2007). PDE inhibitors prevent cAMP hydrolysis in immune cells, decreasing production of pro-inflammatory cytokines such as interleukin-1β and tumor necrosis factor-α (Zhang et al. 2002a, Verghese et al. 1995). However, the utility of general PDE4 inhibitors as anti-inflammatory agents are hampered by the constraint of side effects such as nausea, emesis, and gastric secretions (Robichaud et al. 2002). In addition, we have recently found that a general PDE4 inhibitor, rolipram, increases hemorrhage in the injured brain (Atkins et al. 2012). Thus, the development of isoform-selective PDE inhibitors for brain trauma would benefit by an understanding of the specific isoforms that are involved in the regulation of cAMP and pathogenesis of TBI.
There are 11 different PDE families and each of these families have multiple genes and splice variants, resulting in over 100 unique PDEs isoforms (Conti & Beavo 2007). PDEs 1, 2, 3, 10, and 11 hydrolyze both cAMP and cGMP, PDEs 4, 7, and 8 are specific for cAMP, and PDEs 5, 6, and 9 are specific for cGMP. An abundant family found in the brain that selectively degrades cAMP is PDE4, although PDE1, 2, 3, 7, 8, and 10 are also found in the brain (Reneerkens et al. 2009).
The PDE4 family is composed of 4 different genes, 4A, B, C, and D and they are distinguished from other PDEs by a unique N terminus, which is involved in intracellular targeting and regulating catalytic activity (Conti & Beavo 2007). Each of the genes is distinguished by conserved domains, dividing them into four groups: long, short, supershort, and dead-short isoforms. Long isoforms contain both an upstream conserved region 1 (UCR1) and UCR2, whereas all other isoforms lack UCR1. UCR1 is phosphorylated by protein kinase A (PKA), activating the enzyme (MacKenzie et al. 2002). The catalytic domain is phosphorylated by phosphoinositide-3-kinase as well as extracellular signal-regulated kinase 1/2 (Hill et al. 2006). PDE4A and 4B are, in particular, implicated in several neurological disorders such as schizophrenia, depression, and bipolar disorder (Fatemi et al. 2008a, Fatemi et al. 2008b).
Although broad, non-specific PDE inhibitors improve outcome in several CNS injury paradigms, the development of more targeted PDE inhibitors could abrogate some of the side effects of non-specific PDE inhibitors encountered in clinical trials (Bruno et al. 2009, Burgin et al. 2010, Bruno et al. 2011, Li et al. 2011a, Block et al. 2001, Nikulina et al. 2004, Pearse et al. 2004). It is still unknown whether specific PDEs are differentially expressed after CNS injury. There are some reports that PDE4B and 4D are upregulated after experimental autoimmune encephalomyelitis, acute ischemic stroke, or spinal cord injury (Reyes-Irisarri et al. 2007, Grond-Ginsbach et al. 2008), but whether levels of other PDEs and which specific PDE4 isoforms change after TBI remain unknown. We chose to study changes in PDE1, 3, 4, 8, and 10 because these isoforms are found in the brain or in cells that infiltrate the brain after injury. The objective of this study was to determine if PDEs are regulated by TBI.
MATERIALS AND METHODS
Fluid-percussion brain injury surgery
For these experiments, 84 adult male Sprague Dawley rats were used (300–400 gm, Charles Rivers Laboratories, Wilmington, MA, USA). All experimental procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals, the Society for Neuroscience Guidelines for the Use of Animals in Neuroscience Research, and approved by the University of Miami Animal Care and Use Committee. The fluid-percussion brain injury (FPI) surgery was performed as previously described (Atkins et al. 2012). Anesthesia was induced with 3% isoflurane, 70% N2O, and 30% O2, and maintained with 0.5% isoflurane, 70% N2O, and 30% O2. A 4.8 mm craniotomy (3.8 mm posterior to bregma, 2.5 mm lateral to midline) was made over the right cortex. FPI was delivered by a 2.0±0.2 atmospheric pressure fluid-percussion pulse at the craniotomy. Head and body temperature were maintained between 36.8–37.2°C. Blood gases (pO2 and pCO2), blood pH, and mean arterial blood pressure (MABP) were measured 15 min before and for at least 30 min after the TBI and maintained within normal ranges. Sham-operated rats received identical manipulations.
Compound 4a (0.3 mg/kg, n=8), compound 7b (0.3 mg/kg, n=8), or vehicle (5% DMSO in saline, 6 ml/kg, n=7) were injected i.p. at 30 min post-injury, then once per day for 2 days (Bruno et al. 2009). Animals were pseudo-randomized so that the drug treatments were interleaved and for three animals produced in one day, each animal received a different drug treatment.
Quantitative PCR (qPCR)
At 30 min after sham surgery (n=3) or FPI (n=3), animals were decapitated and the ipsilateral parietal cortex was dissected. Total RNA was isolated by sonication (Branson 250 Sonifier, Branson Ultrasonics Corporation, Danbury, CT, USA) in 1.2 mL of Trizol (Invitrogen, Carlsbad, CA, USA). Chloroform (240 μL) was added and samples were centrifuged. The aqueous layer was transferred to a fresh tube with isopropanol (600 μL), followed by centrifugation. The RNA pellet was rinsed with 75% ethanol, resuspended in 100 μL RNase-free H20 and treated with a RNA clean-up kit (SABiosciences, Frederick, MD, USA). RNA concentration and quality were confirmed via NanoDrop (ThermoFisher Scientific, Pittsburgh, PA, USA). Reverse transcription was performed with 2 μg RNA per sample (SABiosciences). Quantitative PCR (qPCR) was performed in a 96-well format using RT2 SYBR Green/ROX qPCR Master Mix (SABiosciences) and a 7300 Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). Primers were: Pde1a: 5′-CATGAGTGATGGGACCTATGC-3′, 5′-AGTTCGTTCACACCTATGGATG-3′; Pde4b2: 5′-CTGCAGCCTAACTACCTGTC-3′, 5′-ACACTTGGTTCCCTGATCTG-3′; Pde4d2: 5′-ATCCGAGCATGGCGGGAGGAG-3′, 5′-TGCCAGACCGACTCATTTCAG-3′; Pde10a: 5′-CATCGGTCTTGTGGGACATC-3′, 5′-ATGGCACAGACAGGCAATTAG-3′; Gapdh: 5′-CGTCTTCACCACCATGGAGA-3′, 5′-CGGCCATCACGCCACAGCTT-3′; Actin: 5′-CTAGGCACCAGGGTGTGATG-3′, 5′-GAAGGTCTCAAACATGATCTGG-3′. The qPCR reaction was performed in triplicate for each sample. The ΔΔCt method was used and the Actin average from sham animals served as a loading control.
Western blot analysis
At 30 min, 1 hr, 3 hr, 6 hr, 24 hr, or 7 days after TBI (n=6/time point) or sham surgery (n=3/time point), animals were decapitated and the ipsilateral parietal cortex was dissected. Tissue was homogenized and western blotted as previously described (Oliva et al. 2012). Antibodies used were β-actin (AC-15, 1:10,000, Sigma-Aldrich, St. Louis, MO, USA), PDE1A (H-105, 1:4000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), PDE1B (ab14600, 1:500, Abcam, Cambridge, MA, USA), PDE1C (H-262, 1:500, Santa Cruz Biotechnology), PDE3A (H-300, 1:250, Santa Cruz Biotechnology), PDE3B (H-300, 1:250, Santa Cruz Biotechnology), phospho-PDE4A (GTZ14610, 1:2000, GeneTex, Irvine, CA, USA), PDE4A5 (ab42094, 1:2000, Abcam), PDE4A8 (GTX14606, 1:1000, GeneTex), PDE4B (H-56, 1:500, Santa Cruz Biotechnology), PDE4D (H-69, 1:500, Santa Cruz Biotechnology), phospho-PDE4D (ab59212, 1:1000, Abcam), PDE8A (H-64, 1:500, Santa Cruz Biotechnology), PDE8B (I-16, 1:500, Santa Cruz Biotechnology), and PDE10A (H-219, 1:250, Santa Cruz Biotechnology). Western blots were developed with HRP-conjugated secondary antibodies (1:1000, Cell Signaling Technology, Danvers, MA, USA), ECL plus or ECL advance (GE Healthcare, Piscataway, NJ) and x-ray film (Phenix Research Products, Hayward, CA, USA). Films were densitized using ImageJ 1.38x (NIH). Levels of each PDE were normalized to β-actin within each sample, and levels of each PDE in TBI animals were normalized to average PDE levels in sham animals. No significant differences in PDE levels were observed in sham animals at 1, 3, or 24 hr, or 7 days post-surgery, so one sham animal for each time point was pooled in the western blot analyses (not shown).
Immunohistochemistry
At 24 hr after TBI (n=3) or sham surgery (n=3), animals were anesthetized and perfused. The brains were sectioned and immunostained as previously described (Oliva et al. 2012). Primary antibodies used were: PDE1A (sc-50480, Santa Cruz Biotechnology), PDE4B2 (ABS181, Millipore), PDE4D (ABS22, Millipore), phospho-PDE4A (GTX14610, GeneTex), Aldh1L1 (75–164, UC Davis/NINDS/NIHM NeuroMab Facility), OX42 (MCA275R, AbD Serotec, Raleigh, NC, USA), MAP2 (M9942, Sigma-Aldrich), and NeuN (Millipore). Secondary antibodies were Alexa 488, Alexa 546, or Alexa-647-labeled anti-rabbit and anti-mouse IgG (Invitrogen). Nuclei were stained using Hoechst 33342 (Invitrogen).
Imaging was performed using a FluoView FV1000 (Olympus America, Center Valley, PA, USA) laser scanning confocal microscope and 10X 0.4 NA air, 20X 0.85 NA oil-immersion, and 60X 1.42 NA oil-immersion objectives, and an LD laser (405 nm), multi-line argon laser, and HeNe(G) laser. Sections from different animals were processed in parallel, and at least 3 sections/animal were imaged. All animals within each group produced similar results.
Histological analysis
At 3 days post-injury, animals were anesthetized and perfused (Atkins et al. 2012). Brains were sectioned stereologically (150 μm apart, 10 μm thick) and stained with hematoxylin and eosin (H&E) as previously described (Atkins et al. 2012). Contusions were indicated by hemorrhage, inflammatory cells, disordered white matter, necrosis, pyknotic cells, and vacuolization. Using Neurolucida (10.50, MicroBrightField, Williston, VT, USA), the contusion was contoured at 4x using an Olympus BX51TRF microscope (Olympus America). Images at bregma level -5.8 mm are shown and were taken at 20x.
Statistical analysis
Western blot data, quantitative RT-PCR, and contusion volume were analyzed using a one-way ANOVA and post-hoc Dunnett’s t-tests. Contusion areas were analyzed with a repeated measures one-way ANOVA and post-hoc Bonferroni t-tests. A repeated measures two-way ANOVA followed by post-hoc Bonferroni t-tests was used for measures of physiological parameters before and after TBI. Significance was designated at P<0.05. Results are mean ± sem.
RESULTS
Expression levels of specific PDE isoforms after TBI
To identify the PDE isoforms regulated after TBI in the cortex, we assessed the ipsilateral parietal cortex for changes in PDEs 1, 3, 4, 8, and 10 after moderate parasagittal FPI. These isoforms were chosen because they all hydrolyze cAMP and are found in the brain (Lakics et al. 2010, Miro et al. 2002). We found that in the ipsilateral parietal cortex, levels of PDE1A were significantly elevated from 30 min post-injury to 24 hr, and PDE10A was elevated at 6 and 24 hr. PDE1B was significantly decreased from 30 min to 7 days post-injury and levels of PDE1C, 3A, 8A, and 8B were unchanged (Fig. 1). Although PDE3B and PDE7A were detected, levels were too variable to ascertain whether there were any significant changes after TBI (not shown).
Fig. 1.

Upregulation of PDEs after TBI. (A) Representative western blots of the ipsilateral parietal cortex for PDE isoforms after moderate parasagittal FPI. β-actin was used for loading control. (B) Densitometry indicated that expression of PDE1A and 10A significantly increased, and PDE1B significantly decreased in the parietal cortex. No changes were observed for PDE1C, PDE3A, PDE8A, or PDE8B. n=6/group, *p<0.05, **p<0.01, ***p<0.001 for sham versus TBI.
To determine if levels of PDE4 isoforms were regulated after TBI, we probed western blots from the ipsilateral parietal cortex with antibodies against various PDE4 isoforms (Fig. 2). In addition, we assessed whether a PKA phosphorylation site on PDE4A and Ser190 phosphorylation on PDE4D were altered. Phosphorylation of PDE4A significantly increased from 6 hr to 7 days post-injury. Levels of PDE4A5 decreased at 6 hr and 7 days post-injury, and levels of PDE4A8 significantly decreased from 3 hr to 7 days post-injury. No changes were observed with PDE4B1/3, 4B4, 4D4, or 4D3. Phosphorylation of PDE4D was significantly decreased at 30 min post-TBI. Interestingly, PDE4B2 and 4D2 dramatically increased from 30 min to 24 hr post-injury. These results indicate that of all the isoforms studied, levels of PDE1A, PDE4B2, and PDE4D2 are significantly upregulated after TBI.
Fig. 2.

Regulation of PDE4 by TBI. The ipsilateral parietal cortex was analyzed for changes in PDE4A (A, B), 4B (C, D), and 4D (E, F) expression levels and phosphorylation of PDE4A (A, B) and 4D (E, F). Phosphorylation of PDE4A and levels of PDE4B2 and 4D2 significantly increased after TBI. PDE4A5, 4A8, and phosphorylated PDE4D significantly decreased, whereas PDE4B1/3, 4B4, 4D4, and 4D3 were unchanged. n=6/group, *p<0.05, **p<0.01, ***p<0.001 for sham versus TBI.
mRNA analysis of PDE isoforms
To determine if these protein changes were due to gene transcription alterations, quantitative RT-PCR was performed on cortical samples at 30 min post-injury for Pde1a, Pde4b2, Pde4d2, and Pde10a, since these had the most robust changes in protein (Fig. 3). This time point was chosen since protein levels of PDE1A, 4B2, and 4D2 were already significantly increased at 30 min post-injury and PDE10A significantly increased by 1 hr post-injury. If these protein changes were due to gene transcription changes, mRNA changes would likely be evident at this time point. We found that expression of Pde4b2 Pde4d2, and Pde10a significantly increased. However, Pde1a mRNA levels did not significantly change at this time point.
Fig. 3.
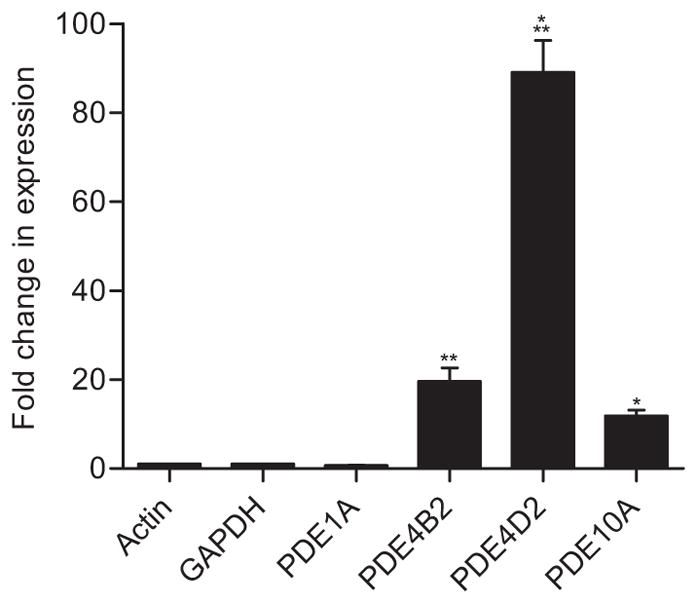
Quantitative PCR of Pde1a, 4b2, 4d2, and 10a at 30 min after TBI in the ipsilateral cortex. Expression of Pde4b2, Pde4d2, and Pde10a were significantly increased, whereas Pde1a was unchanged. n=3/group, *p<0.05, **p<0.01, ***p<0.001 for sham versus TBI.
Localization of PDE expression in the injured brain
We next examined the localization of PDEs in the brain. Since the PDE isoforms that increased after TBI were all statistically significant at 24 hr post-injury, this time point was chosen for immunohistochemistry. PDE1A was expressed in both the ipsilateral and contralateral cortex, as well as in the sham, non-injured brain, and restricted mainly to pyramidal neurons of layers IV/V (Fig. 4). At a subcellular level, PDE1A was strongly expressed in the somata and proximal, apical neurites, but predominantly nuclear excluded. Neurons of the ipsilateral cortex at 24 hr post-injury were qualitatively more intensely labeled as compared to the contralateral cortex, consistent with the western blots.
Fig. 4.

PDE1A is expressed in neurons at 24 hr post-TBI. (B) PDE1A expression (green) in the ipsilateral cortex. NeuN immunostaining (red) revealed that PDE1A was expressed in neurons predominantly in layers IV/V. PDE1A was found in the cytoplasm of the somata and proximal dendrites, but absent from the nucleus. (B) Higher magnification image of the boxed region in panel A. The contralateral cortex (C) and cortex from sham-surgery animals (D) showed expression patterns for PDE1A similar to the ipsilateral cortex (neuronal somata and proximal dendrites, and nuclear excluded). Scale bars 50 μm.
Phospho-PDE4A was expressed in neurons in both the ipsilateral and contralateral cortex at 24 hr post-TBI, and in the sham, non-injured cortex (Fig. 5). In contrast to PDE1A, phospho-PDE4A expression was found in the nucleus of neurons throughout all cortical layers.
Fig. 5.

Phospho-PDE4A is expressed in neuronal nuclei. (A) Phospho-PDE4A (pPDE4A) expression (green) in the ipsilateral cortex at 24 hr after TBI. NeuN immunostaining (red) revealed that phospho-PDE4A expression was highly restricted to the nuclei of neurons throughout all cortical layers. (B) Higher magnification image of the boxed region in the right panel of A. The contralateral cortex (C) and cortex from sham-surgery animals (D) showed expression patterns for phospho-PDA4A similar to the ipsilateral cortex (neuronal nuclei throughout all layers). Scale bars 50 μm.
PDE4B2 was expressed in neurons in both the ipsilateral and contralateral cortex at 24 hr after trauma, and in sham, uninjured brains (Fig. 6). Unlike PDE1A and phospho-PDE4A however, PDE4B2 expression was almost exclusively dendritic, co-localizing with MAP2.
Fig. 6.

PDE4B2 is expressed in dendrites. (A) PDE4B2 expression (green) in the ipsilateral cortex at 24 hr after injury. MAP2 immunostaining (red) revealed that PDE4B2 was expressed in dendrites. (B) Higher magnification image of the boxed region in the right panel of A. The contralateral cortex (C) and cortex from sham-surgery animals (D) showed expression patterns for PDA1A similar to that of the ipsilateral cortex (neuronal somata and proximal dendrites, and nuclear excluded). E shows parallel immunostaining with no primary antibodies. Scale bars 50 μm.
Immunostaining for PDE4D provided a highly reliable marker for the TBI brain, in which intensely labeled cells were present throughout the ipsilateral cortex, but absent contralaterally (Fig. 7), and absent from the cortex of sham, non-injured brains (not shown). In contrast to PDE1A, phospho-PDE4A, and PDE4B, PDE4D was not expressed in neurons. Furthermore, PDE4D did not co-localize with Aldh1L1, an astrocyte marker, or OX42, a microglia/macrophage marker.
Fig. 7.

PDE4D immunoreactivity distinguishes the ipsilateral cortex at 24 hr after brain trauma. (A) PDE4D expression in the ipsilateral (A1) and contralateral (A2) cortex. Only the ipsilateral cortex showed intensely-labeled cells. (B) Higher magnification image of the boxed region in A, showing PDE4D labeled cells (green) and Hoechst staining (blue in B2). The cells intensely labeling for PDE4D did not colocalize with NeuN to label neurons (C), Aldh1L1 to identify astrocytes (D), or OX42 to detect microglia/macrophages (E). Scale bars 400 μm (A), 40 μm (B–E).
Inhibition of PDE4D increased contusion volume
Based on these results, we hypothesized that PDE4B and 4D may be likely targets to reduce PDE signaling and restore cAMP levels after TBI. Rolipram inhibits 4B and 4D equally, and reduces inflammation when given before TBI, but causes vascular perturbations when given post-injury (Atkins et al. 2012, Atkins et al. 2007b). To determine whether this was mediated by PDE4B or 4D, we administered the novel compounds 4a or 7b, which are somewhat more selective against PDE4D as compared to PDE4A4, 4B2, or 4C2, to animals after TBI (Bruno et al. 2009). We found that in comparison to vehicle-treated animals, both compounds 4a and 7b increased hemorrhage in the injured parietal cortex (Fig. 8). This increase in hemorrhage was reflected in a significant increase in contusion volume for both compound 4a and 7b and in particular, at the peak area of pathology, bregma level -5.8 mm, for compound 4a. Physiological parameters for MABP, blood oxygenation levels, and blood pH were not significantly different between treatment groups although some TBI-induced changes were noted (Table S1). These results indicate that inhibition of PDE4 with compounds somewhat more selective for PDE4D than PDE4A4, 4B2, or 4C2 significantly worsen histopathological outcome after TBI.
Fig. 8.
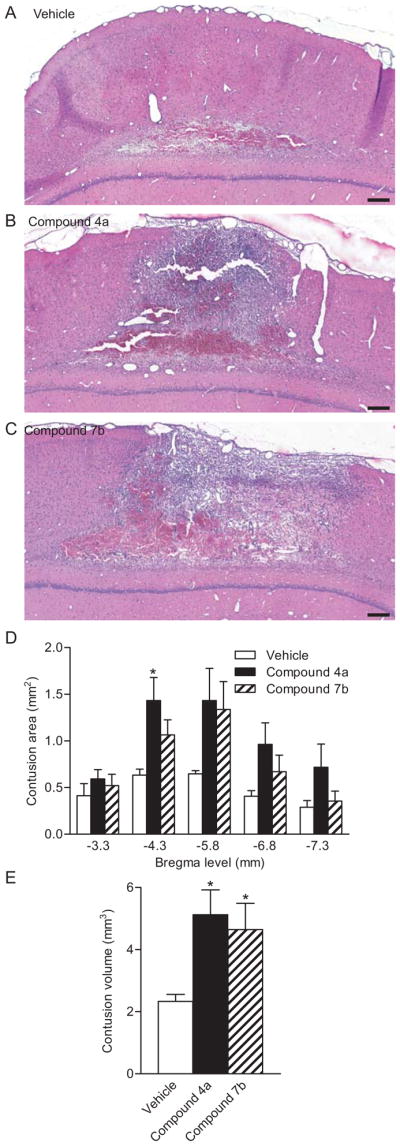
Inhibition of PDE4 worsened histopathology after TBI. Images of the contused cortex stained with H&E at 3 days post-injury for vehicle (A), compound 4a (B) and compound 7b-treated (C) animals. Scale bars 500 μm. Contusion volume (C) and contusion area at bregma level -5.8 mm (D) were significantly greater in animals treated with compound 4a as compared to animals treated with vehicle. n=7–8 per group, *p<0.05 for TBI-vehicle versus TBI-drug treatment.
DISCUSSION
Understanding the regulation of PDEs is important for the rational development of targeted therapies to restore cAMP levels after TBI. Four genes encode the PDE4 families, A–D. Although PDE4A, B, and D are found in the brain, PDE4C is absent, and the expression pattern of each isoform varies greatly, suggesting non-redundancy in their functioning (Lakics et al. 2010, Miro et al. 2002, Conti & Beavo 2007). In this study, we found that protein levels of PDE1A, PDE4B2, PDE4D2, and PDE10A, in particular, were significantly elevated after moderate parasagittal FPI. Correspondingly, mRNA levels of Pde4b2, Pde4d2, and Pde10a were also significantly increased, whereas Pde1A mRNA levels did not significantly change. The increase in PDE1A protein, but not mRNA at 30 min post-injury was surprising. We speculate that the increase in PDE1A protein may be due to regulation at the mRNA level rather than gene transcription. Alternatively, the increase in PDE1A protein could reflect changes in antigenicity due to posttranslational modifications or a change in a particular isoform that was missed with the primers used for the qPCR analysis.
In accordance with our findings, previous studies have shown that PDE4B2 and 4D2 are low in the non-injured brain, although their mRNAs are present (Iona et al. 1998, D’Sa et al. 2002, Miro et al. 2002). While PDE1A and PDE4B2 were expressed in neurons, the induced PDE4D expression after TBI was unclear. Based on the immunostaining pattern, we speculate that PDE4D may be present in inflammatory cells not detected with the OX42 immunostaining. Other cell types that may also express PDE4D not assayed in these studies include oligodendrocytes, pericytes, endothelial cells, and smooth muscle cells. PDE4D has been found in both endothelial and smooth muscle cells (Mehats et al. 2003, Tilley & Maurice 2002). The results with compounds 4a and 7b on increasing hemorrhage within the contused cortex suggest the possibility that PDE4D is involved in the neurovascular unit. Given the pronounced induction of PDE4D2 specifically in the injured hemisphere, it appears that PDE4D2 has the potential to be an injury-specific biomarker.
Increased periods of neuronal activity increase PDE4 activity in the brain and specifically protein levels of PDE4B2 (Suda et al. 1998, Ahmed & Frey 2005). Both PDE4B2 and 4D2 have cAMP-response elements (CRE) on their promoters and cAMP selectively upregulates both of these isoforms (D’Sa et al. 2002, Verghese et al. 1995, Manning et al. 1999). CREB is activated during FPI; however, this activation is more delayed than the upregulation of PDE4B2 and 4D2 (Atkins et al. 2007a). Besides CRE elements, an NF-κB canonical site is present on the PDE4B2 promoter (Verghese et al. 1995, Jin & Conti 2002). We speculate that the early increase in these isoforms occurs through the NF-κB pathway, whereas the more sustained increase could also be explained by CREB-mediated gene transcription.
We also observed a significant increase in phosphorylation at the PKA site on PDE4A, although at the earliest time point assessed (i.e. 30 min), there was a small, but non-significant decrease. Given the decrease in cAMP levels and PKA activation observed after TBI, this may reflect a compensatory mechanism: PKA phosphorylation of the long isoforms increases PDE activity, which may further decrease cAMP levels (MacKenzie et al. 2002).
The functional consequences of an increase in PDE10A in the cortex after TBI remain to be explored. PDE10A is predominantly expressed in the striatum, but is found at lower levels in the cortex (Seeger et al. 2003, Lakics et al. 2010). Whether PDE10A is altered in the striatum after FPI remains to be determined. Motor deficits and striatal dysfunction occur after TBI, and PDE10A inhibitors currently in use for Huntington’s disease are a potential area of exploration for amelioration of gait deficits after TBI (Giampa et al. 2010, Shin et al. 2011, Williams et al. 2009).
Therapeutic Strategies
Of all the isoforms studied, the most pronounced changes were with PDE4B2 and 4D2. These specific isoforms are highly inducible, and their expression is upregulated within inflammatory cells which increases release of pro-inflammatory cytokines (Manning et al. 1999, Jin & Conti 2002, Reyes-Irisarri et al. 2007, Ariga et al. 2004). Isoform-specific inhibitors for PDE4s are in development, and a novel set of compounds have been generated that are more selective for PDE4D as compared to PDE4B2, PDE4A4, and PDE4C2 (Bruno et al. 2011, Bruno et al. 2009). At 10 μM, compound 4a inhibits PDE4Ds1-3 at 58%, PDE4A4 at 34%, PDE4B2 at 39% and PDE4C2 at 19%. Compound 7b inhibits PDE4Ds1-3 at 67%, PDE4A4 at 34%, PDE4B2 at 23%, and PDE4C2 at 25%. In contrast, rolipram has similar selectivity for all of these isoforms (Bruno et al. 2009). Thus, we capitalized upon these novel inhibitors to determine if they would alter histopathological outcome after TBI. We found that both compounds significantly worsened outcome and increased hemorrhage in the cortex. These results indicate that isoform-specific inhibitors against PDE4D are not a viable acute therapeutic route for TBI.
The increase in PDE1A suggests that PDE1-selective inhibitors may provide a potential therapeutic avenue (Medina 2011). The PDE1 family is encoded by 3 genes, and all are calcium/calmodulin-dependent. This results in crosstalk between calcium and cyclic nucleotide signaling, and thus this PDE family may potentially decrease cyclic nucleotide levels by responding to the calcium homeostatic dysregulation induced by TBI (Sun et al. 2008). PDE1A hydrolyzes cGMP with higher affinity than cAMP, suggesting that besides cAMP, cGMP may also decrease after TBI (Temple et al. 2001). PDE1A was selectively expressed in neurons, suggesting a neuronal-specific dysregulation of cGMP. In these studies, we used a pan-PDE1A antibody, precluding identification of which PDE1A splice variant increased after TBI (Michibata et al. 2001). Other localization studies of PDE1A have shown that PDE1A1 is found in heart and lung, whereas PDE1A2 is expressed in the hippocampus and cortex (Yan et al. 1994). Interestingly, several non-specific PDE1 inhibitors, such as caffeine and amantadine, have already been found to improve outcome in TBI (Li et al. 2008, Medina 2011, Dixon et al. 1999).
The finding that a PDE4D inhibitor worsened outcome needs to be interpreted with caution. Compounds 4a and 7b are somewhat more selective towards 4D1-3, with less inhibition of 4A4, 4B2, or 4C2 (Bruno et al. 2009). Recent studies in vivo showed that compound 7b improves late-phase memory consolidation, as well as increases cAMP levels in the hippocampus (Bruno et al. 2011). While very low doses have no effect on memory, higher doses show a decrease in efficacy. These behavioral results provide further evidence that PDE4D plays a critical role in memory consolidation while further investigation is required to assess other effects at higher doses, particularly in inflammation and vasodilation (Burgin et al. 2010, Li et al. 2011b, Zhang et al. 2002b).
Further studies with a thorough dose-response curve of compounds 4a and 7b are needed to determine if lower doses do confer an anti-inflammatory response in the traumatized brain. At the dose tested in these studies, we speculate that PDE4D inhibition may have increased vasodilation by affecting PDE4s present in endothelial cells (Mehats et al. 2003, Tilley & Maurice 2002). In our previous experiments with the PDE4 inhibitor rolipram which also aggravated histopathology after TBI, rolipram increased blood-brain barrier permeability. However, it was unclear whether this increased inflammation was due to a direct effect on blood-brain barrier permeability, or another physiological response such as vasodilation. Compounds 4a and 7b may have acted on PDE4 isoforms found in endothelial and smooth muscle cells, to increase dilation and bleeding in the vulnerable cortex, thus aggravating the inflammatory response (Miwa et al. 2009, Mehats et al. 2003, Willette et al. 1997).
In summary, using an experimental model of TBI, we found an acute upregulation of PDE1A, PDE4B2, PDE4D2, phosphorylated PDE4A, and PDE10A. These results suggest that specific inhibitors against these particular isoforms may be potential therapeutics for TBI. To begin to test more selective inhibitors, we utilized PDE4 inhibitors somewhat more selective against PDE4D than PDE4A or PDE4B and found that these inhibitors worsened TBI pathology. Our results suggest that an exploration of PDE1A, PDE4A, PDE4B2, or PDE10A inhibitors may be more fruitful in improving outcome after TBI.
Supplementary Material
Acknowledgments
This work was supported by The Miami Project to Cure Paralysis, and National Institutes of Health grants NS069721, AG033266, and NS056072. We thank Dr. Beata Frydel and Dayaris Morffi for technical assistance.
Abbreviations used
- cAMP
cyclic AMP
- CRE
cAMP-response elements
- FPI
fluid-percussion brain injury
- H&E
hematoxylin and eosin
- i.p
intraperitoneally
- MABP
mean arterial blood pressure
- PDE
phosphodiesterase
- PKA
protein kinase A
- qPCR
quantitative PCR
- TBI
traumatic brain injury
- UCR
upstream conserved region
Footnotes
The authors declare no conflicts of interest.
References
- Ahmed T, Frey JU. Phosphodiesterase 4B (PDE4B) and cAMP-level regulation within different tissue fractions of rat hippocampal slices during long-term potentiation in vitro. Brain Res. 2005;1041:212–222. doi: 10.1016/j.brainres.2005.02.023. [DOI] [PubMed] [Google Scholar]
- Ariga M, Neitzert B, Nakae S, Mottin G, Bertrand C, Pruniaux MP, Jin SL, Conti M. Nonredundant function of phosphodiesterases 4D and 4B in neutrophil recruitment to the site of inflammation. J Immunol. 2004;173:7531–7538. doi: 10.4049/jimmunol.173.12.7531. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Kang Y, Furones C, Truettner JS, Alonso OF, Dietrich WD. Postinjury treatment with rolipram increases hemorrhage after traumatic brain injury. J Neurosci Res. 2012;90:1861–1871. doi: 10.1002/jnr.23069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Oliva AA, Jr, Alonso OF, Chen S, Bramlett HM, Hu BR, Dietrich WD. Hypothermia treatment potentiates ERK1/2 activation after traumatic brain injury. Eur J Neurosci. 2007a;26:810–819. doi: 10.1111/j.1460-9568.2007.05720.x. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Oliva AA, Jr, Alonso OF, Pearse DD, Bramlett HM, Dietrich WD. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp Neurol. 2007b;208:145–158. doi: 10.1016/j.expneurol.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block F, Schmidt W, Nolden-Koch M, Schwarz M. Rolipram reduces excitotoxic neuronal damage. Neuroreport. 2001;12:1507–1511. doi: 10.1097/00001756-200105250-00041. [DOI] [PubMed] [Google Scholar]
- Bruno O, Fedele E, Prickaerts J, et al. GEBR-7b, a novel PDE4D selective inhibitor that improves memory in rodents at non-emetic doses. Br J Pharmacol. 2011;164:2054–2063. doi: 10.1111/j.1476-5381.2011.01524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno O, Romussi A, Spallarossa A, Brullo C, Schenone S, Bondavalli F, Vanthuyne N, Roussel C. New selective phosphodiesterase 4D inhibitors differently acting on long, short, and supershort isoforms. J Med Chem. 2009;52:6546–6557. doi: 10.1021/jm900977c. [DOI] [PubMed] [Google Scholar]
- Burgin AB, Magnusson OT, Singh J, et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 2010;28:63–70. doi: 10.1038/nbt.1598. [DOI] [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- D’Sa C, Tolbert LM, Conti M, Duman RS. Regulation of cAMP-specific phosphodiesterases type 4B and 4D (PDE4) splice variants by cAMP signaling in primary cortical neurons. J Neurochem. 2002;81:745–757. doi: 10.1046/j.1471-4159.2002.00878.x. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Chatzipanteli K, Vitarbo E, Wada K, Kinoshita K. The role of inflammatory processes in the pathophysiology and treatment of brain and spinal cord trauma. Acta Neurochir Suppl. 2004;89:69–74. doi: 10.1007/978-3-7091-0603-7_9. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Kraus MF, Kline AE, Ma X, Yan HQ, Griffith RG, Wolfson BM, Marion DW. Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Restorative neurology and neuroscience. 1999;14:285–294. [PubMed] [Google Scholar]
- Fatemi SH, King DP, Reutiman TJ, et al. PDE4B polymorphisms and decreased PDE4B expression are associated with schizophrenia. Schizophr Res. 2008a;101:36–49. doi: 10.1016/j.schres.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Lee S. Phosphodiesterase-4A expression is reduced in cerebella of patients with bipolar disorder. Psychiatr Genet. 2008b;18:282–288. doi: 10.1097/YPG.0b013e3283060fb8. [DOI] [PubMed] [Google Scholar]
- Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS One. 2010;5:e13417. doi: 10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grond-Ginsbach C, Hummel M, Wiest T, et al. Gene expression in human peripheral blood mononuclear cells upon acute ischemic stroke. J Neurol. 2008;255:723–731. doi: 10.1007/s00415-008-0784-z. [DOI] [PubMed] [Google Scholar]
- Hill EV, Sheppard CL, Cheung YF, Gall I, Krause E, Houslay MD. Oxidative stress employs phosphatidyl inositol 3-kinase and ERK signalling pathways to activate cAMP phosphodiesterase-4D3 (PDE4D3) through multi-site phosphorylation at Ser239 and Ser579. Cell Signal. 2006;18:2056–2069. doi: 10.1016/j.cellsig.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Iona S, Cuomo M, Bushnik T, Naro F, Sette C, Hess M, Shelton ER, Conti M. Characterization of the rolipram-sensitive, cyclic AMP-specific phosphodiesterases: identification and differential expression of immunologically distinct forms in the rat brain. Mol Pharmacol. 1998;53:23–32. doi: 10.1124/mol.53.1.23. [DOI] [PubMed] [Google Scholar]
- Jin SL, Conti M. Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-α responses. Proc Natl Acad Sci U S A. 2002;99:7628–7633. doi: 10.1073/pnas.122041599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakics V, Karran EH, Boess FG. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharm. 2010;59:367–374. doi: 10.1016/j.neuropharm.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Li LX, Cheng YF, Lin HB, Wang C, Xu JP, Zhang HT. Prevention of cerebral ischemia-induced memory deficits by inhibition of phosphodiesterase-4 in rats. Metab Brain Dis. 2011a;26:37–47. doi: 10.1007/s11011-011-9235-0. [DOI] [PubMed] [Google Scholar]
- Li W, Dai S, An J, et al. Chronic but not acute treatment with caffeine attenuates traumatic brain injury in the mouse cortical impact model. Neuroscience. 2008;151:1198–1207. doi: 10.1016/j.neuroscience.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Li YF, Cheng YF, Huang Y, Conti M, Wilson SP, O’Donnell JM, Zhang HT. Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J Neurosci. 2011b;31:172–183. doi: 10.1523/JNEUROSCI.5236-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie SJ, Baillie GS, McPhee I, et al. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1) Br J Pharmacol. 2002;136:421–433. doi: 10.1038/sj.bjp.0704743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning CD, Burman M, Christensen SB, Cieslinski LB, Essayan DM, Grous M, Torphy TJ, Barnette MS. Suppression of human inflammatory cell function by subtype-selective PDE4 inhibitors correlates with inhibition of PDE4A and PDE4B. Br J Pharmacol. 1999;128:1393–1398. doi: 10.1038/sj.bjp.0702911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina AE. Therapeutic utility of phosphodiesterase type I inhibitors in neurological conditions. Front Neurosci. 2011;5:21. doi: 10.3389/fnins.2011.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehats C, Jin SL, Wahlstrom J, Law E, Umetsu DT, Conti M. PDE4D plays a critical role in the control of airway smooth muscle contraction. FASEB J. 2003;17:1831–1841. doi: 10.1096/fj.03-0274com. [DOI] [PubMed] [Google Scholar]
- Michibata H, Yanaka N, Kanoh Y, Okumura K, Omori K. Human Ca2+/calmodulin-dependent phosphodiesterase PDE1A: novel splice variants, their specific expression, genomic organization, and chromosomal localization. Biochim Biophys Acta. 2001;1517:278–287. doi: 10.1016/s0167-4781(00)00293-1. [DOI] [PubMed] [Google Scholar]
- Miro X, Perez-Torres S, Puigdomenech P, Palacios JM, Mengod G. Differential distribution of PDE4D splice variant mRNAs in rat brain suggests association with specific pathways and presynaptical localization. Synapse. 2002;45:259–269. doi: 10.1002/syn.10100. [DOI] [PubMed] [Google Scholar]
- Miwa T, Mori A, Nakahara T, Ishii K. Intravenously administered phosphodiesterase 4 inhibitors dilate retinal blood vessels in rats. Eur J Pharmacol. 2009;602:112–116. doi: 10.1016/j.ejphar.2008.10.060. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A. 2004;101:8786–8790. doi: 10.1073/pnas.0402595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA, Jr, Kang Y, Sanchez-Molano J, Furones C, Atkins CM. STAT3 signaling after traumatic brain injury. J Neurochem. 2012;120:710–720. doi: 10.1111/j.1471-4159.2011.07610.x. [DOI] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10:610–616. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- Reneerkens OA, Rutten K, Steinbusch HW, Blokland A, Prickaerts J. Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharm (Berl) 2009;202:419–443. doi: 10.1007/s00213-008-1273-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Irisarri E, Sanchez AJ, Garcia-Merino JA, Mengod G. Selective induction of cAMP phosphodiesterase PDE4B2 expression in experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol. 2007;66:923–931. doi: 10.1097/nen.0b013e3181567c31. [DOI] [PubMed] [Google Scholar]
- Robichaud A, Savoie C, Stamatiou PB, Lachance N, Jolicoeur P, Rasori R, Chan CC. Assessing the emetic potential of PDE4 inhibitors in rats. Br J Pharmacol. 2002;135:113–118. doi: 10.1038/sj.bjp.0704457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger TF, Bartlett B, Coskran TM, et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003;985:113–126. doi: 10.1016/s0006-8993(03)02754-9. [DOI] [PubMed] [Google Scholar]
- Shin SS, Bray ER, Zhang CQ, Dixon CE. Traumatic brain injury reduces striatal tyrosine hydroxylase activity and potassium-evoked dopamine release in rats. Brain Res. 2011;1369:208–215. doi: 10.1016/j.brainres.2010.10.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda S, Nibuya M, Ishiguro T, Suda H. Transcriptional and translational regulation of phosphodiesterase type IV isozymes in rat brain by electroconvulsive seizure and antidepressant drug treatment. J Neurochem. 1998;71:1554–1563. doi: 10.1046/j.1471-4159.1998.71041554.x. [DOI] [PubMed] [Google Scholar]
- Sun DA, Deshpande LS, Sombati S, Baranova A, Wilson MS, Hamm RJ, DeLorenzo RJ. Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur J Neurosci. 2008;27:1659–1672. doi: 10.1111/j.1460-9568.2008.06156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple MD, Delahunty TM, Hamm RJ, Phillips LL, Lyeth BG, Povlishock JT. Subtle alterations in NMDA-stimulated cyclic GMP levels following lateral fluid percussion brain injury. J Neurotrauma. 2001;18:47–55. doi: 10.1089/089771501750055767. [DOI] [PubMed] [Google Scholar]
- Tilley DG, Maurice DH. Vascular smooth muscle cell phosphodiesterase (PDE) 3 and PDE4 activities and levels are regulated by cyclic AMP in vivo. Mol Pharmacol. 2002;62:497–506. doi: 10.1124/mol.62.3.497. [DOI] [PubMed] [Google Scholar]
- Verghese MW, McConnell RT, Strickland AB, Gooding RC, Stimpson SA, Yarnall DP, Taylor JD, Furdon PJ. Differential regulation of human monocyte-derived TNF α and IL-1 β by type IV cAMP-phosphodiesterase (cAMP-PDE) inhibitors. J Pharmacol Exp Ther. 1995;272:1313–1320. [PubMed] [Google Scholar]
- Willette RN, Shiloh AO, Sauermelch CF, Sulpizio A, Michell MP, Cieslinski LB, Torphy TJ, Ohlstein EH. Identification, characterization, and functional role of phosphodiesterase type IV in cerebral vessels: effects of selective phosphodiesterase inhibitors. J Cereb Blood Flow Metab. 1997;17:210–219. doi: 10.1097/00004647-199702000-00011. [DOI] [PubMed] [Google Scholar]
- Williams G, Morris ME, Schache A, McCrory PR. Incidence of gait abnormalities after traumatic brain injury. Arch Phys Med Rehabil. 2009;90:587–593. doi: 10.1016/j.apmr.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Yan C, Bentley JK, Sonnenburg WK, Beavo JA. Differential expression of the 61 kDa and 63 kDa calmodulin-dependent phosphodiesterases in the mouse brain. J Neurosci. 1994;14:973–984. doi: 10.1523/JNEUROSCI.14-03-00973.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Yang L, Konishi Y, Maeda N, Sakanaka M, Tanaka J. Suppressive effects of phosphodiesterase type IV inhibitors on rat cultured microglial cells: Comparison with other types of cAMP-elevating agents. Neuropharm. 2002a;42:262–269. doi: 10.1016/s0028-3908(01)00174-5. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Jin SL, Frith SA, Suvarna N, Conti M, O’Donnell JM. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology. 2002b;27:587–595. doi: 10.1016/S0893-133X(02)00344-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.