

Abstract
Typically, F1-hybrids are more vigorous than their homozygous, genetically distinct parents, a phenomenon known as heterosis. In the present study, the transcriptomes of the reciprocal maize (Zea mays L.) hybrids B73×Mo17 and Mo17×B73 and their parental inbred lines B73 and Mo17 were surveyed in primary roots, early in the developmental manifestation of heterotic root traits. The application of statistical methods and a suitable experimental design established that 34,233 (i.e., 86%) of all high-confidence maize genes were expressed in at least one genotype. Nearly 70% of all expressed genes were differentially expressed between the two parents and 42%–55% of expressed genes were differentially expressed between one of the parents and one of the hybrids. In both hybrids, ∼10% of expressed genes exhibited nonadditive gene expression. Consistent with the dominance model (i.e., complementation) for heterosis, 1124 genes that were expressed in the hybrids were expressed in only one of the two parents. For 65 genes, it could be shown that this was a consequence of complementation of genomic presence/absence variation. For dozens of other genes, alleles from the inactive inbred were activated in the hybrid, presumably via interactions with regulatory factors from the active inbred. As a consequence of these types of complementation, both hybrids expressed more genes than did either parental inbred. Finally, in hybrids, ∼14% of expressed genes exhibited allele-specific expression (ASE) levels that differed significantly from the parental-inbred expression ratios, providing further evidence for interactions of regulatory factors from one parental genome with target genes from the other parental genome.
Heterosis describes the superior vigor of heterozygous F1-progeny as compared to the average of their genetically distinct, homozygous parents. The superior vigor of hybrid plants can be measured for a diverse set of agronomically important traits such as stress resistance, maturity, biomass, and grain yield (for review, see Falconer and Mackay 1996). The degree of heterosis in a hybrid can differ for distinct traits of a genotype or different genotype combinations (e.g., Hoecker et al. 2006).
Heterosis is extensively exploited in commercial plant breeding programs (Duvick 1999). Virtually all maize (Zea mays L.) grown commercially in North America and western Europe is hybrid (Duvick 1999, 2005). Despite its agronomic importance, the molecular principles underlying heterosis are still only poorly understood. Plant growth and development are determined by complex, tightly regulated global gene expression networks (Long et al. 2008). Hence, the superior vigor of highly heterozygous hybrid plants compared to their homozygous parental inbred lines is assumed to be a result of global differences in gene expression between inbred lines and hybrids. Such differences have been observed for many genes in different tissues and genotypes (for review, see Birchler et al. 2010). These studies revealed tissue- and stage-specific differential gene expression patterns between inbred lines and hybrids (for review, see Hochholdinger and Hoecker 2007) and the consistent differential expression of key genes (Hoecker et al. 2008a). In addition, for some traits a correlation between the number of differentially expressed genes and the degree of heterosis was suggested (Li et al. 2009; Riddle et al. 2010). Significantly, gene expression divergence in hybrids seems to correlate with the genetic divergence of the parental inbred lines (Birchler and Veitia 2010). Recently, some initial experiments correlating parental gene expression with hybrid vigor have been performed (Frisch et al. 2010; Thiemann et al. 2010).
Phenotypically, the degree of heterosis for a specific trait in hybrids is defined with respect to the quantitative deviation of a trait from the average parental value, designated mid-parent value (MPV). Mid-parent heterosis (MPH) is defined as hybrid performance of a trait that exceeds the MPV, whereas hybrid traits that display best-parent heterosis (BPH) significantly exceed the better performing parent (Hochholdinger and Hoecker 2007). In addition to high levels of phenotypic variation, heterosis is also often associated with transcriptional variation, and the classification of nonadditive gene expression is usually based on the terms describing the phenotypic degrees of heterosis-related traits reviewed in Hochholdinger and Hoecker (2007). Accordingly, hybrid gene expression levels that equal that of one parent but are significantly different from that in the second parent are classified as high- or low-parent heterosis, while hybrid gene expression levels significantly exceeding parental values are defined as above high-parent or below low-parent heterosis.
In the past, several classical genetic concepts were considered to explain hybrid vigor (for review, see Hochholdinger and Hoecker 2007). Among those, the dominance model explains the better performance of hybrids by the complementation of deleterious recessive alleles by superior dominant alleles at multiple loci (Jones 1917). Recently, it was suggested that a combination of different genetic principles might best explain the manifestation of hybrid vigor (Swanson-Wagner et al. 2006; Lippman and Zamir 2007). To better understand the processes underlying the manifestation of heterosis for different phenotypic traits, versatile molecular data were collected at different regulatory levels including the genome (Lai et al. 2005; Swanson-Wagner et al. 2010), the proteome (Hoecker et al. 2008b; Marcon et al. 2010), the metabolome (Römisch-Margl et al. 2010), and the transcriptome (for review, see Paschold et al. 2010). Nevertheless, association of multilevel molecular data with phenotypic variation of hybrids remains challenging. Consequently, thus far most studies focused on distinct regulatory levels.
Maize is a highly polymorphic species with a level of structural genomic diversity that is, perhaps, unique among higher eukaryotes (Springer et al. 2009). Different maize genotypes display significant copy number variation (CNV) and presence/absence variation (PAV) of genes, which have been hypothesized to contribute to the unusual level of phenotypic diversity in this important cereal (Lai et al. 2010; Swanson-Wagner et al. 2010). The regulatory machinery controlling gene expression was considered to play a key role in the manifestation of heterosis. The recent availability of high-throughput gene expression surveys has fostered transcriptome analyses of inbred lines and hybrids in search of nonadditive gene expression patterns in highly heterozygous heterotic hybrids (Guo et al. 2006; Stupar and Springer 2006; Swanson-Wagner et al. 2006; Wang et al. 2006; Meyer et al. 2007; Uzarowska et al. 2007; Hoecker et al. 2008b; Wei et al. 2009; He et al. 2010; Jahnke et al. 2010). The fraction of nonadditively expressed genes in these studies greatly differed depending on the analyzed species, tissue, experimental procedures, and surveyed genes. This observation is consistent with studies suggesting varying degrees of heterosis in different tissues and organs of plants (Springer and Stupar 2007). Moreover, allele-specific gene expression levels have been determined in hybrids in search of genes displaying asymmetric activity ratios of their alleles compared to their parental inbred lines (Guo et al. 2004; Stupar and Springer 2006; Stupar et al. 2007; Zhuang and Adams 2007; Guo et al. 2008; Swanson-Wagner et al. 2009; He et al. 2010).
Based on gene expression profiling studies, specific genes were suggested to be involved in the manifestation of heterosis for a number of traits in maize, rice, and tomato (Hoecker et al. 2008a; Wei et al. 2009; Guo et al. 2010; Krieger et al. 2010). However, whether such genes contribute to general molecular mechanisms underlying the formation of hybrid vigor remains to be determined.
The recent development of high-throughput sequencing technologies, including the availability of protocols for sequencing mRNA populations (e.g., RNA-seq) has enabled unprecedented resolution in global analyses of gene expression (Marioni et al. 2008; Z Wang et al. 2009). Next generation sequencing (NGS) approaches such as sequencing-by-synthesis (SBS) allow unbiased and highly reproducible deep-sequencing of entire transcriptomes (Metzker 2010). A major advantage of NGS approaches in contrast to closed platforms such as microarrays is that they allow for sequencing all expressed genes in a specific tissue, irrespective of accurate genome annotation or even without a priori knowledge of the genome (Hurd and Nelson 2009). Moreover, in contrast to microarray-based assays, NGS approaches allow for the detection of single nucleotide polymorphisms (SNPs) which are of particular interest when studying allele-specific expression patterns. The transcriptomes of several plant species and different tissues have been sequenced by NGS (Swanson-Wagner et al. 2009; Filichkin et al. 2010; Lai et al. 2010; Larsen et al. 2010; Wang et al. 2010; Zenoni et al. 2010; Zhang et al. 2010) and a few studies have used RNA-seq to compare the transcriptomes of inbred and hybrids, providing initial insights into their regulatory landscapes (XF Wang et al. 2009; He et al. 2010). However, to date a replicated and unbiased survey of gene expression patterns using NGS approaches related to plant heterosis is not at hand.
The aim of the present study was to globally survey the transcriptome of the two inbred lines B73 and Mo17 in comparison to their reciprocal F1-progeny in an unbiased approach via RNA-seq using four biological replicates to increase the power for detecting differential expression while maintaining a low false discovery rate (FDR). For this purpose, primary roots of 3.5-d-old seedlings were surveyed, as young primary roots display a very simple structure. This stage was previously demonstrated to precede the first detectable developmental changes between inbred lines and hybrids during heterosis manifestation for numerous root-related traits (Hoecker et al. 2006). By choosing this particular stage, we intended to identify transcriptional patterns and regulators possibly determining the processes leading to heterosis for these early root system traits.
Results
Next generation sequencing and mapping of maize inbred and hybrid transcriptomes
Global gene expression profiles of 3.5-d-old primary roots of the reciprocal hybrids B73×Mo17 and Mo17×B73 and their parental inbred lines B73 and Mo17 were analyzed via RNA-seq using four biological replicates. Between 31 and 37 million 40-bp reads were obtained for each of the replicates and genotypes (Supplemental Table S1). Among these, 53%–68% mapped to unique positions in the maize B73 reference genome (ZmB73_RefGen_v2; Supplemental Table S1). After removal of “stacked reads” (duplicate reads removal, see Methods), 91%–96% of the remaining reads mapped to the “filtered gene set” (ZmB73_5B_FGSv2), a set of 39,656 high-confidence, predicted gene models defined via a combination of evidence-based and ab initio approaches followed by stringent TE filtering (Schnable et al. 2009; http://www.maizesequence.org). In total, 34,233 genes of the FGSv2 were expressed in the analyzed tissue (i.e., 86% of genes in the FGSv2) in at least one of the four genotypes (sum of the number of expressed genes included in the Venn diagram in Figure 2B, see below) based on a Bayesian data-augmented Markov chain Monte Carlo approach (see Methods).
Figure 2.

Analysis of transcriptomic expression complementation by single parent expression (SPE) among the genes expressed in the two inbred lines. (A) The different genotypes are indicated with numbers 1–4, and each square field of the four-way Venn diagram represents a different expression class. (B) The numbers of genes of the various expression classes are indicated.
Differential gene expression is more common between parental inbred lines than between parents and hybrids or between reciprocal hybrids
To achieve a comprehensive overview of differential gene expression, all possible (N = 6) pairwise comparisons of the four genotypes were performed (Fig. 1A) based on the criteria defined in the Methods section. By controlling the false discovery rate at 5%, the expected ratio of false discoveries divided by the number of genes declared to be differentially expressed is kept at ∼5%. However, the number of false negative results (genes that are truly differentially expressed but are not declared to be so) can be quite high. Fortunately, it is possible to estimate the number of false negatives that result for any particular significance threshold (Nettleton 2006). Because it is not possible to determine which genes are false negatives without additional experimentation, it is not possible to make a list of such genes. Even so, the total number of differentially expressed genes (m1 = true positives plus false negatives) can be estimated by comparing the observed distribution of P-values for any comparison of interest to a uniform distribution (Nettleton et al. 2006). An estimate of m1 provides an important measure of the full extent of differential expression and supplements a list of specific genes that can be confidently declared differentially expressed. Thus, in this report we have usually provided estimates of the total number of differentially expressed genes (m1) along with the number of genes declared to be differentially expressed while controlling FDR at the 5% level.
Figure 1.

Tests for differential gene expression were conducted on the six pairwise comparisons between the four analyzed genotypes. (A) The transcriptomes of primary roots (2–4 cm long) of 3.5-d-old seedlings of two inbred lines (B73 and Mo17) and their reciprocal hybrids were compared. (B) The estimated total number of differentially expressed genes (i.e., m1; dark gray) and the number declared to be differentially expressed while controlling the false discovery rate (FDR) at 5% (light gray) are shown.
When controlling FDR at 5%, 19,382 (Fc [Fold change] ≥ 2: 6959) genes were declared to be differentially expressed between the inbred lines B73 and Mo17 (Fig. 1B). The overall number of genes differentially expressed between the inbred lines B73 and Mo17 (m1) was estimated to be 21,290, which corresponds to ∼70% of all expressed genes in these genotypes (Table 1). In the comparison between the reciprocal hybrids, 341 (Fc ≥ 2: 39) specific genes were declared to be differentially expressed when controlling the FDR at 5%. The overall number of genes differentially expressed between reciprocal hybrids (m1) was estimated to be 4585 genes (15% of all expressed genes). The four hybrid-inbred line comparisons revealed between 7416 (Fc ≥2 : 2225) and 11,207 (Fc ≥ 2: 2777) differentially expressed genes when controlling the FDR at 5%, and estimates of m1 ranged from 12,793 to 16,870 genes which corresponds to 42% and 55% of all expressed genes. Hence, inbred-hybrid comparisons revealed fewer differentially expressed genes than the comparison of the two parental inbred lines but more than the comparison of the reciprocal hybrids. Plotting fold-changes versus gene frequency for the FDR 5% genes (Supplemental Fig. S2) for which we did not introduce a fold-change cut-off demonstrated that many of statistically significant changes in gene expression were modest fold-changes. Even so, several thousand genes exhibited greater than twofold changes in expression (see numbers in brackets above).
Table 1.
Classification of the genes expressed in the two reciprocal hybrids according to their expression levels in comparison to the corresponding parental values (defined as lower parent [LP] and higher parent [HP])
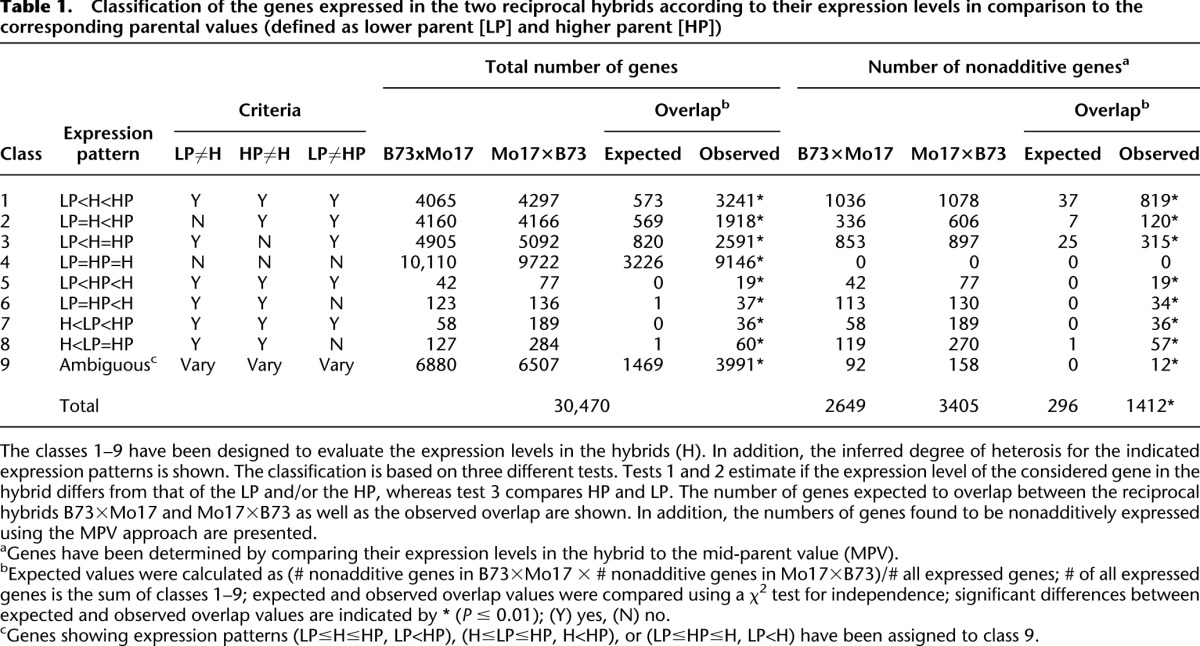
Many genes exhibit nonadditive expression patterns which are often conserved between reciprocal hybrids
To determine relative expression levels of genes in hybrids as compared to their parental inbred lines and to identify genes displaying expression levels that differ from additivity, nine gene expression classes were defined (Table 1) based on similar classifications published previously (Stupar and Springer 2006; Hoecker et al. 2008a). First, for any given gene, differential expression values in the hybrid relative to the parental inbred lines displaying the lower (low-parent, LP) and higher (high-parent, HP) expression levels, were estimated by combining three pairwise t-tests (“Criteria” in Table 1). According to this classification scheme, 4065 (B73×Mo17) and 4297 (Mo17×B73) genes were expressed in hybrids at levels between the parental values (class 1, “Total number of genes,” Table 1; Supplemental Table S3). Furthermore, 4160 (B73×Mo17) and 4166 (Mo17×B73) genes exhibited low-parent expression levels in a hybrid (class 2), whereas 4905 (B73×Mo17) and 5092 genes (Mo17×B73) exhibited high-parent expression levels in a hybrid (class 3). Class 4 represents the largest class, containing 10,110 (B73×Mo17) and 9722 (Mo17×B73) genes that do not exhibit differential expression among the parents and hybrids. Finally, 350 (B73×Mo17) and 686 (Mo17×B73) genes display extreme expression levels above the HP or below the LP (classes 5–8).
Within these classes, a subset of genes displayed nonadditive gene expression, i.e., expression levels in a hybrid that were significantly different from the average of the parental values (mid-parent value). Testing for expression levels deviating significantly from the MPV (“Number of nonadditive genes,” Table 1) identified 2649 and 3405 nonadditively expressed genes in the reciprocal hybrids B73×Mo17 and Mo17×B73, respectively, with an overlap of 1412 (53%) genes between the two hybrids. The observed overlap significantly exceeds the number of 296 genes expected purely by chance (P ≤ 0.01) (see footnote b, Table 1).
Extreme expression complementation
An extreme case of differential expression occurs when some genotypes express a gene, but others do not. We determined whether or not a gene is expressed in each of the four genotypes using a Bayesian data-augmented Markov chain Monte Carlo approach (see Methods). The results of this experiment were summarized in a four-way Venn diagram (Fig. 2). The 15 possible genotype combinations of the two inbred lines B73 (1) and Mo17 (2) and the reciprocal hybrids B73×Mo17 (3) and Mo17×B73 (4) are summarized in Figure 2A. The number of genes for each of the 15 classes that displayed such expression patterns is depicted in Figure 2B. In total, 34,233 genes were expressed in at least one of the 16 classes depicted in the Venn diagram. This number in Figure 2B differs from the total number of genes displayed in Table 1. This discrepancy is due to the application of different statistical approaches (see Methods and Discussion). Only eight genes were expressed in only one or two genotypes, and no gene was expressed in both parents but in only one of the two reciprocal hybrids (Fig. 2). In contrast, hundreds of genes (N = 358 and N = 766) (Fig. 2; Supplemental Table S4) were expressed in both hybrids but in only one of the two parental inbred lines. Such single parent expression (SPE) patterns represent a special instance of the dominance (i.e., complementation) model. As a consequence of complementation, both hybrids expressed more genes (34,225 and 34,226 in B73×Mo17 and Mo17×B73, respectively) than did either parental inbred (33,460 and 33,874 in B73 and Mo17, respectively) (Fig. 2; Supplemental Table S4). To exclude the possibility that SPE is simply the result of the complete absence of affected genes from the genomes of one of the parental inbred lines (rather than differentially regulated gene expression), all SPE genes were compared to a previously published set of presence/absence variation genes (Swanson-Wagner et al. 2010). For technical reasons, only PAVs that are present in the B73 reference genome but absent from the Mo17 genome were detected by Swanson-Wagner et al. (2010). Hence, in our analysis we only considered the SPE class 1-3-4 (Fig. 2). Of the 358 SPE genes in this class, 65 matched genomic PAVs identified by Swanson-Wagner et al. (2010) and C Rustenholz, C-T Yeh, Y Huang, and PS Schnable, pers. comm. (Supplemental Table S4 and Methods). Hence, the remaining 293 genes of the 1-3-4 class (Fig. 2B) represent cases of genotype-dependent differential gene expression. For all of these genes, expression in the hybrids was dominant over lack of expression in one of the parental inbreds.
To determine whether complementation of gene expression in hybrids is associated with the reactivation of the “silenced” allele or if the hybrids only express the allele contributed from the “active” parent, we used a set of more than 4 million single nucleotide polymorphisms between the B73 and Mo17 transcriptomes (C-T Yeh and PS Schnable, unpubl.; Methods) to ascertain allele-specific expression levels in the SPE genes (classes 1-3-4 and 2-3-4 in Fig. 1B, respectively). It was possible to assay for allele-specific expression in 174 of the 358 class 1-3-4 genes and for 373 of the 766 class 2-3-4 genes due to the presence of SNPs in these genes. In the hybrids, both parental alleles of 33/174 class 1-3-4 genes and 139/373 class 2-3-4 genes were expressed (Supplemental Table S5), suggesting that regulatory factors from the active parent successfully interacted with and activated the allele from the silent parent. In the remaining instances, the allele from the silent parent may be defective in some manner.
Hybrids exhibit a high frequency of novel allele-specific expression patterns
For each gene, we determined the allelic expression ratios in hybrids and compared these ratios to the expression ratio of the two inbred parents. Overall, an estimated 4239 (B73×Mo17) and 4245 (Mo17×B73) genes exhibited allelic expression levels in the hybrids that differed significantly from the expression ratio of the parental inbred lines (Table 2). When controlling the FDR at 5%, 516 (B73×Mo17) and 600 (Mo17×B73) of these genes could be identified. The overlap (N = 276) between the gene lists from the two hybrids is significantly larger than expected by chance (N = 10; P ≤ 0.01) (footnote b, Table 2). Of these overlapping genes, 249 displayed conserved allelic directionality between the two hybrids, while for 23 genes, the two hybrids exhibited opposite directionality (Supplemental Table S5). Ninety six (B73×Mo17) and 111 (Mo17×B73) genes showed nonadditive gene expression, and for 32 genes, the patterns of unexpected allelic expression and nonadditive expression were found in both hybrids (Table 2). Again, the observed overlap significantly exceeded the expected values (N = 0; P ≤ 0.01).
Table 2.
Genes whose allelic expression levels in hybrids differ from the expected values
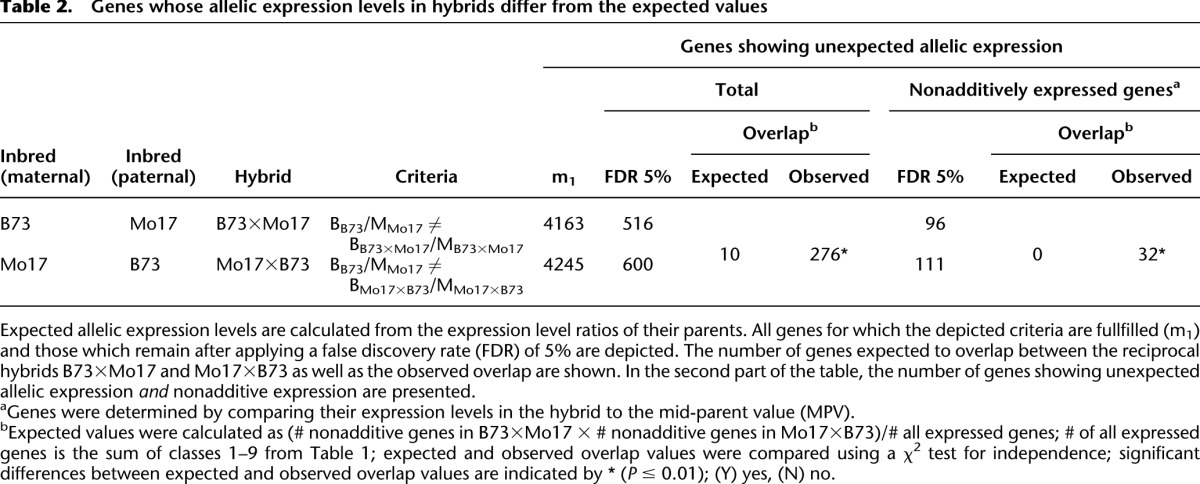
To analyze if allele-specific expression levels exhibit reciprocal effects, we searched for genes showing differences in the ratio of allelic contributions between the two hybrids. In each of the two hybrids, 10,462 genes expressed the parental alleles in a ratio different from 1 (Supplemental Table S2). Controlling the FDR at 5% resulted in the identification of 8305 (B73×Mo17) and 8261 (Mo17×B73) genes for which the two alleles were not equally expressed. Due to the slight mapping bias of Mo17 reads (Mo17 reads that contain polymorphisms relative to B73 are at a disadvantage when being aligned to the B73 reference genome), more genes displayed a preferential expression of the B73 allele. For such genes, an increase in allelic expression cannot be separated from artifacts introduced by the mapping bias. Hence, we considered only genes for which the Mo17 allele was expressed at a higher level than the B73 allele. Using this conservative strategy, significantly higher expression of the Mo17 allele was found for 1480 (B73×Mo17) and 1551 (Mo17×B73) genes. For 1020 of these genes, a stronger expression of the Mo17 allele was found in both hybrids (Supplemental Table S2). This overlap significantly exceeds the number expected purely by chance (N = 75; P ≤ 0.01) (see footnote a, Supplemental Table S2).
Discussion
Heterosis is manifested early in seedling development for a number of root traits (Hoecker et al. 2006). To account for this in the present study, global gene expression patterns of the inbred lines B73 and Mo17 and their reciprocal hybrids B73×Mo17 and Mo17×B73 were compared very early in development, i.e., 3.5 d after germination. While in the past, global gene expression surveys related to heterosis were mainly conducted via microarray analyses (for review, see Hochholdinger and Hoecker 2007; Goff 2010; Paschold et al. 2010); the present study was performed using RNA-seq technology. Statistical power provided by fourfold biological replication of each genotype allowed a fully quantitative survey of overall and allele-specific gene expression profiles of all annotated genes that are expressed in seedling roots.
Single parent expression is complemented in reciprocal hybrids
Approximately 70% of all expressed genes were differentially expressed between the two inbred parents. This value is significantly higher than in previous reports. For example, microarray-based analyses reported that 4%–18% of maize genes were differentially expressed in different tissues of the maize inbred lines B73 and Mo17 (Stupar and Springer 2006). Moreover, high density SNP tiling array profiling of Arabidopsis seedlings revealed that 31% of all analyzed genes were differentially expressed between the parental inbred lines (Zhang and Borevitz 2009). Furthermore, in an RNA-seq experiment of 4-wk-old rice shoots, 24% of all expressed genes were differentially expressed (He et al. 2010). These distinct rates of differential gene expression between parental inbred lines might be species-, genotype-, tissue-, or stage-specific, but most likely they are, in part, a consequence of differences in experimental design, technology platforms, and statistical analysis procedures. For instance, microarray experiments tend to underestimate the number of differentially expressed genes. Moreover, differentially expressed genes are typically calculated with reference to the total number of genes on a chip; hence, it is difficult to determine how many genes are actually expressed in such experiments. Finally, in microarray experiments, often arbitrarily introduced fold-change cut-offs such as Fc ≥ 2 are introduced. By contrast, RNA-seq experiments allow full quantification of gene expression. In addition, deep sequencing allows for the detection and quantification of weakly expressed genes, which contribute significantly to the total number of expressed genes (Supplemental Figs. S1, S2). Therefore, we did not introduce any fold-change cut-offs because even small fold-changes of genes with low expression levels could be biologically relevant. While in the present study, 70% of all expressed genes were differential between B73 and Mo17, only 36% of the differential genes displayed Fc ≥ 2, which is similar to numbers observed in previous studies. In the present study, it was possible to determine that 86% of all high-confidence maize genes were expressed in young maize roots, while in 4-wk-old rice shoots, 52% of non-transposable element (TE) genes were shown to be expressed (He et al. 2010). The observation of significant rates of differential gene expression between parental lines supports the notion that transcriptome diversity in young maize hybrid roots could be mainly conditioned by transcriptomic differences of the distantly related parental inbred lines (Stupar et al. 2008). As suggested by the dominance model, the combination of two diverse inbred genomes in hybrids might lead to their integration or complementation promoting the “selection” of superior alleles and supporting the formation of more vigorous plants (Jones 1917). A special instance of the complementation model is single parent expression, i.e., the expression of a gene in reciprocal hybrids, which is expressed only in one parental inbred line. This expression pattern was observed for 1124 genes (Fig. 2B). Some SPE genes were also observed in microarray analyses of different maize tissues (Stupar and Springer 2006). Although maize exhibits high rates of presence/absence variation (Springer et al. 2009; Swanson-Wagner et al. 2010), only 65 of 358 B73-expressed SPEs detected in this study were the result of PAVs. Hence, most of the SPE patterns observed in this study are probably the consequence of differential expression of genes, rather than presence/absence variation in genomic DNA. As a result of complementation, the absolute number of expressed genes in both hybrids was higher than in their parental inbred lines. The expression of more genes in hybrids as compared to their parental inbred lines might provide a phenotypic advantage to hybrid plants.
Compared to the parental inbred lines B73 and Mo17, fewer gene expression differences were observed between the reciprocal hybrids B73×Mo17 and Mo17×B73 harboring identical nuclear genomes (Fig. 1B). Similar tendencies have been reported previously in maize (Stupar and Springer 2006) and Arabidopsis (Vuylsteke et al. 2005), while larger reciprocal effects than in the present study have been documented in rice (He et al. 2010) and maize (Swanson-Wagner et al. 2009). The four pairwise comparisons of gene expression in inbred lines versus hybrids revealed intermediate numbers of differentially expressed genes with reference to the hybrid and inbred line comparisons. Hence, the degree of genomic difference correlates with differential gene expression. In each of the four comparisons the hybrid displayed a 50% genomic identity with the respective inbred line to which it was compared. Moreover, the observation that each hybrid showed less differential expression when compared to its mother (Fig. 1B) suggests that the maternal influence on the hybrid transcriptome exceeds the paternal influence, independent of their genotype. In the maize inbred lines B73 and Mo17, hybrid gene expression patterns that resemble the transcriptome of the maternal (Guo et al. 2003) or paternal (Swanson-Wagner et al. 2009) parent have been reported previously, while transcriptomic surveys of other genotypes did not support such a trend (Uzarowska et al. 2007).
Differential and nonadditive gene expression shape the hybrid transcriptome
Gene expression in hybrids compared to their parental inbred lines can be classified as differential (Springer and Stupar 2007) or nonadditive (Hoecker et al. 2008a). Differential gene expression is defined as expression in a hybrid that is significantly different from expression in at least one parental inbred line. Differential expression between inbred lines and hybrids was observed for the majority of genes in the present study (classes 1–3 and 5–8 in Table 1). In contrast, nonadditive gene expression is defined as expression of a gene in a hybrid that is significantly different from the average value of the parental inbred lines (mid-parent value). Hence, while nonadditive gene expression is observed in the same classes as differential expression (Table 1), nonadditively expressed genes represent a subset of differentially expressed genes. Classes 5–8 represent extreme expression classes. In classes 5 and 6, the hybrid displays a significantly higher expression than both parents, and in classes 7 and 8, the hybrid displays a significantly lower expression than both parents. These instances have been previously designated above high-parent and below low-parent expression (Hoecker et al. 2008a) or under- and overdominance (Swanson-Wagner et al. 2006). Therefore, almost all genes belonging to these classes (in classes 5 and 7, even all genes) display nonadditive expression. The numbers of genes showing expression that exceeds or is below both parental expression values is lower than those found in rice shoots (He et al. 2010) and in meristems of other maize genotypes (Uzarowska et al. 2007). Similar numbers of such genes were reported from different tissues of the rice hybrid LYP9 (Wei et al. 2009), whereas other studies found fewer such genes in maize embryos, endosperm, seedling shoots, and immature ears of hybrids (Stupar and Springer 2006; Swanson-Wagner et al. 2006; Meyer et al. 2007; Stupar et al. 2007; Uzarowska et al. 2007). Although it is possible that there are organ-specific differences in nonadditive gene expression, it is more likely that differences in experimental approaches play a larger role in explaining the differences among reports. Among the genes classified in the eight classes displayed in Table 1, ∼10% were nonadditively expressed. Compared with previous studies, these numbers (considered as a ratio of all expressed or all analyzed genes) are in the range of nonadditively expressed genes detected in inbred lines and hybrids of rice (He et al. 2010), Arabidopsis (Vuylsteke et al. 2005), and maize (Stupar and Springer 2006; Swanson-Wagner et al. 2006; Stupar et al. 2007).
Another notable finding was the high degree of conservation of all nonadditively expressed gene classes between the two reciprocal hybrids (Table 1). The number of such genes significantly exceeded the expected values, hence underscoring the importance of nuclear genome composition in reciprocal hybrids irrespective of the parental origin of the two alleles.
Different statistical approaches were applied to determine the expression classes in Table 1 and expression complementation in Figure 2B (see Methods). This explains the different numbers of expressed genes in these two experiments. Briefly, in Table 1, a conservative approach was used in determining what genes were expressed in order to have high confidence that the genes it was examining were truly “ON” and to have better power for differential expression detection. In doing so, we used a strict total read cut-off, and genes which did not make this threshold were eliminated from the analysis.
In Figure 2B, we did not make any inference regarding the relative ratios of the expression, and the inference was strictly limited to whether genes were expressed at all. As such, we used a more flexible model that did not include any prior censoring of the data.
Differential allele-specific expression affects nonadditivity
For more than 4100 genes, allelic ratios in the hybrids were observed that differed from the parental ratios (Table 2). These expression patterns might be predetermined by the parental genomes or, alternatively, could also be the result of epigenetic regulation. For example, reduced levels of 24-nt sRNAs in reciprocal hybrids compared to the parental lines have been suggested to be involved in the altered epigenetic landscape of hybrids thereby promoting the formation of heterotic phenotypes (Groszmann et al. 2011). Controlling the FDR at 5% left only ∼13% of the genes with unexpected allelic ratios, a number which is lower than that previously reported in Arabidopsis F1-hybrids (Zhang and Borevitz 2009). Interestingly, ∼19% of these genes also show nonadditivity in their total gene expression levels. This considerable fraction implies that altered allelic activity in hybrids contributes to nonadditive gene expression patterns.
In summary, RNA-seq sequencing technology allows for unprecedented accuracy and coverage in transcriptome studies. The results presented in this study allow for statistically substantiated conclusions about the quantitative transcriptomic complexity of maize hybrids in comparison to their parental inbred lines. Single parent expression seems to play a major role in shaping transcriptomes of hybrids. The ability to compensate for alleles not expressed in one of the two parents might be a mechanism contributing to hybrid vigor. Furthermore, it was demonstrated that the parental inbred lines greatly differ in gene expression levels and that most of these differences are no longer present in reciprocal hybrids. Nonadditivity seems to be the rule rather than the exception in hybrids, and analyses of the allelic gene expression landscape revealed numerous deviations from the parental ratios. Finally, for a large number of genes, these classification patterns are highly conserved between reciprocal hybrids, indicating that they are determined by the nuclear genomes of their parents rather than by cytoplasmic or epigenetic factors.
Methods
Plant material and growth conditions
Seeds of the inbred lines B73 and Mo17 and the two reciprocal hybrids B73×Mo17 and Mo17×B73 were germinated in paper rolls in distilled water as previously described (Hoecker et al. 2006). For each genotype, 18 paper rolls each containing 10 kernels were prepared. All material was germinated in a single 10-L bucket in which the 72 paper rolls were arranged randomly. Three and a half d after germination primary roots of 2–4-cm-length were harvested. Ten primary roots were pooled for each of the four biological replicates per genotype.
Even at this very early developmental stage, hybrid primary roots were longer than those of their parental inbred lines. To avoid differential gene expression patterns based simply on root length, pools of roots having the same lengths (2–4 cm) were analyzed for all genotypes and replicates.
RNA isolation and sequencing library construction
The pooled primary roots were ground under liquid nitrogen, and RNA was isolated as previously described (Winz and Baldwin 2001). RNA quality was assayed via agarose gel electrophoresis and a Bioanalyzer (Agilent Technologies, Boeblingen, Germany). Only samples with an RIN (RNA integrity number) (Schroeder et al. 2006) ≥8.7 were used for downstream analyses. The cDNA libraries for sequencing-by-synthesis were constructed according to the protocol of the manufacturer (Illumina). The 16 samples were loaded on two flow cells in a predefined order (Supplemental Table S1). Cluster generation and sequencing (40-bp, single read) were performed as per the instructions of the manufacturer (Genome Analyzer II, Illumina).
Analysis of raw sequencing data
Illumina's RTA software version 1.6.32.0 was used for real time image analysis and the Genome Analyzer data analysis pipeline OLB version 1.6 for base calling and run statistics on all 16 lanes (flow cells EAS517_0031_FC611DEAAXX and EAS517_0032_FC611DLAAXX). On average, a raw cluster density of 300,000 clusters per tile was achieved yielding between ∼0.95 and ∼1.2 Gb (∼31 to ∼37 million reads) per lane and sample post filtering (see Supplemental Table S1 for sample allocation and sequencing performance per lane). Sequencing error rates per lane were estimated using ELAND alignments against the maize cDNA release ZMGI 19.0 (http://compbio.dfci.harvard.edu/cgi-bin/tgi/gimain.pl?gudb=maize) suggesting per base error probabilities of 0.76%–1.41% and 0.52%–0.84% for the two flow cells, respectively. Further analyses were performed based on OLB's fastq output files.
Read mapping and allele-specific read calling
The maize B73 reference genome (RefGen_v2) (Schnable et al. 2009) was indexed using novoindex (hash length 14 and step size 1), and all high quality reads were aligned to the whole reference genome using the short reads aligner NOVOALIGN (http://www.novocraft.com, version 2.07.11). Only those reads uniquely mapping to the B73 reference genome (Schnable et al. 2009) with a maximum of two mismatches out of 40-bp read length were subsequently analyzed. In addition, redundant reads mapping at the same starting coordinate and mapping orientation (“stacked reads”) were removed from the set of uniquely mapping RNA-seq reads. The remaining reads of all samples were projected to the filtered gene set (FGSv2; http://ftp.maizesequence.org/current/filtered-set/, Release 5b.60) of the B73 reference genome derived from the maize genome sequencing project (MGSP) using a Perl script. Allele-specific gene expression was analyzed with a set of 4,034,683 single nucleotide polymorphisms (identified between the B73 and Mo17 genotypes via 123SNP software which is available for download from http://schnablelab.plantgenomics.iastate.edu/software). B73-Mo17 SNPs were called by aligning B73 RefGen_v2 sequences and Mo17 sequences retrieved from NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) records (accession numbers: SRA009756, SRA049859, SRA048055, SRA053592, SRA050451) and MaizeGDB (http://ftp.maizegdb.org/MaizeGDB/FTP/Mo17/).
The allele-specific reads were extracted from the reads uniquely projected onto the FGSv2 and associated with any B73-Mo17 SNP using a customized Perl script. Since the mapping procedure presented here did not allow for quantifying sequence reads mapping to repeats throughout the genome, only uniquely mapping genes were considered. In addition, transcript isoforms of a given gene locus could not be distinguished.
Expression complementation
Classification of the genewise transcriptional activity (presence/absence) for each genotype was determined by a Bayesian data-augmented Markov chain Monte Carlo approach (Tanner and Wing 1987), treating the status of transcription for each genotype within each gene as missing information. We modeled the status of transcription for all four genotypes for a given gene simultaneously as a state vector, composed of four binary variables, resulting in 16 total possible classes. The reads were modeled via a negative binomial distribution using the mean-overdispersion parameterization, with the median read count across transcripts selected as an offset for each experimental unit. A log-link was used to model the mean read counts, with random effects used for overall gene effects as well as gene-specific genotype effects, which we assumed were both normally distributed. Each gene was assumed to have its own unique dispersion parameter, which was treated as random and gamma distributed. A background error rate was included in order to account for falsely mapped reads. This parameter was estimated from the data using an initial “naive” classification of the transcriptional statuses in which the expression of a gene was classified as absent for a particular genotype if at least half of the observed counts for that gene-by-genotype combination were zero. Given this initial “naive” classification, the maximum likelihood estimate of the background error rate parameter was obtained. The background error rate was then fixed at the value of the maximum likelihood estimate during the model fitting process to avoid identifiability complications. With respect to prior distribution selection, we modeled the marginal probability of state vector class membership using a Dirichlet distribution and selected the remaining priors either to take advantage of conditional conjugacy or to be noninformative. The model was defined using JAGS (Plummer 2011), an extension of the BUGS language (Gilks et al. 1994), and fit in R using the R2jags library package (Su and Yajima 2011). We ran two Markov chains at a total of 15,000 iterations each, treating the first 5000 as a burn-in and then thinning the remaining iterations by saving every 10th iteration to avoid issues with autocorrelation, resulting in parameter traces of length 1000 for our posterior sample. We also analyzed the sample to verify that the model had converged by visually inspecting the mixture of the chains and using posterior checking procedures from the coda output diagnostics package (Plummer et al. 2010) in R. Traces of the state vector were used to estimate the posterior probability of class membership, and we determined classification based upon the state vector which had the largest proportion of instances within the trace.
Statistical procedures for analyzing differential gene expression and differential allele-specific expression
To estimate differences in total and allele-specific gene expression between the four genotypes, two different models within the GLIMMIX procedure in SAS were used. In both cases, data were modeled one gene at a time, and the observed counts were treated as Poisson random variables conditional on their means, which were modeled using the log link. In both models, differences among library sizes for the experimental units were accounted for with the offset option in Proc GLIMMIX. Following Bullard et al. (2010), the log of the upper quartile of the experimental-unit-specific total count distribution across genes was used as the offset. The model for total counts included fixed effects for flow cells and genotypes. Additionally, a normally distributed random effect was included for each experimental unit to account for overdispersion (extra Poisson variation). The model for allele-specific counts included fixed effects for flow cells, genotypes, alleles, and the interaction between genotype and allele. Additionally, a random effect for each experimental unit was included to account for overdispersion (extra Poisson variation) and to account for correlation between the two allele-specific counts arising from each experimental unit within a hybrid genotype. Each model was fit twice for each gene, once using the observed counts and a second time using the observed counts added to one. There were 1176 (714) genes in the allele-specific (total count) analysis for which adding one to the observed counts allowed SAS to converge and produce results when using the original counts failed to converge. In these cases, results from the analysis using the observed counts plus one were used. In all other cases, results from the analysis using the original counts were used.
Hypotheses tests of interest were conducted using the contrast statement within Proc GLIMMIX. The resulting P-values for each hypothesis test were used to estimate the total number of differentially expressed genes for each comparison (Nettleton et al. 2006). The P-values for each comparison were converted to q-values that were then used to identify differentially expressed genes while controlling the FDR at the 5% level (Storey and Tibshirani 2003).
Genes with few or no observed reads may have inadequate power to detect differential expression; that is, genes with very few reads will produce large P-values for tests of differential expression regardless of how those reads are distributed across treatments. The inclusion of such genes and their corresponding large P-values can distort the distribution of P-values from which the number of true null hypotheses is estimated, which in turn affects estimated false discovery rates (q-values). To address this issue, the total count (allele-specific) analysis described in this paper was applied to genes that had at least 20 total (allele-specific) reads summed across the 16 samples and for which at least six samples had at least one total (allele-specific) read.
Classification of gene expression patterns
To classify gene expression in the hybrids relative to their parental inbred lines, nine gene expression classes were postulated (Table 1). Moreover, each gene was classified according to the expression level of each hybrid versus both parental lines. For each gene, the degree of estimated expression levels for a hybrid and the two parental lines directly follows from the estimated coefficients from the GLIMMIX analysis of total counts. If the contrast that compared the expression of two genotypes produced a q-value less than 0.05, a strict inequality was used to describe their relative expression. For each gene, the parental line with the larger estimated expression was denoted high-parent, and the other parental line was denoted low-parent. In a second approach to identify nonadditively expressed genes, gene expression levels in hybrids were compared to the mid-parent values. To conduct MPV analyses, two contrast statements were used for each gene to compare the estimated (log-scale) expression of each hybrid to the average (log-scale) expression of the parental lines.
Presence/absence variation calling
To identify the PAVs summarized in Supplemental Table S4, comparative genomic hybridization probes from Springer et al. (2009) were mapped to the B73 reference genome (AGPv2). Probes that exhibited 100% identity over 100% of their length at three or fewer locations in the genome were used for further analyses. Segmentation and PAV calling were performed as described by Springer et al. (2009).
Data access
Raw sequencing data are stored at the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA049019.
Acknowledgments
We thank Korbinian Schneeberger (Max Planck Institute for Developmental Biology, Tuebingen, Germany) for initial data analysis, Sanzhen Liu (Iowa State University, Ames, IA) for mapping support, and Gerd Müller (University of Bonn, Germany) for assistance in statistical analyses. This work was supported by a DFG (Deutsche Forschungsgemeinschaft) grant HO2249/9-1 to F.H., and NSF (National Science Foundation) grant IOS-1027527 to P.S.S.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.138461.112.
References
- Birchler JA, Veitia RA 2010. The gene balance hypothesis: Implications for gene regulation, quantitative traits and evolution. New Phytol 186: 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Yao H, Chudalayandi S, Vaiman D, Veitia RA 2010. Heterosis. Plant Cell 22: 2105–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S 2010. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11: 94 doi: 10.1186/1471-2105-11-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvick DN. 1999 Heterosis: Feeding people and protecting natural resources. In The genetics and exploitation of heterosis in crops (ed. JG Coors, S Pandey), pp. 19–29. American Society of Agronomy, Crop Science Society of America, and Soil Science Society of America, Madison, WI. [Google Scholar]
- Duvick DN 2005. The contribution of breeding to yield advances in maize (Zea mays L.). Adv Agron 86: 83–145 [Google Scholar]
- Falconer DS, Mackay TFC 1996. Introduction to quantitative genetics. Longman Harlow, Essex, UK [Google Scholar]
- Filichkin SA, Priest HD, Givan SA, Shen RK, Bryant DW, Fox SE, Wong WK, Mockler TC 2010. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res 20: 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M, Thiemann A, Fu JJ, Schrag TA, Scholten S, Melchinger AE 2010. Transcriptome-based distance measures for grouping of germplasm and prediction of hybrid performance in maize. Theor Appl Genet 120: 441–450 [DOI] [PubMed] [Google Scholar]
- Gilks WR, Thomas A, Spiegelhalter DJ 1994. A language and program for complex Bayesian modeling. Statistician 43: 169–177 [Google Scholar]
- Goff SA 2010. A unifying theory for general multigenic heterosis: Energy efficiency, protein metabolism, and implications for molecular breeding. New Phytol 189: 923–937 [DOI] [PubMed] [Google Scholar]
- Groszmann M, Greavesa IK, Albertyn ZI, Scofield GN, Peacock WJ, Dennis ES 2011. Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc Natl Acad Sci 108: 2617–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Rupe MA, Danilevskaya ON, Yang XF, Hut ZH 2003. Genome-wide mRNA profiling reveals heterochronic allelic variation and a new imprinted gene in hybrid maize endosperm. Plant J 36: 30–44 [DOI] [PubMed] [Google Scholar]
- Guo M, Rupe MA, Zinselmeier C, Habben J, Bowen BA, Smith OS 2004. Allelic variation of gene expression in maize hybrids. Plant Cell 16: 1707–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Rupe MA, Yang XF, Crasta O, Zinselmeier C, Smith OS, Bowen B 2006. Genome-wide transcript analysis of maize hybrids: Allelic additive gene expression and yield heterosis. Theor Appl Genet 113: 831–845 [DOI] [PubMed] [Google Scholar]
- Guo M, Yang S, Rupe M, Hu B, Bickel DR, Arthur L, Smith O 2008. Genome-wide allele-specific expression analysis using Massively Parallel Signature Sequencing (MPSS) reveals cis- and trans-effects on gene expression in maize hybrid meristem tissue. Plant Mol Biol 66: 551–563 [DOI] [PubMed] [Google Scholar]
- Guo M, Rupe MA, Dieter JA, Zou JJ, Spielbauer D, Duncan KE, Howard RJ, Hou ZL, Simmons CR 2010. Cell Number Regulator1 affects plant and organ size in maize: Implications for crop yield enhancement and heterosis. Plant Cell 22: 1057–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He GM, Zhu XP, Eiling AA, Chen LB, Wang XF, Guo L, Liang MZ, He H, Zhang HY, Chen FF, et al. 2010. Global epigenetic and transcriptional trends among two rice subspecies and their resiprocal hybrids. Plant Cell 22: 17–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochholdinger F, Hoecker N 2007. Towards the molecular basis of heterosis. Trends Plant Sci 12: 427–432 [DOI] [PubMed] [Google Scholar]
- Hoecker N, Keller B, Piepho HP, Hochholdinger F 2006. Manifestation of heterosis during early maize (Zea mays L.) root development. Theor Appl Genet 112: 421–429 [DOI] [PubMed] [Google Scholar]
- Hoecker N, Keller B, Muthreich N, Chollet D, Descombes P, Piepho HP, Hochholdinger F 2008a. Comparison of maize (Zea mays L.) F1-hybrid and parental inbred line primary root transcriptomes suggests organ-specific patterns of nonadditive gene expression and conserved expression trends. Genetics 179: 1275–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoecker N, Lamkemeyer T, Sarholz B, Paschold A, Fladerer C, Madlung J, Wurster K, Stahl M, Piepho HP, Nordheim A, et al. 2008b. Analysis of nonadditive protein accumulation in young primary roots of a maize (Zea mays L.) F1-hybrid compared to its parental inbred lines. Proteomics 8: 3882–3894 [DOI] [PubMed] [Google Scholar]
- Hurd PJ, Nelson CJ 2009. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief Funct Genomics Proteomics 8: 174–183 [DOI] [PubMed] [Google Scholar]
- Jahnke S, Sarholz B, Thiemann A, Kuhr V, Gutierrez-Marcos JF, Geiger HH, Piepho HP, Scholten S 2010. Heterosis in early seed development: A comparative study of F1 embryo and endosperm tissues 6 days after fertilization. Theor Appl Genet 120: 389–400 [DOI] [PubMed] [Google Scholar]
- Jones DF 1917. Dominance of linked factors as a means of accounting for heterosis. Genetics 2: 466–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger U, Lippman ZB, Zamir D 2010. The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nat Genet 42: 459–465 [DOI] [PubMed] [Google Scholar]
- Lai JS, Li YB, Messing J, Dooner HK 2005. Gene movement by Helitron transposons contributes to the haplotype variability of maize. Proc Natl Acad Sci 102: 9068–9073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JS, Li RQ, Xu X, Jin WW, Xu ML, Zhao HN, Xiang ZK, Song WB, Ying K, Zhang M, et al. 2010. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet 42: 1027–1030 [DOI] [PubMed] [Google Scholar]
- Larsen PE, Trivedi G, Sreedasyam A, Lu V, Podila GK, Collart FR 2010. Using deep RNA sequencing for the structural annotation of the Laccaria bicolor mycorrhizal transcriptome. PLoS ONE 5: e9780 doi: 10.1371/journal.pone.0009780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XH, Wei YL, Nettleton D, Brummer EC 2009. Comparative gene expression profiles between heterotic and non-heterotic hybrids of tetraploid Medicago sativa. BMC Plant Biol 9: 107 doi: 10.1186/1471-2229-9-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman ZB, Zamir D 2007. Heterosis: Revisiting the magic. Trends Genet 23: 60–66 [DOI] [PubMed] [Google Scholar]
- Long TA, Rady SM, Benfey PN 2008. Systems approaches to identifying gene regulatory networks in plants. Annu Rev Cell Dev Biol 24: 81–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcon C, Schützenmeister A, Schütz W, Madlung J, Piepho H-P, Hochholdinger F 2010. Non-additive protein accumulation patterns in maize (Zea mays L.) hybrids during embryo development. J Proteome Res 9: 6511–6522 [DOI] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y 2008. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18: 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker ML 2010. Sequencing technologies—the next generation. Nat Rev Genet 11: 31–46 [DOI] [PubMed] [Google Scholar]
- Meyer S, Pospisil H, Scholten S 2007. Heterosis associated gene expression in maize embryos 6 days after fertilization exhibits additive, dominant and overdominant pattern. Plant Mol Biol 63: 381–391 [DOI] [PubMed] [Google Scholar]
- Nettleton D 2006. A discussion of statistical methods for design and analysis of microarray experiments for plant scientists. Plant Cell 18: 2112–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettleton D, Hwang JTG, Caldo RA, Wise RP 2006. Estimating the number of true null hypotheses from a histogram of p values. J Agric Biol Environ Stat 11: 337–356 [Google Scholar]
- Paschold A, Marcon C, Hoecker N, Hochholdinger F 2010. Molecular dissection of heterosis manifestation during early maize root development. Theor Appl Genet 120: 383–388 [DOI] [PubMed] [Google Scholar]
- Plummer M. 2011. http://sourceforge.net/projects/mcmc-jags/files/ JAGS Version 3.0.0 User manual.
- Plummer M, Best N, Cowles K, Vines K 2010. coda: Output analysis and diagnostics for MCMC. http://CRAN.R-project.org/package=coda.R package version 014-2 [Google Scholar]
- Riddle NC, Jiang HM, An LL, Doerge RW, Birchler JA 2010. Gene expression analysis at the intersection of ploidy and hybridity in maize. Theor Appl Genet 120: 341–353 [DOI] [PubMed] [Google Scholar]
- Römisch-Margl L, Spielbauer G, Schützenmeister A, Schwab W, Piepho HP, Genschel U, Gierl A 2010. Heterotic patterns of sugar and amino acid components in developing maize kernels. Theor Appl Genet 120: 369–381 [DOI] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei FS, Pasternak S, Liang CZ, Zhang JW, Fulton L, Graves TA, et al. 2009. The B73 maize genome: Complexity, diversity, and dynamics. Science 326: 1112–1115 [DOI] [PubMed] [Google Scholar]
- Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T 2006. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7: 3 doi: 10.1186/1471-2199-7-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer NM, Stupar RM 2007. Allele-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize. Plant Cell 19: 2391–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer NM, Ying K, Fu Y, Ji TM, Yeh CT, Jia Y, Wu W, Richmond T, Kitzman J, Rosenbaum H, et al. 2009. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet 5: e1000734 doi: 10.1371/journal.pgen.1000734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci 100: 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupar RM, Springer NM 2006. Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 173: 2199–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupar RM, Hermanson PJ, Springer NM 2007. Nonadditive expression and parent-of-origin effects identified by microarray and allele-specific expression profiling of maize endosperm. Plant Physiol 145: 411–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupar RM, Gardiner JM, Oldre AG, Haun WJ, Chandler VL, Springer NM 2008. Gene expression analyses in maize inbreds and hybrids with varying levels of heterosis. BMC Plant Biol 8: 33 doi: 10.1186/1471-2229-8-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Yajima M 2011. R2jags: A package for running jags from R. http://CRAN.R-project.org/package=R2jags.R package version 002-17 [Google Scholar]
- Swanson-Wagner RA, Jia Y, DeCook R, Borsuk LA, Nettleton D, Schnable PS 2006. All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc Natl Acad Sci 103: 6805–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson-Wagner RA, DeCook R, Jia Y, Bancroft T, Ji TM, Zhao XF, Nettleton D, Schnable PS 2009. Paternal dominance of trans-eQTL influences gene expression patterns in maize hybrids. Science 326: 1118–1120 [DOI] [PubMed] [Google Scholar]
- Swanson-Wagner RA, Eichten SR, Kumari S, Tiffin P, Stein JC, Ware D, Springer NM 2010. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res 20: 1689–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner MA, Wing HW 1987. The calculation of posterior distributions by data augmentation. J Am Stat Assoc 82: 528–540 [Google Scholar]
- Thiemann A, Fu JJ, Schrag TA, Melchinger AE, Frisch M, Scholten S 2010. Correlation between parental transcriptome and field data for the characterization of heterosis in Zea mays L. Theor Appl Genet 120: 401–413 [DOI] [PubMed] [Google Scholar]
- Uzarowska A, Keller B, Piepho HP, Schwarz G, Ingvardsen C, Wenzel G, Lubberstedt T 2007. Comparative expression profiling in meristems of inbred-hybrid triplets of maize based on morphological investigations of heterosis for plant height. Plant Mol Biol 63: 21–34 [DOI] [PubMed] [Google Scholar]
- Vuylsteke M, van Eeuwijk F, Van Hummelen P, Kuiper M, Zabeau M 2005. Genetic analysis of variation in gene expression in Arabidopsis thaliana. Genetics 171: 1267–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZK, Ni ZF, Wu HL, Nie XL, Sun QX 2006. Heterosis in root development and differential gene expression between hybrids and their parental inbreds in wheat (Triticum aestivum L.). Theor Appl Genet 113: 1283–1294 [DOI] [PubMed] [Google Scholar]
- Wang XF, Elling AA, Li XY, Li N, Peng ZY, He GM, Sun H, Qi YJ, Liu XS, Deng XW 2009. Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell 21: 1053–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M 2009. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li PH, Brutnell TP 2010. Exploring plant transcriptomes using ultra high-throughput sequencing. Briefings in Functional Genomics 9: 118–128 [DOI] [PubMed] [Google Scholar]
- Wei G, Tao Y, Liu GZ, Chen C, Luo RY, Xia HA, Gan Q, Zeng HP, Lu ZK, Han YN, et al. 2009. A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc Natl Acad Sci 106: 7695–7701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winz RA, Baldwin IT 2001. Molecular interactions between the specialist herbivore Manduca sexta (Lepidoptera, Sphingidae) and its natural host Nicotiana attenuata. IV. Insect-induced ethylene reduces jasmonate-induced nicotine accumulation by regulating putrescine N-methyltransferase transcripts. Plant Physiol 125: 2189–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, Malerba G, Bellin D, Pezzotti M, Delledonne M 2010. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol 152: 1787–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Borevitz JO 2009. Global analysis of allele-specific expression in Arabidopsis thaliana. Genetics 182: 943–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GJ, Guo GW, Hu XD, Zhang Y, Li QY, Li RQ, Zhuang RH, Lu ZK, He ZQ, Fang XD, et al. 2010. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res 20: 646–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Adams KL 2007. Extensive allelic variation in gene expression in Populus F-1 hybrids. Genetics 177: 1987–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]