

Abstract
L1 retrotransposons comprise 17% of the human genome and are its only autonomous mobile elements. Although L1-induced insertional mutagenesis causes Mendelian disease, their mutagenic load in cancer has been elusive. Using L1-targeted resequencing of 16 colorectal tumor and matched normal DNAs, we found that certain cancers were excessively mutagenized by human-specific L1s, while no verifiable insertions were present in normal tissues. We confirmed de novo L1 insertions in malignancy by both validating and sequencing 69/107 tumor-specific insertions and retrieving both 5′ and 3′ junctions for 35. In contrast to germline polymorphic L1s, all insertions were severely 5′ truncated. Validated insertion numbers varied from up to 17 in some tumors to none in three others, and correlated with the age of the patients. Numerous genes with a role in tumorigenesis were targeted, including ODZ3, ROBO2, PTPRM, PCM1, and CDH11. Thus, somatic retrotransposition may play an etiologic role in colorectal cancer.
Over two-thirds of our genome may stem from “jumping genes” (de Koning et al. 2011). Three classes of retroelements are known to be currently active and a source of human disease: long interspersed elements (LINEs), the prototype of which is the RNA polymerase II transcribed L1; short interspersed elements (SINEs), consisting essentially of RNA polymerase III transcribed Alus; and SVAs (SINE-R/VNTR/Alus) that are intermediate in size relative to Alus and L1s, and are likely transcribed by RNA polymerase II. A fourth class of retroelements in our genome, human endogenous retroviruses (HERVs) is considered immobile. Full-length L1s are not only responsible for mobilizing themselves, but also for mobilizing the nonautonomous Alu (Dewannieux et al. 2003) and SVA retrotransposons (Ostertag et al. 2003; Hancks et al. 2011; Raiz et al. 2011), inactive L1s (Moran et al. 1996), small RNAs (Gilbert et al. 2005), and classical mRNAs, thereby creating processed pseudogenes (Esnault et al. 2000; Wei et al. 2001; Ohshima et al. 2003).
Although there are about half a million L1s in the human genome, only the human-specific L1s (L1Hs) are currently active, represented in each individual by about 800 germline copies (Ewing and Kazazian 2010), including ∼200 full-length sequences (Boissinot et al. 2000). According to conservative estimates there are only about 100 active L1Hs in any human diploid genome that are retrotranspositionally competent, of which six from the reference genome and 37 from six other genomes are known to be highly active (“hot”) (Brouha et al. 2003; Beck et al. 2010). L1s retrotranspose through a process called target-primed reverse transcription (TPRT) (Luan et al. 1993; Cost et al. 2002) with the help of the L1-encoded proteins open reading frame 1 protein (ORF1p) and ORF2p. Endonuclease and reverse transcriptase activities for L1 integration are provided by ORF2p (Mathias et al. 1991; Feng et al. 1996). The hallmarks of TPRT are the addition of a new poly(A) tail to the integrated sequence and target-site duplication (TSD), usually 6–20 bp in length. A fraction of retrotransposition events are also associated with 3′ transduction, the comobilization of 3′ flanking DNA sequences (Holmes et al. 1994; Moran et al. 1999; Goodier et al. 2000; Pickeral et al. 2000), resulting from transcriptional read-through of the weak L1 poly(A) signal and preferential use of a stronger downstream poly(A) signal. Most de novo L1 retrotransposition events are 5′ truncated (Gilbert et al. 2005), with one extreme truncation described where the whole L1 sequence was missing and only the 3′ transduced sequence was present (Solyom et al. 2012).
Active mobile elements are not only a significant source of intra- and interindividual variation, but can also act as insertional mutagens. There are 97 known disease-associated retrotransposon insertions into protein-coding genes (Hancks and Kazazian 2012; van der Klift et al. 2012), which is an underestimate, as conventional mutation screening methods are not designed to amplify large insertions. Of these nearly 100 cases, 25 are caused by L1s, 60 by Alus, eight by SVAs, and four by poly(A) sequence originating from an unidentifiable source (Hancks and Kazazian 2012; van der Klift et al. 2012). Of these insertions, 30 occur in cancer cases, including four in colon cancer patients (Miki et al. 1992; Su et al. 2000; Kloor et al. 2004; van der Klift et al. 2012). While three of the four colon cancer cases involve predicted germline or early somatic insertions, a somatic L1 insertion occurred in the APC gene in colon cancer (Miki et al. 1992).
In addition to acting as insertional mutagens, retrotransposons can disrupt gene function and genomic integrity in many other ways. These include recombination-mediated gene rearrangements, genetic instability, transcriptional interference, alternative splicing, gene breaking, epigenetic effects, the generation of DNA double-strand breaks, and the expression of small noncoding RNAs (for review, see Goodier and Kazazian 2008; Beck et al. 2011). All of these mechanisms are compatible with a tumorigenic potential of these elements. Retrotransposon overdose is another potential scenario in malignancy and could result in increased insertional mutagenesis, toxicity, or other oncogenic effects. Indeed, the overexpression of L1 ORF1p was observed in certain tumors (Bratthauer and Fanning 1992; Asch et al. 1996; Su et al. 2007; Harris et al. 2010), and RNAi-mediated silencing of L1s resulted in reduced proliferation and differentiation of tumorigenic cell lines (Oricchio et al. 2007). In addition, overexpression of Alu elements may exert disease through RNA toxicity (Kaneko et al. 2011). Thus, the cell likely has intrinsic defense mechanisms to prevent retrotransposon overexpression, including methylation (Yoder et al. 1997; Bourc'his and Bestor 2004) and the expression of several host proteins, such as APOBEC3 family members (Bogerd et al. 2006; Chen et al. 2006; Muckenfuss et al. 2006; Stenglein and Harris 2006) or DNA repair enzymes (Gasior et al. 2006; Suzuki et al. 2009; Coufal et al. 2011).
Here we applied two high-throughput L1-targeted resequencing methods to discover retrotransposon activity in colorectal cancers. We identified numerous nonreference L1 insertions not present in paired normal tissue and report a high retrotransposon insertion rate in tumors. We characterized insertion size and TSDs in cancer tissue, confirming that L1s primarily mobilize in cancer via TPRT. The data suggest the importance of retrotransposition in the biology of colorectal tumorigenesis.
Results
L1 display through high-throughput sequencing
We applied two next generation resequencing methods—hemi-specific PCR coupled to Illumina sequencing (L1-seq) (Ewing and Kazazian 2010) and retrotransposon capture sequencing (RC-seq) (Baillie et al. 2011) to interrogate the retroelement load of colorectal tumors. Approximately 800 nonreference L1Hs copies had been located from individual blood or lymphoblastoid cell lines by L1-seq—the same number as represented by the hg18 reference genome assembly, indicating its capacity to recover essentially all germline L1Hs elements (Ewing and Kazazian 2010). Here, we applied this method to recover somatic insertions from malignant tissues. RC-seq has previously been used to identify somatic mosaicism associated with L1, Alu, and SVA mobilization in the brain (Baillie et al. 2011). Its use of sequence capture for retrotransposon enrichment contrasts with the use of PCR by L1-seq; as a result, RC-seq is expected to cover a broader range of insertions, but with less depth per insertion than L1-seq. A highly multiplexed version of RC-seq was applied to assess whether somatic L1Hs insertions were identified by both approaches.
We sequenced DNA from 16 colorectal tumors and matched normal colons using a pooled L1-seq-based approach. The 16 tumor/normal pairs (32 samples total) were separated into four libraries of eight samples each denoted “colo1/tumor,” “colo1/normal,” “colo2/tumor,” and “colo2/normal.” We sequenced one lane for each library on an Illumina HiSeq 2000 instrument with the exception of colo1/normal, where two lanes of data were generated. The total number of reads generated for each library can be found in Supplemental Table S1.
Using computational methods outlined in Ewing and Kazazian (2010), we identified clusters of reads localized 3′ of predicted insertion sites. We required 100 reads spanning at least 100 bp (“high stringency”) as a minimum for L1 detection, which yields a specificity of >90% based on recovery of reference L1 insertions and nonreference sites discovered in previous studies (see Supplemental Figs. S1, S2 for an exploration of cutoff parameters). Using these criteria, we identified 764 reference L1Hs insertion sites present in NCBI36/hg18 and 400 nonreference insertion sites from the colo1 data. From the colo2 data we identified 816 reference and 433 nonreference insertions. Combining the data, we found 819 reference L1Hs elements and 635 nonreference elements, 336 of which had not been previously cataloged. Many of these uncataloged elements are new somatic insertions in the tumor. In total, 38% of reference and 35% of nonreference insertions were in gene annotations based on UCSC Known Genes. The distribution of L1 insertions detected by L1-seq in this study is shown in Figure 1.
Figure 1.

Genomic distribution of L1 insertions. Outer rings show the density of detected insertion sites for reference (gray) and nonreference (black) L1s. The approximate locations of the 72 PCR-validated somatic insertions are indicated by dots inside the circle. Note that 69 of the 72 insertions were successfully sequenced.
Our primary interest in generating these data was in finding insertions present either in a cancer pooled library or in a normal pooled library and not present in the corresponding paired normal or tumor library. Turning to these, with the same stringency cutoffs as above, we found 35 putative insertions only in colo1/tumor, four only in colo1/normal, 50 predictions only in colo2/tumor, and eight only in colo2/normal. Decreasing the requirements for predicted insertions to 10 reads spanning at least 100 bp (“low stringency”), we found 69 potential insertions only in colo1/tumor, 173 only in colo1/normal, 75 only in colo2/tumor, and 42 only in colo2/normal. The dramatic increase in predictions for colo1/normal only with decreasing stringency is an effect of the higher coverage in the colo1/normal versus colo1/tumor (Supplemental Table S1), as two lanes of sequence were generated for colo1/normal.
Five of the L1-sequenced colorectal tissue pairs were barcoded, pooled, and analyzed by shallow, multiplexed RC-seq (10 libraries, ∼75 million paired-end Illumina GAIIx reads). A total of 26,903 nonreference genomic insertions were detected by at least one read (Supplemental Table S2). Of these, 358 were (1) found in only one donor, (2) were not identified in RC-seq previously performed on pooled blood (Baillie et al. 2011) or databases of retrotransposon polymorphisms (Huang et al. 2010; Iskow et al. 2010; Ewing and Kazazian 2011), and (3) could be annotated with high confidence due to detection by multiple unique amplicons. Of this set, 96 were only found in tumor, including eight L1, 83 Alu, and five SVA. A total of 39 insertions were found only in nontumor samples and 223 were found in both tumor and nontumor. The tumor:nontumor ratios for L1, Alu and SVA, were ∼8:1, 2.5:1, and 2:1, respectively. AluY and L1-Ta/pre-Ta were detected, but no HERVs were detected.
PCR validation
We applied a step-wise PCR amplification scheme to validate insertion sites from L1-seq data and to determine both 5′ and 3′ junctions of L1Hs elements identified by L1-seq. Primary validations focused on confirming the presence of de novo L1 inserts by amplifying their 3′ junction and determining which of the eight patients carried the insertion within a DNA pool (PCR scheme and primer design performed according to Ewing and Kazazian 2010) (Fig. 2A,B). An L1 insertion was considered to be validated as tumor specific if the filled site (L1-containing) PCR product was present in the tumor, but not in the paired normal tissue, and in the case of heterozygous autosomal insertions, the empty site PCR product (WT allele) was amplified from both members of the tissue pair. Using a single PCR condition to amplify the 3′ junctions, we PCR-validated 26/40 and 37/51 insertions from the colo1 and colo2 high-stringency data sets, respectively. We also set out to PCR amplify 16 colo1 insertions represented by less than 100 Illumina reads and were able to validate nine. Thus, we PCR-validated before sequencing the 3′ ends of 72 of 107 putative insertions (Supplemental Table S3; Supplemental Text S1).
Interestingly, among 12 high-stringency putative insertions from normal colon of the combined colo1 and colo2 data sets, none could be validated. Possible explanations for false positives in the L1-seq data include PCR artifacts arising during library preparation, suboptimal PCR conditions used for validation, or L1 insertion into repetitive sequences, refractory to successful primer design.
Our stepwise PCR amplification scheme continued by retrieving the 5′ junctions of tumor-specific L1 insertion events. Several empty-site PCRs had already yielded a higher molecular weight band exclusively in the tumor, which in each case was verified to be a highly truncated L1 element (Fig. 2B). In the remaining cases, long-range PCR and a PCR specifically designed to amplify the 5′ end of a full-length L1 were used to retrieve the 5′ junction.
Figure 2.

PCR validation scheme of L1-seq results. (A) The three-step PCR validation scheme and location of primers used. Triangles symbolize TSD. (B) PCR validation of the 3′ junction (ins. 7). This insertion is in tumor 1 of the eight DNA samples that had been pooled for Illumina sequence analysis (left), while the right panel shows it is present exclusively in the tumor, but not in the normal colon. The higher molecular weight band visible above the ins. 7 empty site PCR product in the tumor is a highly truncated L1. (T) Tumor; (N) normal colon; (FS) filled site PCR product; (ES) empty site PCR product.
Altogether, out of 72 cases where the insertions were PCR-validated to be tumor specific, we successfully sequenced either the 3′ or the 5′ junction in 69 cases (Supplemental Table S3; Supplemental Text S1). For 35 insertions, we sequenced both junctions, enabling us to characterize TSDs and L1 insertion size in cancer tissue (Table 1). Surprisingly, all of the tumor-specific insertions were highly truncated, the mean L1 insertion size being 585 bp, excluding the poly(A) tail (Table 1).
Table 1.
L1Hs insertions in cancer with both 5′ and 3′ junctions characterized
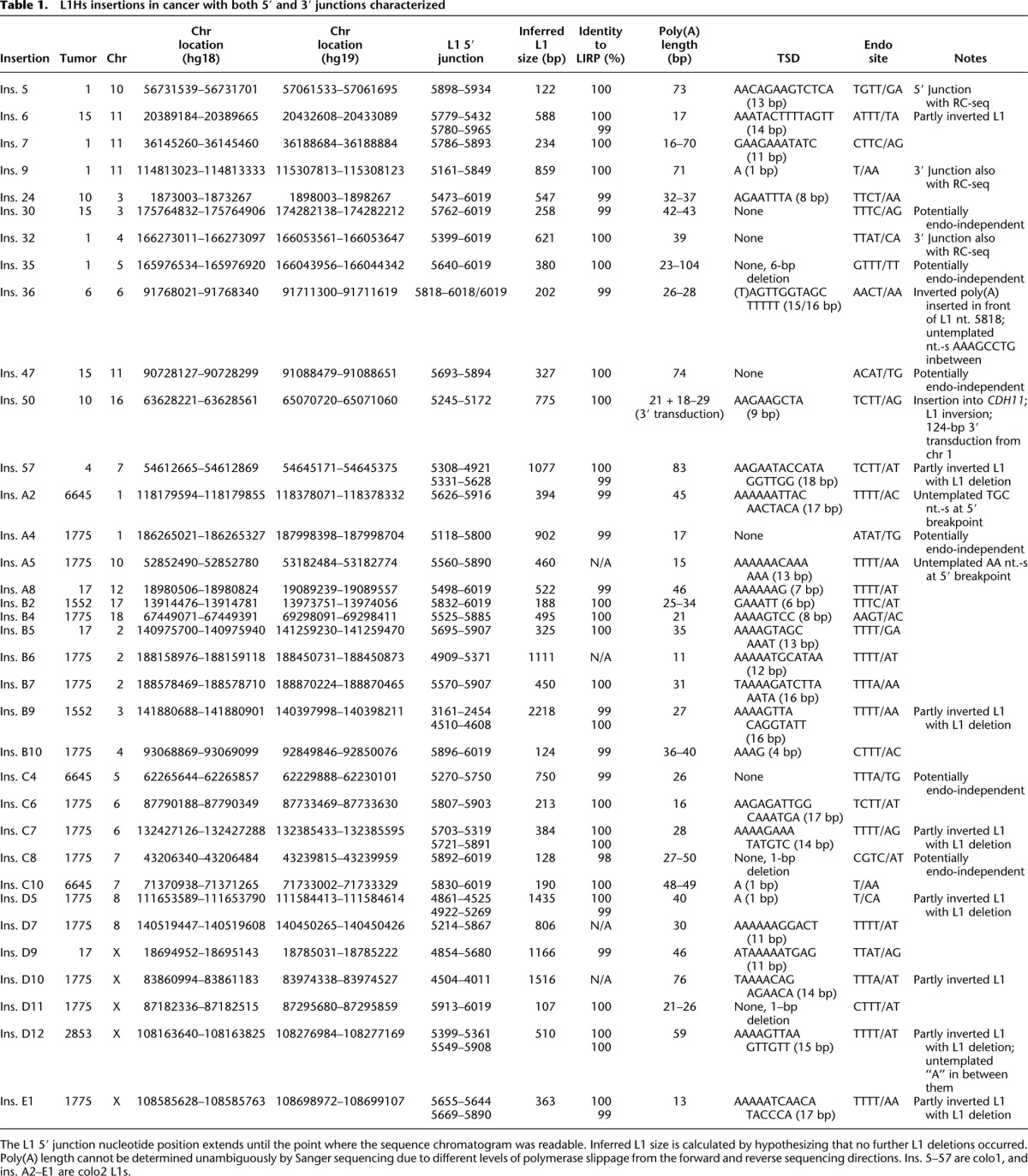
Using a PCR designed to amplify full-length L1 insertions, we failed to amplify the 5′ end of any of the remaining tumor-specific insertions where 5′ junctions could not be identified with the previous PCR approaches. On the other hand, three of 10 germline polymorphic insertions had an intact 5′ end, in agreement with 30% of reference L1Hs elements being full length (Pavlicek et al. 2002). This difference between full-length L1 insertions in tumors (∼0%) versus full-length insertions among polymorphic germline L1Hs (∼30%) is statistically significant (P = 0.016, Fisher's exact test) and is a clear departure from what is observed from the reference genome and from heritable nonreference insertions (Ewing and Kazazian 2010).
L1-seq results on five tumors were corroborated by RC-seq. Eleven high-confidence L1Hs hits were found in these cancers by L1-seq, out of which four were also detected by RC-seq at either the 5′ or 3′ junction (ins. 5, 9, 14, and 32) (Supplemental Tables S2, S3; Supplemental Text S1). Among eight high-confidence L1 insertions within genes detected by RC-seq, but missed by L1-seq, one was validated as present in tumor 10, targeting the DGKI gene (Supplemental Table S2) (Fig. 3). Of the remaining putative tumor-specific insertions that could be PCR-amplified, six of eight L1s, 30 of 57 Alus, and six of 11 SVAs were present in both tumor and paired normal tissue. No other confirmed tumor-specific insertions from the L1-seq data were found by RC-seq.
Figure 3.

Distribution of somatic L1 insertions in tumors. Insertions in black were detected by L1-seq, while the insertion in tumor 10 in white was detected by RC-seq only.
In order to determine whether L1s are frequently mobilized in other nonmalignant somatic tissues, we performed L1-seq on genomic DNA extracted from cerebrum, liver, and testis samples from two other individuals (cadaver samples) who had died from arteriosclerotic cardiovascular disease. None of the Illumina high-stringency sequence peaks suggestive of somatic L1 insertion could be PCR-validated, implying that the high rate of somatic L1 insertions observed in colon cancer was specific to the malignant tissue. Thus, we have no evidence from our data of somatic insertions in normal colon, liver, testis, or cerebrum.
Characterization of tumor-specific insertions
Intriguingly, the number of validated L1 insertions varied widely from tumor to tumor with up to 17 insertions in some and none in three others (Fig. 3). Most retrotransposition events showed hallmarks of TPRT, namely TSD (27/35), L1 endonuclease cleavage site, the presence of L1 poly(A) tail, frequent 5′ inversion (10/35), and in one case, a 3′ transduction (Table 1). However, we note that a substantial fraction of these somatic insertions (8/35) lacked a TSD and six of these lacked a discernible endonuclease cleavage site, suggesting that they were endonuclease-independent insertions (Morrish et al. 2002). Two insertions (“3” and “21”) contained 3′ sequence from other chromosomes, but lacked a poly(A) tail in between the two sequences that would indicate a 3′ transduction. Thus, they are likely cancer-associated recombination events (Supplemental Table S3; Supplemental Text S1).
Numerous genes were targets for insertional mutagenesis in colon tumors by L1s that are represented in the COSMIC database (Catalogue of Somatic Mutations in Cancer, http://www.sanger.ac.uk/genetics/CGP/cosmic/). Examples include PTPRM (protein tyrosine phosphatase, receptor type, M), ODZ3 (odd Oz/ten-m homolog 3), ROBO2 (roundabout, axon guidance receptor, homolog 2), PCM1 (pericentriolar material 1), and CDH11 (cadherin-11). PCM1 and CDH11 are also represented in Sanger's Cancer Gene Census (http://www.sanger.ac.uk/genetics/CGP/Census/). Interestingly, according to COSMIC, in the large intestine these genes were mutated with the following high frequencies: PTPRM (50%), ODZ3 (100%), ROBO2 (15%), PCM1 (12%), CDH11 (52%). All of our hits were intronic and were PCR validated as well as sequenced. Additional interesting genes with a potential role in malignancy were also targeted, for instance, RUNX1T1 (runt-related transcription factor 1), a member of the myeloid translocation genes. Interestingly, somatic RUNX1T1 point mutations were not only found in colorectal cancers (Wood et al. 2007), but the product of the related RUNX3 gene regulates L1 expression (Yang et al. 2003).
Cellular timing of L1 retrotransposition
In an effort to determine at what point in tumorigenesis the L1 insertions occurred, we developed three lines of evidence: analysis of SNPs in sequence flanking the L1 insertion and the empty site, the number of empty site X chromosome alleles in males who had an L1 insertion into the X, and the presence/absence of the L1 insertion in a second section of a particular tumor in which an L1 insertion occurred.
First, we found three insertions (C2, C4, and insertion 31) with flanking heterozygous SNPs. For C2 (ODZ3 gene) there was one SNP, for C4 there were four SNPs, and for insertion 31 there was one flanking SNP (Supplemental Fig. S3A). The presence of both alleles of the particular SNP in the empty site chromosomes is informative in case of no aneuploidy at the respective alleles. If the insertion occurred at the initiation of the tumor (the one-cell stage), the filled site would contain one allele and the empty site would contain the other allele only. If both alleles are present in the empty-site chromosomes, then the insertion likely occurred after the one-cell stage of the tumor. The data showed that all six SNPs near three different insertions in the empty-site chromosomes were heterozygous, suggesting that the insertions occurred after the initiation of tumorigenesis. We carried out array comparative genomic hybridization (aCGH) and found no copy-number gain or loss in the chromosomal arm of these SNPs (data not shown), although small chromosomal aberrations at the respective alleles cannot be ruled out.
Second, we found insertions (D9, D12, and E3) into the X chromosome in two males. If the insertion occurred at the one-cell stage and the male did not have X-chromosome aneuploidy, the tumor should lack an empty site. However, in all cases, we found an empty-site band by PCR, indicating again that the insertions occurred after the one-cell stage of tumorigenesis (Supplemental Fig. S3B contains data on D9 and D12). Again, aCGH showed that both males had a single X chromosome.
Third, we obtained a second portion of tissue from a number of tumors and determined whether the insertions found in the first tumor tissues could be confirmed in the second tumor sample. In three of seven instances (insertions C2, D7, and E3) we were able to confirm the insertion in a second tumor sample, suggesting a relatively early event in tumorigenesis. Four other insertions (“31,” “A10,” “C4,” “D12”) were present in the first tumor section, but not in the second (Supplemental Fig. S3C). In tumor 2853, two insertions were studied: E3 was present in both tumor portions, while D12 was not, suggesting that these two insertions occurred at different times and in different cells of the tumor (data not shown). Furthermore, heterozygosity for L1 flanking SNPs in insertions 31 and C4 in the original tumor sample, as well as the absence of the insertion from the second tumor portion both suggest late insertion events. Thus, from the combination of these data on a small sample size we conclude that most, if not all, of the studied L1 insertions occurred after the initiation of the tumor. However, a minority of the insertions may have occurred at an early stage of tumorigenesis. Furthermore, we cannot exclude the possibility of tumor blood vessels or infiltrating lymphocytes as contributing alternative explanations for some of the results.
Effects of L1 retrotransposition on measures of cellular instability
To address the increased rate of somatic L1 retrotransposition in tumors, we assessed the genomic landscape of the tissue samples by aCGH, microsatellite instability (MSI), and L1 promoter methylation status. A previous report demonstrated a correlation of genome-wide DNA methylation status of tumors with increased de novo L1 insertions (Iskow et al. 2010). We assessed the methylation status of the L1 promoter at four different CpG sites. Although L1 promoter hypomethylation was found in tumor samples compared with paired normal tissue, no correlation was observed between L1 methylation status and the number of L1 insertions (Fig. 4A).
Figure 4.

Analysis of factors influencing L1 activity. (A) L1 CpG promoter methylation status performed by quantitative bisulfite PCR analysis. (N) Normal tissue; (T) tumor tissue; (*) MSI. Replicates of four were done for each data point. (Error bars) Standard deviations. (B) MSI analysis. 6% TBE gel depicting the status of five microsatellite repeats (BAT25, BAT26, D2S123, D17S250, D17346 in descending order) in normal and tumor tissue from two different patients. Tumor tissue “6” contained additional bands and gel shifts compared with the normal tissue, indicating MSI. Samples from “8” demonstrated no differences suggestive of MSI. (C) Correlation of L1 activity with age of the patient at time of surgery. See text for details.
For aCGH, we analyzed normal and tumor tissue from six patient samples, two that possessed the greatest number of de novo tumor insertions (12 and 1775), two with no tumor insertions (8 and 7647), and the two males mentioned above with both empty and filled insertion sites in X chromosomes (17 and 2853). Tumor samples 8, 12, 17, and 1775 contained complex chromosomal changes, including entire and partial chromosomal gains and losses. Tumor samples 2853 and 7647 presented no detectable aberrations relative to normal tissue. Interestingly, three of the samples (12, 17, and 1775) with complex chromosomal rearrangements had a high number of validated insertions. Likewise, patient 7647 with no validated L1 insertions had no detectable chromosomal changes. The outliers from the trend of a direct relationship between chromosomal aberrations and L1 insertions were patients 8 and 2853. Interestingly, these latter patients are potentially genetically predisposed to colon cancer due to familial cancer aggregation or a very young age of diagnosis.
In addition to analysis of gross genomic abnormalities, we assessed the status of the mismatch DNA repair pathway by assessing microsatellite expansions in the genome. Seven of the 16 patients were MSI positive. Although two samples with the highest number of somatic L1 insertions were MSI positive, MSI status did not correlate with the number of de novo L1 insertions for each tumor (Fig. 4B).
Interestingly, a statistically significant correlation was observed between the number of insertions and the age of the investigated patients (P = 0.01425, R2 = 0.3128, where age is the time of surgical sample removal). Eight or more validated insertions were observed only in the tumors of patients 78 yr old or older. An outlier in the correlation was a 72-yr-old with no validated insertions. However, he was the only proband with rectal cancer, but no colon tumor diagnosis. When this patient was excluded from the analysis, as well as cases with a presumed genetic predisposition to colon cancer (familial polyposis case and a 17-yr-old male), an even more significant correlation was observed between the age of sporadic colon cancer patients and L1 activity (P = 0.001548, R2 = 0.578) (Fig. 4C).
Discussion
Our L1-seq method has revealed a high rate of L1Hs retrotransposition in certain colorectal cancer genomes. The neighboring matched normal colon sample in these 16 cases, as well as cerebrum, liver, and testis from two other individuals yielded no L1 insertions that could be validated, indicating few or no retrotransposition events in these normal tissues.
Iskow et al. (2010) used 454 pyrosequencing to search for de novo L1 insertions in five glioblastomas, five medulloblastomas, as well as leukemia and breast cancer cell lines, but they found no insertions in these cases. However, they identified nine somatic L1 insertions in six of 20 lung tumors. Since TSDs were not reported, the question of whether L1 integration in lung cancer occurs through TPRT remained open.
Here we report that evolutionarily young L1Hs retrotransposons can mobilize themselves through the classical TPRT mechanism in colon cancer genomes at a high frequency. The true retrotransposition rate is likely to be even higher, as our method does not detect insertions mobilized by L1 elements in trans, such as Alus, SVAs, most inactive L1s, and processed pseudogenes. In addition, there are likely other L1 insertions in our data set that have not been subjected to validation. Longer tumor-specific 3′ transductions will be missed as well, as it is difficult to differentiate between the progenitor and the transduced sequence by their 3′ flank with L1-seq.
In order to determine why the rate of somatic cell retrotransposition was high in some tumors compared with the normal tissue, we assessed how the following factors correlate with retrotransposition rate: age of colorectal cancer patients, chromosomal aberrations, L1 methylation status, and mismatch DNA repair as reflected by MSI. A clear correlation of retrotransposition activity was observed with the age of colon cancer patients in sporadic cases. Furthermore, we also analyzed genetic instability by aCGH in six samples and found a modest association between the number of chromosomal aberrations and the age of the sporadic colon cancer patients. Altogether, it is possible that the hypomethylated microenvironment of the tumors, together with genetic instability as reflected by MSI and gross chromosomal changes, have a cumulative effect in older patients. Our results are in agreement with the correlation of retrotransposition activity with genome instability during yeast chronological aging (Maxwell et al. 2011).
In this study, we found that genes with a known driver function in cancer are mutagenized by L1Hs elements. The accumulation of retrotransposon sequences is predicted to cause further genetic instability through recombination. Thus, an elevated insertion rate is expected to contribute to tumor evolution. As exemplified by a somatic L1 insertion into the APC gene (Miki et al. 1992), it is clear that in some fraction of colorectal cancers retrotransposon insertions can be etiologically significant. Yet, it remains unclear in what fraction of cases retrotransposons initiate malignant transformation and in how many instances they contribute solely to a more aggressive phenotype. SNP, X chromosome, and secondary sampling data from tumor samples suggest that L1 insertions likely occurred at various times after the initiation of the tumor. Although we found no evidence for L1 insertion into colon cancer tumor-suppressor genes or oncogenes that would be indicative of driver mutation-induced tumor clonality, analysis of a larger number of tumors or a deeper sequencing of retrotransposon insertions could uncover such events. We propose that it is possible to estimate insertion timing and tumor heterogeneity more precisely by evaluating pure tumor samples. Our findings are in agreement with a very recent report on retrotransposon insertions in epithelial cancers (Lee et al. 2012). Intriguingly, an intronic L1 integration event was found in that study as well in the ROBO2 gene in a colon tumor. Additionally, they detected intronic L1 insertions in CDH12, while we characterized an insertion into the CDH11 gene. Thus, the role of cell-adhesion genes in retrotransposon insertion-mediated colorectal tumorigenesis may deserve further investigation.
An unexpected finding of our PCR-based validation is the severely truncated nature of all validated L1 insertions in colon cancer. It was not possible to assess whether this is a general characteristic of the malignant phenotype, of all somatic tissues, or of the gastrointestinal tract in particular, as no de novo L1 insertions could be uncovered from normal colon, liver, testis, and brain. In a transgenic mouse model, 30 of 33 somatic L1 insertions were 5′ truncated (Babushok et al. 2006), raising the possibility of a gradual decrease in L1 size from germline to somatic to malignant insertions. In cultured HeLa cells, 94/100 insertions were 5′ truncated (Gilbert et al. 2005). This might indicate that some cancer tissues or cultured cells could allow full-length insertions to accumulate. The overexpression of an exogenous L1 element, coupled with the bias toward recovering larger inserts in that assay, and the unknown effects of cell culture conditions on retrotransposition complicate transferring conclusions on the L1 5′ truncation rate to cancer tissue. Likewise, it is not understood why the majority of germline L1 insertions are 5′ truncated as opposed to Alu and SVA insertions that are mostly full length, yet also mobilized by L1s (Hancks et al. 2011).
The truncated structure of L1 elements in colorectal cancer may be useful in understanding the mechanism of 5′ truncation both in normal and tumor cells. We propose two possible explanations: (1) If TPRT timing is coupled to the cell cycle, the elevated cell division rate of malignant cells may not leave sufficient time to complete integration of long mobile elements; (2) a DNA repair pathway might monitor and remove de novo mobile element insertions in healthy tissues. Once this presumed surveillance pathway is down-regulated in cancer, retrotransposon insertions are not removed efficiently and are allowed to accumulate. At the same time, another or the same DNA repair pathway might specialize in truncating fresh integrants or prohibiting them from completing retrotransposition. If this process is up-regulated, the truncation rate increases. The efficiency of such a pathway might correlate with insertion size or be sequence specific, thus preferentially targeting L1 elements over Alus and SVAs. Interestingly, nonhomologous end joining (NHEJ) is an important DNA repair pathway candidate with a reported conflicting dual role in regulating retrotransposon insertions, offering an explanation for parallel L1 up-regulation and truncation (Suzuki et al. 2009). We propose that by comparing the genome or transcriptome of tumors with a high rate of retrotransposition to their paired normal tissues, we may discern clues to cellular factors causing L1 mobilization and 5′ truncation.
To conclude, the cancerous colon of many patients is the second reported organ beside the brain (Baillie et al. 2011) in which a high rate of retrotransposition occurs. Lung, prostate, and ovarian tumors are also reported to allow a lower level of L1 mobilization (Iskow et al. 2010; Lee et al. 2012), but many other cancer types appear to be nonpermissive for a detectable rate of retrotransposition. All L1 insertions in the colorectal tumors of this study were highly truncated, potentially indicating the footprint of a defective or hyperactive DNA repair pathway in cancer. The cause and effect of retrotransposon mobilization in cancers warrants further investigation.
Methods
Human DNA samples
DNA was extracted from human patient tissue samples acquired from the University of Minnesota Tissue Procurement Facility from BioNet (IRB#0805E32181). See Supplemental Table S4 for patient data. Briefly, 2 mg of tissue was digested overnight at 55°C on a rotating platform in 710 μL of digest buffer (1 M Tris at pH 8.0, 1 mM EDTA, 1× SSC, 1% SDS, 1 Mm NaCl, 10 μg/mL Proteinase K). Following digest, DNA was purified using phenol-chloroform-isoamyl alcohol (Life Sciences) isolation protocol.
Human frozen tissue from two Caucasian cadavers with arteriosclerotic cardiovascular disease were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore. DNA isolation was done utilizing the AllPrep DNA/RNA Mini Kit (Qiagen).
Library construction, sequencing, and analysis
L1-seq
The library for L1Hs elements was made according to Ewing and Kazazian (2010), while the library for L1, Alu, and SVA elements (RC-seq) was constructed according to Baillie et al. (2011). L1Hs elements were TOPO-TA cloned (Invitrogen) and Sanger-sequenced for quality control of the library preparation, and were subsequently sequenced on an Illumina HiSeq 2000 at the Johns Hopkins University Genetic Resources Core Facility High Throughput Sequencing Center.
Pooled L1-seq library sequence data was analyzed as described previously (Ewing and Kazazian 2010), and compared against insertion sites of known reference and nonreference transposable elements (Beck et al. 2010; Ewing and Kazazian 2010, 2011; Huang et al. 2010; Iskow et al. 2010; Witherspoon et al. 2010; Hormozdiari et al. 2011; Stewart et al. 2011). Gene annotations were obtained from UCSC Known Genes (Hsu et al. 2006).
RC-seq
The library for retrotransposon capture and sequencing was created utilizing DNA from five pairs of colorectal and normal tissue (samples 1; 4; 6; 8; 10) by the same method as published previously (Baillie et al. 2011). One significant change was made to the technique, in that liquid phase hybridization was performed as opposed to solid surface, chip-based hybridization. The libraries were then sequenced on an Illumina Genome Analyzer II and aligned to the genome by a computational pipeline that utilized SOAP2 to align reads to the genome and used much the same method as published (Ballie et al. 2011; R Shukla, KR Upton, M Muñoz-Lopez, DJ Gerhardt, JK Baillie, ME Fisher, PM Brennan, A Collino, S Ghisletti, S Sinha, et al., in prep.).
PCR validation of the Illumina results
A three-step PCR validation protocol was used to validate the next-generation sequencing reads and to retrieve 3′ and 5′ junctions. As the first step, L1 3′ ends together with flanking genomic regions were amplified using the same AC dinucleotide–specific primer of L1Hs as used for Illumina sequencing (L1Hs primer: GGGAGATATACCTAATGCTAGATGACAC) and a primer selected from the 3′ flanking region based on the reference genome sequence (FS primer). PCR reactions were carried out in 12.5 μL of 2× GoTaq Green master mix (Promega) in a total volume of 25 μL, with 0.8 μL of FS primer, 1.5 μL of L1Hs primer, and 25 ng of DNA to amplify the filled site. The empty site was amplified with the same conditions, except that 1.5 μL of FS primer, 1.5 μL of ES primer, and 12.5 ng of DNA were used. Primers were 20 pmol/μL and their location is depicted in Figure 2A. Reactions were incubated for 2 min at 95°C, followed by 30 cycles of 30 sec at 95°C, 30 sec at 57°C, and 1.5 min at 72°C, followed by a final extension of 5 min at 72°C on a PTC-200 Peltier Thermal Cycler. Long-range PCR to recover longer L1 insertions was performed with the Expand Long Template PCR System (Roche) according to the manufacturer's instructions in buffer 1, with 1 μL of 20 μM FS and ES primers each, and 25 ng of tumor DNA. 5′ junctions were PCR amplified using the same conditions as for the 3′ junction, except that a primer hybridizing to the L1 5′UTR was used (L1nt112out: GATGAACCCGGTACCTCAGA) together with the respective ES primer, and primer extension time was only 45 sec. FS and ES primer sequences are included in the Supplemental Material (Supplemental Table S1). PCR products were cut out of the gel, extracted with the QIAquick Gel Extraction Kit (Qiagen), and sequenced. See Supplemental Text S1 for Sanger sequence data on insertions.
Microsatellite instability assays
To assess the MSI status, we utilized five markers recommended by the National Cancer Institute (Bethesda markers): BAT25 and BAT26 to assess mononucleotide repeats (A)n and D2S123, D5S346, and D17S250 to assess dinucleotide repeats (CA)n. MSI status was determined using previously established protocols (Ashktorab et al. 2003; Muller et al. 2004). Primers were developed by the NCI for screening patients in the clinic.
L1 methylation status
The methylation level of L1 promoters was performed according to Wilhelm et al. (2010). Briefly, each sample was amplified three times and each amplification was pyrosequenced once. The average of the three was utilized to determine the value of CpG methylation for each of the four positions analyzed for an L1.
aCGH
DNA from patients 8, 12, 17, 1775, 2853, and 7647 were restriction digested and labeled with fluorochrome Cyanine-5 using random primers and exo-Klenow fragment DNA polymerase. DNA from a sex-matched control was labeled concurrently with Cyanine-3. The sample and control DNA were combined and array-based comparative genomic hybridization (aCGH) and single nucleotide polymorphism analysis (SNP) was performed with a 180K Cancer CGH+SNP microarray constructed by Agilent Technologies, Inc. that contains ∼115,000 distinct biological oligonucleotides and 55,000 SNP sites, spaced at an average interval of 25 KB (for 20,000 cancer-associated CGH probes: one probe/0.5–1 KB). The ratio of sample to control DNA for each oligo was calculated using Feature Extraction software 10.10 (Agilent Technologies). The abnormal threshold was applied using Cytogenomics 2.060 (Agilent Technologies). A combination of several statistical algorithms was applied. A minimum of three oligos that have a minimum absolute ratio value of 0.1 (based on a log(2) ratio) is required for reporting of a copy-number loss or gain. Analysis was performed using Human Genome Build 19 (Feb 2009) as the reference.
Data access
The sequence and phenotypic data from this study have been deposited in dbGaP (http://www.ncbi.nlm.nih.gov/dbgap) under accession number phs000536.v1.p1. The dbGaP accession number assigned to this study is phs000536.v1.p1.
Acknowledgments
We thank Ricardo Linares, John L. Goodier, and Prabhat K. Mandal for their great insights into the project and for their comments on the manuscript. We thank David Haussler for advice. Ling Cheung is acknowledged for excellent technical assistance. The cytogenetic analyses were performed in the Cytogenetics Core Laboratory at the University of Minnesota with support from the comprehensive Masonic Cancer Center NIH Grant #P30 CA077598-09. Research in the Kazazian laboratory is funded by grants from the National Institutes of Health awarded to H.H.K. Human tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore (NICHD Contract no. N01-HD-4-3368 and NO1-HD-4-3383). The role of the NICHD Brain and Tissue Bank is to distribute tissue, and therefore, cannot endorse the studies performed or the interpretation of results.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.145235.112.
References
- Asch HL, Eliacin E, Fanning TG, Connolly JL, Bratthauer G, Asch BB 1996. Comparative expression of the LINE-1 p40 protein in human breast carcinomas and normal breast tissues. Oncol Res 8: 239–247 [PubMed] [Google Scholar]
- Ashktorab H, Smoot DT, Carethers JM, Rahmanian M, Kittles R, Vosganian G, Doura M, Nidhiry E, Naab T, Momen B, et al. 2003. High incidence of microsatellite instability in colorectal cancer from African Americans. Clin Cancer Res 9: 1112–1117 [PubMed] [Google Scholar]
- Babushok DV, Ostertag EM, Courtney CE, Choi JM, Kazazian HH Jr 2006. L1 integration in a transgenic mouse model. Genome Res 16: 240–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, et al. 2011. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479: 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV 2010. LINE-1 retrotransposition activity in human genomes. Cell 141: 1159–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Garcia-Perez JL, Badge RM, Moran JV 2011. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet 12: 187–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O'Shea KS, Moran JV, Cullen BR 2006. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci 103: 8780–8785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Chevret P, Furano AV 2000. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol 17: 915–928 [DOI] [PubMed] [Google Scholar]
- Bourc'his D, Bestor TH 2004. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431: 96–99 [DOI] [PubMed] [Google Scholar]
- Bratthauer GL, Fanning TG 1992. Active LINE-1 retrotransposons in human testicular cancer. Oncogene 7: 507–510 [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr 2003. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci 100: 5280–5285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD 2006. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol 16: 480–485 [DOI] [PubMed] [Google Scholar]
- Cost GJ, Feng Q, Jacquier A, Boeke JD 2002. Human L1 element target-primed reverse transcription in vitro. EMBO J 21: 5899–5910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Marchetto MC, Muotri AR, Mu Y, Carson CT, Macia A, Moran JV, Gage FH 2011. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc Natl Acad Sci 108: 20382–20387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD 2011. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 7: e1002384 doi: 10.1371/journal.pgen.1002384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewannieux M, Esnault C, Heidmann T 2003. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet 35: 41–48 [DOI] [PubMed] [Google Scholar]
- Esnault C, Maestre J, Heidmann T 2000. Human LINE retrotransposons generate processed pseudogenes. Nat Genet 24: 363–367 [DOI] [PubMed] [Google Scholar]
- Ewing AD, Kazazian HH Jr 2010. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res 20: 1262–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Kazazian HH Jr 2011. Whole-genome resequencing allows detection of many rare LINE-1 insertion alleles in humans. Genome Res 21: 985–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH Jr, Boeke JD 1996. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87: 905–916 [DOI] [PubMed] [Google Scholar]
- Gasior SL, Wakeman TP, Xu B, Deininger PL 2006. The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol 357: 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Lutz S, Morrish TA, Moran JV 2005. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol 25: 7780–7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Kazazian HH Jr 2008. Retrotransposons revisited: The restraint and rehabilitation of parasites. Cell 135: 23–35 [DOI] [PubMed] [Google Scholar]
- Goodier JL, Ostertag EM, Kazazian HH Jr 2000. Transduction of 3′-flanking sequences is common in L1 retrotransposition. Hum Mol Genet 9: 653–657 [DOI] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH 2012. Active human retrotransposons: Variation and disease. Curr Opin Genet Dev 22: 191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Goodier JL, Mandal PK, Cheung LE, Kazazian HH Jr 2011. Retrotransposition of marked SVA elements by human L1s in cultured cells. Hum Mol Genet 20: 3386–3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Normart R, Yang Q, Stevenson E, Haffty BG, Ganesan S, Cordon-Cardo C, Levine AJ, Tang LH 2010. Association of nuclear localization of a long interspersed nuclear element-1 protein in breast tumors with poor prognostic outcomes. Genes Cancer. 1: 115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes SE, Dombroski BA, Krebs CM, Boehm CD, Kazazian HH Jr 1994. A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat Genet 7: 143–148 [DOI] [PubMed] [Google Scholar]
- Hormozdiari F, Alkan C, Ventura M, Hajirasouliha I, Malig M, Hach F, Yorukoglu D, Dao P, Bakhshi M, Sahinalp SC, et al. 2011. Alu repeat discovery and characterization within human genomes. Genome Res 21: 840–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D 2006. The UCSC known genes. Bioinformatics 22: 1036–1046 [DOI] [PubMed] [Google Scholar]
- Huang CR, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T, et al. 2010. Mobile interspersed repeats are major structural variants in the human genome. Cell 141: 1171–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE 2010. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 141: 1253–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, et al. 2011. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471: 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloor M, Sutter C, Wentzensen N, Cremer FW, Buckowitz A, Keller M, von Knebel Doeberitz M, Gebert J 2004. A large MSH2 Alu insertion mutation causes HNPCC in a German kindred. Hum Genet 115: 432–438 [DOI] [PubMed] [Google Scholar]
- Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ III, Lohr JG, Harris CC, Ding L, Wilson RK, et al. 2012. Landscape of somatic retrotransposition in human cancers. Science 337: 967–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH 1993. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell 72: 595–605 [DOI] [PubMed] [Google Scholar]
- Mathias SL, Scott AF, Kazazian HH Jr, Boeke JD, Gabriel A 1991. Reverse transcriptase encoded by a human transposable element. Science 254: 1808–1810 [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Burhans WC, Curcio MJ 2011. Retrotransposition is associated with genome instability during chronological aging. Proc Natl Acad Sci 108: 20376–20381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y 1992. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res 52: 643–645 [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH Jr 1996. High frequency retrotransposition in cultured mammalian cells. Cell 87: 917–927 [DOI] [PubMed] [Google Scholar]
- Moran JV, DeBerardinis RJ, Kazazian HH Jr 1999. Exon shuffling by L1 retrotransposition. Science 283: 1530–1534 [DOI] [PubMed] [Google Scholar]
- Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, Moran JV 2002. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet 31: 159–165 [DOI] [PubMed] [Google Scholar]
- Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, Flory E, Schumann GG, Munk C 2006. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem 281: 22161–22172 [DOI] [PubMed] [Google Scholar]
- Muller A, Edmonston TB, Dietmaier W, Buttner R, Fishel R, Ruschoff J 2004. MSI-testing in hereditary non-polyposis colorectal carcinoma (HNPCC). Dis Markers 20: 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima K, Hattori M, Yada T, Gojobori T, Sakaki Y, Okada N 2003. Whole-genome screening indicates a possible burst of formation of processed pseudogenes and Alu repeats by particular L1 subfamilies in ancestral primates. Genome Biol 4: R74 doi: 10.1186/gb-2003-4-11-r74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oricchio E, Sciamanna I, Beraldi R, Tolstonog GV, Schumann GG, Spadafora C 2007. Distinct roles for LINE-1 and HERV-K retroelements in cell proliferation, differentiation and tumor progression. Oncogene 26: 4226–4233 [DOI] [PubMed] [Google Scholar]
- Ostertag EM, Goodier JL, Zhang Y, Kazazian HH Jr 2003. SVA elements are nonautonomous retrotransposons that cause disease in humans. Am J Hum Genet 73: 1444–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicek A, Paces J, Zika R, Hejnar J 2002. Length distribution of long interspersed nucleotide elements (LINEs) and processed pseudogenes of human endogenous retroviruses: Implications for retrotransposition and pseudogene detection. Gene 300: 189–194 [DOI] [PubMed] [Google Scholar]
- Pickeral OK, Makalowski W, Boguski MS, Boeke JD 2000. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res 10: 411–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiz J, Damert A, Chira S, Held U, Klawitter S, Hamdorf M, Lower J, Stratling WH, Lower R, Schumann GG 2011. The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res 40: 1666–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyom S, Ewing AD, Hancks DC, Takeshima Y, Awano H, Matsuo M, Kazazian HH Jr 2012. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Hum Mutat 33: 369–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenglein MD, Harris RS 2006. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem 281: 16837–16841 [DOI] [PubMed] [Google Scholar]
- Stewart C, Kural D, Stromberg MP, Walker JA, Konkel MK, Stutz AM, Urban AE, Grubert F, Lam HY, Lee WP, et al. 2011. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet 7: e1002236 doi: 10.1371/journal.pgen.1002236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LK, Steinbach G, Sawyer JC, Hindi M, Ward PA, Lynch PM 2000. Genomic rearrangements of the APC tumor-suppressor gene in familial adenomatous polyposis. Hum Genet 106: 101–107 [DOI] [PubMed] [Google Scholar]
- Su Y, Davies S, Davis M, Lu H, Giller R, Krailo M, Cai Q, Robison L, Shu XO, Children's Oncology Group 2007. Expression of LINE-1 p40 protein in pediatric malignant germ cell tumors and its association with clinicopathological parameters: A report from the Children's Oncology Group. Cancer Lett 247: 204–212 [DOI] [PubMed] [Google Scholar]
- Suzuki J, Yamaguchi K, Kajikawa M, Ichiyanagi K, Adachi N, Koyama H, Takeda S, Okada N 2009. Genetic evidence that the non-homologous end-joining repair pathway is involved in LINE retrotransposition. PLoS Genet 5: e1000461 doi: 10.1371/journal.gen.1000461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Klift HM, Tops CM, Hes FJ, Devilee P, Wijnen JT 2012. Insertion of an SVA element, a nonautonomous retrotransposon, in PMS2 intron 7 as a novel cause of lynch syndrome. Hum Mutat 33: 1051–1055 [DOI] [PubMed] [Google Scholar]
- Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, Moran JV 2001. Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol 21: 1429–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm CS, Kelsey KT, Butler R, Plaza S, Gagne L, Zens MS, Andrew AS, Morris S, Nelson HH, Schned AR, et al. 2010. Implications of LINE1 methylation for bladder cancer risk in women. Clin Cancer Res 16: 1682–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherspoon DJ, Xing J, Zhang Y, Watkins WS, Batzer MA, Jorde LB 2010. Mobile element scanning (ME-Scan) by targeted high-throughput sequencing. BMC Genomics 11: 410 doi: 10.1186/1471-2164-11-410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. 2007. The genomic landscapes of human breast and colorectal cancers. Science 318: 1108–1113 [DOI] [PubMed] [Google Scholar]
- Yang N, Zhang L, Zhang Y, Kazazian HH Jr 2003. An important role for RUNX3 in human L1 transcription and retrotransposition. Nucleic Acids Res 31: 4929–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13: 335–340 [DOI] [PubMed] [Google Scholar]