

Abstract
Thousands of long noncoding RNAs (lncRNAs) have been found in vertebrate animals, a few of which have known biological roles. To better understand the genomics and features of lncRNAs in invertebrates, we used available RNA-seq, poly(A)-site, and ribosome-mapping data to identify lncRNAs of Caenorhabditis elegans. We found 170 long intervening ncRNAs (lincRNAs), which had single- or multiexonic structures that did not overlap protein-coding transcripts, and about sixty antisense lncRNAs (ancRNAs), which were complementary to protein-coding transcripts. Compared to protein-coding genes, the lncRNA genes tended to be expressed in a stage-dependent manner. Approximately 25% of the newly identified lincRNAs showed little signal for sequence conservation and mapped antisense to clusters of endogenous siRNAs, as would be expected if they serve as templates and targets for these siRNAs. The other 75% tended to be more conserved and included lincRNAs with intriguing expression and sequence features associating them with processes such as dauer formation, male identity, sperm formation, and interaction with sperm-specific mRNAs. Our study provides a glimpse into the lncRNA content of a nonvertebrate animal and a resource for future studies of lncRNA function.
Since the discovery of Xist, a long noncoding RNA (lncRNA) required for mammalian X chromosome inactivation (Borsani et al. 1991; Brockdorff et al. 1992; Brown et al. 1992), thousands of other lncRNAs have been reported in mammals and other vertebrates (Okazaki et al. 2002; Numata et al. 2003; Carninci et al. 2005; Guttman et al. 2009; Gerstein et al. 2010; Guttman et al. 2010; Kim et al. 2010; Orom et al. 2010; Grabherr et al. 2011; Pauli et al. 2011b; Ulitsky et al. 2011; Y Wang et al. 2011). When considering their genomic origins relative to annotated protein-coding genes, most lncRNAs are classified either as long intervening ncRNAs (lincRNAs), which derive from loci that do not overlap the exons of protein-coding genes, or as antisense ncRNAs (ancRNAs), which derive from the opposite strand of the protein-coding gene such that they have potential to pair to the mature mRNA. lincRNAs are also called long intergenic RNAs, and ancRNAs are also called natural antisense transcripts (NATs). Most lncRNA gene models resemble those of protein-coding genes in terms of the CpG islands, multiexonic structures, and poly(A)-signals, but they have no more than chance potential to code for protein and are translated poorly from relatively short reading frames, if at all (Numata et al. 2003; Guttman et al. 2010; Ingolia et al. 2011).
Although for most lncRNAs, functions have not yet been investigated, some are known to play gene-regulatory roles or other biological roles in cells or during embryonic development (Goodrich and Kugel 2006; Mercer et al. 2009; Huarte and Rinn 2010; Koziol and Rinn 2011; Pauli et al. 2011a; Tsai et al. 2011). For example, HOTAIR is a 2.2-kb lincRNA that recruits the polycomb complex to modify the chromatin state of HOX genes to repress their transcription (Rinn et al. 2007; Gupta et al. 2010; Tsai et al. 2010), and TP53COR1 (also known as lincRNA-p21) is induced by TP53 upon DNA damage or oncogenic stress and causes the widespread suppression of numerous genes by recruiting the repressor protein HNRNPK, thereby acting as a potential tumor suppressor (Huarte et al. 2010). Additional lincRNAs are also associated with transcriptional regulation (Martianov et al. 2007; Wang et al. 2008; Zhao et al. 2008), whereas Malat1 can regulate genes at the post-transcriptional level by titrating an SR protein that regulates alternative mRNA splicing (Ji et al. 2003; Tripathi et al. 2010). Other examples include the megamind and cyrano lincRNAs, which are conserved from human to fish and play important roles in embryonic development (Ulitsky et al. 2011).
Compared to most mRNAs, lincRNAs generally accumulate to lower levels, and although some have detectable sequence conservation, many have no more conservation than expected by chance, implying that a large subset of lncRNAs are either biochemical noise or play newly evolved, species-specific roles (Carninci et al. 2005; Guttman et al. 2010; Cabili et al. 2011; Ulitsky et al. 2011). However, some lincRNAs without detectable sequence conservation derive from syntenic loci and have conserved gene structure (conserved exon size and number), suggesting that the apparent lack of conservation might reflect technical difficulties, such as greater challenges in accurate sequence alignment (Ulitsky et al. 2011).
lncRNAs are also found in invertebrates, as illustrated by the roX1 and roX2 lincRNAs, which are required for dosage compensation in flies (Larschan et al. 2011). In Caenorhabditis elegans, a subgroup of the modENCODE consortium carried out RNA-seq on poly(A)-selected RNA, which enabled annotation of 64,824 transcripts from 21,733 genes that would be expected to include some with little coding potential (Hillier et al. 2009; Gerstein et al. 2010). In parallel, using orthologous criteria (tiling array data, predicted RNA secondary structures, and sequence conservation), another subgroup of the consortium predicted ∼7000 noncoding RNA (ncRNA) candidates, 1678 of which did not overlap with annotated protein-coding genes (Gerstein et al. 2010; Lu et al. 2010). However, we noticed that the overlap between these 1678 ncRNA candidates and the 64,824 transcripts identified by RNA-seq included only 24 transcripts, which is smaller than the chance expectation of 120 ± 8 (mean ± SD for 10 cohorts of length-matched loci falling between protein-coding genes), implying that the confident identification of lncRNAs in C. elegans might benefit from additional analyses.
One potential function of noncoding transcripts is to serve as precursors or templates for the production of endogenous guide RNAs for RNAi or related silencing pathways. For example, the BIC proto-oncogene ncRNA was later reannotated as the primary transcript of the mammalian miR-155 miRNA (Lagos-Quintana et al. 2002). C. elegans has PIWI-interacting RNAs (21U-RNAs) and many endogenous small interfering RNAs (endo-siRNAs), including 22G-RNAs and 26G-RNAs (which tend to start with a G and be 22 and 26 nt long, respectively) (Ruby et al. 2006; Batista et al. 2008). The most abundant class of endo-siRNAs, 22G-RNAs, are produced by RRF-1 and EGO-1, RNA-dependent RNA polymerases (RDRPs) acting on template transcripts, and then become associated with worm-specific argonautes (WAGO proteins and CSR-1) (Ruby et al. 2006; Claycomb et al. 2009; Gu et al. 2009). CSR-1-associated 22G-RNAs target thousands of germline-specific genes, tend to map to the exons of those mRNAs, and are implicated in chromosome segregation (Claycomb et al. 2009). By contrast, WAGO-1-associated 22G-RNAs often map to both introns and exons of pre-mRNAs and have unknown biological roles (Gu et al. 2009). In addition, some 22G-RNAs map to clusters of loci lacking annotated transcripts. Because they did not correspond to known transcripts, such RNAs were initially annotated as a unique class of small-RNAs (tiny noncoding RNAs, or tncRNAs), distinct from endogenous siRNAs (Ambros et al. 2003). However, as high-throughput sequencing revealed their similarities to endo-siRNAs, tncRNAs were reclassified as siRNAs, with the presumption that they derive from ncRNA template transcripts that still needed to be identified (Ruby et al. 2006; Pak and Fire 2007).
In this study, we identify lncRNA genes, starting with a pipeline that constructs transcript annotations de novo by combining data from RNA-seq and poly(A)-site mapping and then removes those with detectable protein-coding potential or experimentally observed ribosome association. Hundreds of lncRNAs that have either single- or multiexonic transcript structures with poly(A) signals were found, thereby providing a glimpse into the lncRNA content of a nonvertebrate animal.
Results
De novo gene annotation using multimodal transcriptome data
We first developed a pipeline for global de novo annotation of transcripts from RNA-seq and poly(A)-site data sets. Because our focus was on lncRNAs, we chose not to consider information helpful for predicting protein-coding transcripts (such as sequence conservation, homology with known genes, codon usage, or coding potential), reasoning that by avoiding the consideration of this information we could use our accuracy for identifying previously annotated mRNAs to indicate accuracy for identifying lncRNAs.
Using TopHat, an alignment program that maps RNA-seq reads to putative exon junctions as well as genomic sequence (Trapnell et al. 2009), we mapped more than 1 billion reads (including 50 million exon-junction reads) from 25 non-strand-specific RNA-seq data sets (Gerstein et al. 2010; Lamm et al. 2011) and more than 80 million reads (including 5 million exon-junction reads) from 10 strand-specific RNA-seq data sets (Fig. 1A; Supplemental Table S1A,B; Lamm et al. 2011). To avoid false-positive exon-junction hits, we required that the inferred introns be ≥40 nt and ≤3058 nt, which would capture all but the shortest and longest 1% of introns within annotated protein-coding genes. Using the Cufflinks program (Trapnell et al. 2010), de novo gene annotations were constructed for non-strand-specific and strand-specific RNA-seq data sets, respectively (Fig. 1A). As expected, the annotations based on larger amounts of data (non-strand-specific RNA-seq) were more sensitive, whereas the annotations based on more informative reads (strand-specific RNA-seq) were more specific (Supplemental Table S1C), especially in instances of convergent overlapping transcripts, which are quite common in C. elegans, where they include a sixth of the mRNAs (Jan et al. 2011).
Figure 1.

Identification of C. elegans lncRNA genes. (A) Pipeline for de novo gene annotation and identification of lncRNAs. See main text and Supplemental Methods for details. (B) Venn diagram showing the overlap between the results of de novo gene annotation and modENCODE gene annotation. (C) Venn diagram showing the overlap of candidate lincRNA loci that passed the indicated filters. (D) Venn diagram showing the overlap of candidate ancRNA loci that passed the indicated filters. (E) The fraction of potential lncRNAs that had 3P-seq supported poly(A)-sites. Shown are the numbers of genes, with the number of splicing/3′ UTR isoforms in parentheses. (F) Diagram of trans-splicing by splice leader 1 (SL1). A chimeric read spanning the SL1-exon junction is diagnostic of trans-splicing. (G) Number of chimeric reads and unique junctions mapping to the upstream regions of lincRNA and protein-coding genes. For protein-coding genes, 100 cohorts, each selected to match the set of lincRNA genes with respect to gene number and expression levels, were used to estimate the 90% confidence interval (error bar).
To maximize both sensitivity and specificity, we designed scripts that (1) used information from the strand-specific reads to correct the non-strand-specific annotations, systematically evaluating each exon for strand-specific support and for transitions in the strand supported (Supplemental Fig. S1A), (2) incorporated information from exon-junction hits that was not incorporated in the original annotations from either the strand-specific or non-strand-specific data (Supplemental Fig. S1B), (3) used poly(A) sites identified using 3P-seq (poly[A] position profiling by sequencing) (Jan et al. 2011) to assign 3′ ends of most transcripts (Supplemental Fig. S1B). The resulting 82,462 transcripts from 24,644 loci included 84.1% of the nucleotides falling within the RefSeq protein-coding transcripts (ce6), and the RefSeq protein-coding transcripts made up 66.3% of the nucleotides within the 82,462 transcripts (Supplemental Table S1C). These percentages did not perfectly reflect annotation sensitivity and specificity, in part because these RefSeq protein-coding transcripts were missing any lncRNAs that might be present in C. elegans as well as the 3′ UTRs of many protein-coding transcripts (Mangone et al. 2010; Jan et al. 2011). Moreover, based on observations in vertebrates, where lncRNAs tend to be expressed at levels lower than those of protein-coding transcripts (Guttman et al. 2010; Cabili et al. 2011; Ulitsky et al. 2011), the sensitivity for lncRNAs was expected to be lower than that of protein-coding transcripts (Supplemental Fig. S2). Nonetheless, the improvement of these percentages over those of our initial de novo annotations suggested even greater sensitivity than that achieved for the annotations from non-strand-specific data and even greater specificity than that achieved for the annotations from strand-specific data (Supplemental Table S1C).
The 82,462 transcript isoforms (including 3′ UTR isoforms) consisted of 78,940 multi-exon transcripts and 3521 single-exon transcripts, which together derived from 24,644 unique loci (Fig. 1A). Most of the loci overlapped with those annotated by modENCODE (Fig. 1B), as expected, because both sets of annotations were based largely on the same non-strand-specific RNA-seq and 3P-seq data sets (Gerstein et al. 2010). Nonetheless, our annotations included 3745 unique loci. In addition, 1347 modENCODE loci were not detected, presumably because we did not include EST data or the additional types of information useful for predicting protein-coding loci.
Genome-wide identification of lncRNAs
To identify lncRNAs, our 24,644 loci were filtered to remove those that overlapped the sense strand of annotated protein-coding genes, pseudogenes, ribosomal RNA, tRNA, miRNA, and other known classes of ncRNAs (Ce6 and Ensembl version 57). Of the 3291 loci (5029 transcript isoforms) that remained after this filtering, 1546 (2522 transcript isoforms) with ≥100 nt of antisense overlap with pre-mRNAs or other annotated transcripts were classified as ancRNA candidates, and the remaining 1745 loci (2507 transcript isoforms) were designated lincRNA candidates (Fig. 1A).
For each of these lncRNA candidates, coding potential was evaluated, removing those with (1) scores ≥ –1.0 when using the coding potential calculation (CPC) program (Supplemental Fig. S3A; Jia et al. 2010), (2) marginal confidence in coding potential (P ≤ 0.01) as evaluated using the RNAcode program (Washietl et al. 2011), or (3) evidence of ribosome association in experiments that sequenced RNA either sedimenting with polyribosomes (polyribosome reads/RNA-seq RPKM ≥ 0.1) (Supplemental Fig. S3B; Lamm et al. 2011) or protected by ribosomes (ribosome RPKM/RNA-seq RPKM ≥ 0.1) (Supplemental Fig. S3C; Stadler and Fire 2011). We also excluded loci that overlapped recently annotated protein-coding genes (through WormBase release WS231). This filtering retained 801 potential lincRNA loci (Fig. 1C) and 344 potential ancRNA loci (Fig. 1D). Further analysis using the 3P-seq data to identify transcripts with evidence of a poly(A) tail recovered 170 lincRNA loci, which were represented by 262 alternative splicing/3′-end isoforms, and 58 ancRNA loci, which were represented by 95 alternative splicing/3′-end isoforms (Fig. 1E; Supplemental Table S2). The lincRNA loci were named using the linc gene classifier (i.e., linc-1 through linc-170), and the ancRNA loci were named using the anr classifier (an acronym for ancRNA that is also the reverse of “RNA”). The search for lncRNA poly(A) sites included more genomic regions than did the previous analysis of UTRs (Jan et al. 2011) and, therefore, identified poly(A) sites that had not been previously recognized (Supplemental Fig. S4). The mean lengths of the lincRNAs and ancRNAs with assigned poly(A) sites were 830 and 756 nt, respectively, which were shorter than the mean length of mRNAs (∼2.2 kb) (Supplemental Table S2A,B).
The potential lncRNAs with assigned poly(A) sites (Supplemental Table S2C,D) were carried forward as our set of C. elegans lncRNAs because they were the ones most confidently annotated as independent transcripts. Of the 170 lincRNA loci, 95 overlapped a modENCODE gene model (Gerstein et al. 2010) and nine overlapped one of the 1678 ncRNA candidates (Lu et al. 2010). Of the 58 ancRNA loci, 24 overlapped a modENCODE gene model (Gerstein et al. 2010). Although identified with less confidence, the potential lncRNAs without 3P-seq support (Supplemental Table S2E,F) are likely to include some interesting transcripts, including canonical lncRNAs that have poly(A) tails but lacked 3P-seq support because they are not highly expressed at the stages with 3P-seq data. Other potentially interesting transcripts, presumably including some enhancer-associated transcripts, might not be polyadenylated. One highly conserved noncoding RNA excluded because it lacked a poly(A) tail was the metazoan signal-recognition particle RNA (Supplemental Table S2E).
mRNA partners of ancRNAs
Of the 58 ancRNAs, 39 were fully embedded within pre-mRNA partners (14 fully within introns), 11 had divergent overlap with their pre-mRNA partner, four had convergent overlap, and four fully encompassed their pre-mRNA or ncRNA partner. About half of the mRNA partners were hypothetical genes without confirmed expression, which raised the possibility that many might not be authentic mRNAs (implying that the corresponding ancRNAs might eventually be reannotated as lincRNAs). For mRNA partners in each subgroup, we found no evidence for common functions (as measured using Gene Ontology enrichment). Expression analysis for each subgroup revealed that only convergent pairs tended to be anti-correlated with each other and that, for convergent pairs, more examples will be needed to establish statistical significance (mean r = –0.17, P = 0.16, one-sample t-test) (Supplemental Fig. S5).
Occasional trans-splicing of lincRNAs
Identifying transcript start sites (TSSs) is challenging in C. elegans because trans-splicing often replaces the 5′ end of pre-mRNAs with a splice-leader sequence (Fig. 1F). Approximately 70% of mRNAs possess splice leaders at their 5′ end, most of which have unknown TSSs. To examine whether lincRNAs also possess splice leaders, we looked for evidence of chimeric RNA-seq reads that did not map to the genome and instead had at least 12 nt of splice leader sequence (choosing from the 10 leaders most frequently observed for mRNA) fused to sequences in the 5′ regions of lincRNAs and mRNAs (Fig. 1F). Of the 343,660 RNA-seq reads that contained the 3′ part of a splice leader but did not match the genome, 87,194 were chimeric reads that resulted from trans-splicing near the 5′ end (within –500 to 100 nt) of mRNAs for 20,587 unique RefSeq genes. This analysis captured 6624 unique junctions with at least two reads (Supplemental Table S3A). In contrast, only 52 chimeric reads capturing only 12 unique junctions with at least two reads (Supplemental Table S3B) represented trans-splicing to the 170 lincRNAs (within –500 to 100 nt of the 5′ end inferred by RNA-seq reads). Even when compared to expression-matched mRNAs, the number of chimeric reads and unique trans-splicing junctions were more abundant for mRNAs than for lincRNAs (Fig. 1G).
Trans-splicing frequently serves to separate polycistronic pre-mRNAs into individual mRNAs. In C. elegans, >1000 operons have been identified, each containing two to eight genes and accounting for 15% of all C. elegans genes (Spieth et al. 1993; Blumenthal and Gleason 2003). Among the 170 lincRNA genes, three tandem clusters were found (Supplemental Table S4), each containing lincRNA genes within 1 kb of each other. For example, linc-41 is next to linc-21, with only 320 bp separating the two genes (Supplemental Fig. S6). The fraction of clustered lincRNA genes (1.8%) was much less than that of protein-coding genes, and none of corresponding lincRNAs were associated with the SL2 sequence, which is used for polycistronic pre-mRNAs (Spieth et al. 1993). These observations suggest that lincRNA genes are less likely than protein-coding genes to have the operon-like transcript structure. Because trans-splicing tends to occur very close to the start codon, the splice leader is also thought to confer more efficient translation initiation (Blumenthal and Steward 1997; Lall et al. 2004). This role for the splice leader would help explain why trans-splicing is more frequent for mRNAs than for lincRNAs. Indeed, we found that for mRNAs, the AUG translational start codon was the codon most enriched in the 30 nt downstream from the junctions (adjusted P = 8 × 10−5), whereas for lincRNAs, other codons were enriched instead (CCG, adjusted P = 8.9 × 10−4 and GAC, adjusted P = 0.012) (Supplemental Table S3C). Taken together, our results show that some C. elegans lincRNA primary transcripts are trans-spliced, but for the few that are trans-spliced, the trans-splicing plays a role apart from separating polycistronic transcripts or enhancing translation initiation.
lincRNAs antisense to endo-siRNA clusters
When mapping published (Batista et al. 2008; Gu et al. 2009) and newly generated small-RNA sequencing data (Supplemental Table S5), we noticed that highly expressed endo-siRNAs often mapped antisense to lincRNAs (examples in Supplemental Fig. S7). Of the 170 lincRNAs, 46 were antisense to either 22G-RNAs (small-RNA RPKM ≥ 5) or 26G-RNAs (small-RNA RPKM ≥ 5) in at least one developmental stage (Supplemental Table S6; Supplemental Fig. S8A). This fraction of lincRNAs (27.1%) was comparable to that of protein-coding genes antisense to endo-siRNAs (32.1%). As observed for mRNAs, more highly expressed lincRNAs tended to map antisense to endo-siRNAs (Supplemental Table S6; Fig. 2A; Supplemental Fig. S8A). For example, 44% of 73 lincRNAs with mean RPKM ≥ 1 mapped antisense to 22G-RNAs (small-RNA RPKM ≥ 5) in L4 or adult stages (Fig. 2A), whereas only 9.3% of lincRNAs with mean RPKM < 1 mapped antisense to 22G-RNAs in the same stages (Supplemental Fig. S8A; Table 1).
Figure 2.

Endo-siRNAs mapping antisense to lincRNAs. (A) Abundance of endo-siRNAs mapping antisense to 73 lincRNAs with mean RPKM ≥ 1. The key indicates the log-scaled RPKM values (endo-siRNA reads per kilobase per million genomic mapping reads). The lincRNAs were sorted by the mean RPKM values (averaging RPKMs calculated from all 35 RNA-seq samples). The data used to make this heat map are presented in Supplemental Table S6. (B) Improved annotations of loci corresponding to the top 30 22G-RNA clusters from the adult stage. (Left panel) Fractions of 22G-RNAs mapping to the antisense strand (red), sense strand (green), and intergenic or intronic regions (gray) of protein-coding genes annotated in ce6. (Right panel) Fractions of 22G-RNAs mapping to the indicated transcripts of the de novo gene annotation, highlighting those mapping antisense to new transcripts (orange). Clusters mapping antisense to either lincRNAs or newly annotated transcripts that satisfied only two of the three lincRNA filtering criteria are indicated (blue and gray asterisks, respectively) as are those mapping antisense to pseudogenes (T09F5.12, Y39E4B.14, and C47G2.6). (C) Improved annotations of loci corresponding to the top 30 26G-RNA clusters from the embryo stage; otherwise, as in B.
Table 1.
lincRNAs antisense to endo-siRNAs (small-RNA RPKM ≥ 5) in L4 and adult stages
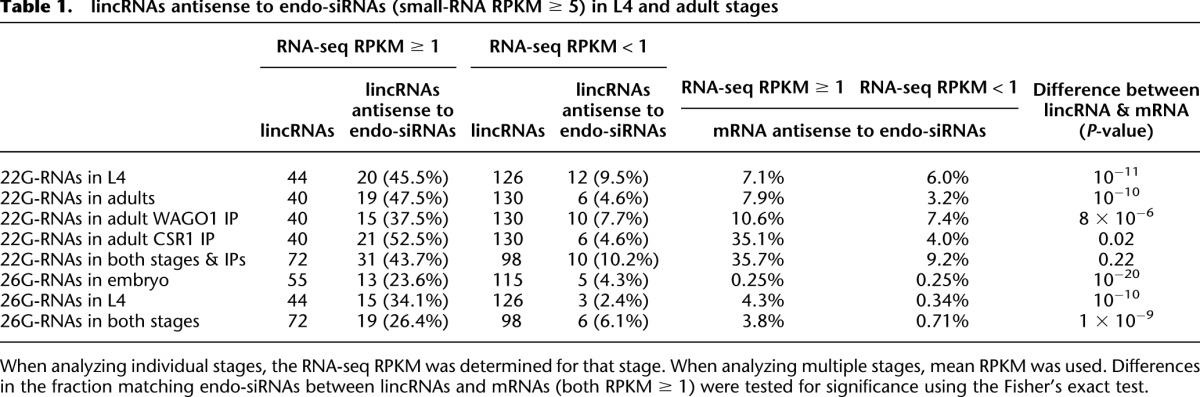
Although, overall, lincRNAs resembled mRNAs in mapping antisense to endo-siRNAs, lincRNAs tended to map to some subclasses of endo-siRNAs more than to others. About 40% of both lincRNAs and mRNAs with RPKM ≥ 1 in adult mapped antisense to 22G-RNAs (P = 0.22, Fisher's exact test), whereas the fraction antisense to WAGO-1-associated 22G-RNAs was much higher for lincRNAs than for mRNAs (37.5% and 10.6%, respectively, P < 8 × 10−6, Fisher's exact test) (Table 1). Moreover, 26% of 72 lincRNAs with mean RPKM ≥ 1 mapped antisense to 26G-RNAs (small-RNA RPKM ≥ 5) in either embryo or L4 stages—a fraction more than seven times higher than that of mRNAs (P < 1 × 10−9), which increases to 100 times higher when considering 26G-RNAs in the embryo stage (P < 10−20) (Table 1).
Many of the endo-siRNAs that mapped to previously unannotated regions of the genome mapped antisense to our newly annotated lincRNAs. After grouping the 22G-RNAs mapping within 100 nt of each other into clusters and ranking the clusters by the number of 22G-RNA reads, we found that the first and seventh clusters mapped antisense to linc-22 and linc-29, respectively (Fig. 2B; Supplenmental Fig. S7A). Three other clusters mapped antisense to pseudogenes, another class of ncRNA gene (Fig. 2B). A similar analysis of 26G-RNA clusters (grouping those mapping within 1000 nt of each other) found that five of the top 30 clusters mapped antisense to lincRNAs and that two mapped antisense to a pseudogene (Fig. 2C; Supplemental Fig. S7B). Moreover, three 22G-RNA and three 26G-RNA clusters mapped antisense to newly annotated transcripts that did not pass our cutoffs for lincRNA annotation because they satisfied only two of the three filtering criteria (Figs. 1C, 2B,C). Overall, the lincRNA annotations and other recent gene-annotation improvements (Gerstein et al. 2010) provided substantial insight into the origins of endo-siRNAs, with most of the siRNA clusters that previously mapped to unannotated regions now mapping antisense to newly annotated transcripts (Supplemental Fig. S8B,C). Of those clusters mapping predominantly (90% of reads) to one annotation, between 3% and 16% (depending on the stage) mapped antisense to lincRNAs, a 12- to 47-fold enrichment compared to mRNAs (P < 0.006 to <10−14) (Supplemental Fig. S8B,C).
In RNAi-treated C. elegans, target mRNAs are down-regulated at both the post-transcriptional and transcriptional levels (Montgomery et al. 1998). RNAi-mediated transcriptional repression occurs cotranscriptionally, whereby nuclear-localized siRNAs inhibit RNA polymerase II elongation and facilitate the establishment of histone H3 lysine 9 methylation (H3K9me3) (Guang et al. 2010; Burkhart et al. 2011; Gu et al. 2012). By analogy, the lincRNAs antisense to endo-siRNAs presumably template the production of the corresponding siRNAs, some of which recruit heterochromatin factors to silence regions from which they originate. Supporting this conjecture, genes for lincRNAs antisense to 22G-RNAs and 26G-RNAs tended to map to the ends of chromosomes, which have a higher density of the H3K9me3 heterochromatic mark (Supplemental Fig. S9A), and these genes had significantly more H3K9me3 signal compared to genes for other lincRNAs (Supplemental Fig. S9B). Moreover, the H3K9me3 signal at these lincRNAs depended on the nrde-2 nuclear RNAi pathway (Supplemental Fig. S9C).
Sequence composition and conservation of nematode lncRNAs
The A/U contents of both lincRNA and ancRNA sequences were comparable to that of sampled intergenic regions, falling between that of CDS/5′ UTRs and that of 3′ UTRs (Fig. 3A), as observed for zebrafish and mammalian lincRNAs (Ulitsky et al. 2011). No base-composition differences were observed between lincRNAs antisense to endo-siRNAs and other lincRNAs (Fig. 3B).
Figure 3.

lincRNA sequence composition and conservation. (A) A/U content of lincRNAs and ancRNAs, compared to that of mRNA 5′ UTRs, 3′ UTRs, and coding regions, and that of intergenic regions. Box and whisker plots indicate the median, interquartile range (IQR) between 25th and 75th percentiles (box), and 1.5 IQR (whisker). (B) A/U content of lincRNAs antisense to abundant 22G-RNAs (≥5 RPKM) and those antisense to less abundant or no 22G-RNAs (<5 RPKM); otherwise, as in A. (C) The fraction of mRNAs containing annotated repeat elements. (D) The fraction of lincRNAs containing annotated repeat elements. (E) Fraction of residues aligned in multiple-genome alignments for the indicated mRNA and lincRNA regions. Control exons were generated by random selection of a length-matched region from intergenic space of the same chromosome; within this control region, exons were assigned to the same relative positions as in the authentic lincRNA locus. Annotated repeats were removed from the control exons, lincRNA exons, and lincRNA introns prior to analysis. (F) Conservation of lincRNA and mRNA introns and exons. Shown are cumulative distributions of mean phastCons scores derived from the six-way whole-genome alignments (Siepel et al. 2005). Control exons were as in E. (G) Relationship between mapping to 22G-RNAs and sequence conservation. lincRNAs were assigned to three groups based on the abundance (RPKM) of antisense-mapping 22G-RNAs. Shown are cumulative distributions of mean phastCons scores (Siepel et al. 2005) for each group. (H) Lengths of conserved regions within exons. For each exon that had an average phastCons score > 0, the maximum length of regions exceeding a phastCons score of 0.5 was measured. For CDS exons, 1000 length-matched exons were randomly selected from coding regions.
The extent of sequence similarity (E-value ≤ 10−10) between lincRNAs (22 out of 170, 12.9%) was much higher than that between mRNAs (1.8 ± 1.6% for 10 random cohorts of 170 mRNAs, P < 1.0 × 10−5, Fisher's exact test) (Supplemental Fig. S10), due in part to the increased presence of repeat elements in lincRNA sequences (50% of lincRNAs with a similar sequence among them harbored an annotated repeat element). The fraction of lincRNAs with repeat sequences (17.6%) was much greater than for mRNAs (2.5%, P < 10−15, Fisher's exact test) (Fig. 3C,D). Repeat elements that lincRNAs shared included helitron, satellite sequences, LINE elements, and transposable repeat elements.
To examine the overall conservation of nematode lincRNAs, we used the phastCons scores (Siepel et al. 2005), focusing on residues that were aligned in the whole-genome sequence alignments but did not map to annotated repeats. The fraction of lincRNA residues aligned in the whole-genome alignments was ∼31.7%, which was much smaller than those of mRNA CDS (88%) and 3′ UTRs (55%) and comparable to those of mRNA introns (25%) and intergenic controls, termed control exons (27%) (Fig. 3E). We compared the conservation of exons and introns of lincRNA to those of length-matched exons and introns of protein-coding genes. The aligned lincRNA exons were more conserved than corresponding lincRNA introns but much less conserved than CDS exons and 3′ UTRs, and about as conserved as mRNA introns and length-matched control exons (Fig. 3F). The conservation of lincRNAs was anti-correlated with the density of corresponding endo-siRNAs (Fig. 3G), such that lincRNAs without antisense 22G-RNAs (46.5% of 170 lincRNAs) were more conserved than both mRNA introns and control exons, whereas those with the most antisense 22G-RNAs (24.1%) were least conserved (Fig. 3F,G). These results suggest the existence of at least two different subclasses of lincRNAs: nonconserved ones associated with endo-siRNAs and moderately conserved ones not associated with endo-siRNAs. Likewise, some protein-coding genes and pseudogenes to which certain classes of endo-siRNAs map appear particularly nonconserved (Fischer et al. 2011). The lower conservation of lincRNAs associated with endo-siRNAs is consistent with origins from heterochromatic regions, which are often poorly conserved.
Some vertebrate lincRNAs contain short conserved regions, which play important roles despite rapid sequence evolution elsewhere in the lincRNAs (Ulitsky et al. 2011). We examined whole-genome sequence alignments (Siepel et al. 2005) to check whether nematode lincRNAs also have short evolutionally conserved sequence elements. Of lincRNA exons that had any conserved element, only ∼30% had a conserved segment >50 nt (phastCons score ≥ 0.5), whereas, of CDS exons that had any conserved element, ∼60% had a conserved segment >50 nt (P < 1.0 × 10−5, KS-test) (Fig. 3H; Supplemental Fig. S11). Thus, as in vertebrates, the conserved lincRNAs tend to have shorter regions of conservation than do conserved mRNAs.
Developmental expression of nematode lincRNAs
In vertebrates, lincRNAs tend to be expressed in a development-specific manner (Cabili et al. 2011; Ulitsky et al. 2011). To examine if this was also the case for C. elegans lincRNAs, we compared for each gene model the maximum expression among 10 developmental stages to the mean expression over the remaining nine stages (Fig. 4A; Supplemental Table S7A). By this metric, lincRNAs tended to be expressed more differentially than did mRNAs, with a median fold difference between maximum and mean RPKMs of 5.6 for lincRNAs, compared to 3.7 for mRNAs (P < 7 × 10−7, KS-test) (Fig. 4A). The most extreme example was linc-3, which had an RPKM of 1002 in the dauer stage, which was 10,000 times greater than the average of the nine other stages and conditions (Fig. 4A,B). Of the 46 unique lincRNAs with maximum RPKM ≥ 8, 20 had ratios exceeding 10, and linc-1 had the highest maximum RPKM, which exceeded 1360 in male-related stages (Supplemental Table S7A,B). The more specific expression of many lincRNAs might partly explain their lower overall expression levels.
Figure 4.

Developmental- and stage-specific expression of lincRNAs. (A) Differential expression of lincRNAs. For each lincRNA and mRNA, the maximum RPKM value from 10 distinct developmental stages (Supplemental Table S1B) is plotted relative to the mean value for the remaining nine stages. If the mean value was 0, a small value (0.1) was added to avoid the log 0 value error. For stages with multiple samples, the median value of RPKMs was used. The inset shows cumulative distributions of log2-scaled ratios of maximum and mean RPKMs for lincRNA and mRNAs. (B) Dauer-specific expression of linc-3. Plotted are the RPKM values of linc-3 in 10 distinct stages. (C) Four large lincRNA expression clusters over 35 different developmental stages/conditions (top key). Colored asterisks indicate lincRNA genes within 10 kb of each other. Within each cluster, lincRNAs are sorted based on their expression level (mean RPKM), with the expression level indicated at the far right. The five columns on the right show the abundance (RPKM) of endo-siRNAs mapping antisense to each lincRNA (bottom key). (D) Correlation between lincRNA expression and that of their closest protein-coding gene. Shown is the average correlation for pairs with the indicated relative orientations (tandem, convergent, and divergent), considering only pairs within 1 kb of each other. As a control, mean correlations were also calculated for number-matched cohorts of random pairs of lincRNA and protein-coding genes. For comparison, mean correlations were calculated for number-matched cohorts of protein-coding gene pairs. For both the controls and comparisons, the average correlation of 1000 cohorts is reported for each orientation, with error bars showing the 95% confident interval.
To study coexpression of lincRNAs in specific conditions or developmental stages, we performed a CLICK clustering analysis (Sharan et al. 2003), based on Pearson correlation of z scores derived from expression distributions for each lincRNA. Four major coexpressed clusters were found, accounting for 24 embryo-specific, 41 early larval-specific, 49 sperm-specific, and 21 dauer-specific lincRNAs (Fig. 4C; Supplemental Table S7C–F). Using a similar approach, we also found one coexpressed cluster comprising 38 L3-specific ancRNAs (Supplemental Fig. S12; Supplemental Table S7G,H). As expected if some lincRNAs were templates of siRNAs, the sperm-specific lincRNAs were more frequently antisense to endo-siRNAs. Although five sperm-specific lincRNAs were transcribed from genes within 10 kb of each other, most lincRNAs in each expression cluster were from genes that were not close together (Supplemental Table S7C–F).
The expression relationships between two neighboring genes can correlate with their orientations with respect to each other, with tandem or divergent genes showing a greater tendency to be coexpressed (Korbel et al. 2004; Chen and Stein 2006; Kensche et al. 2008; Chen et al. 2010; GZ Wang et al. 2011). We examined the extent to which lincRNA genes were coexpressed with their closest neighboring protein-coding gene (limiting the analysis to genes within 1 kb of each other, which excluded 74 of the 170 lincRNAs) (Supplemental Fig. S13). Expression of the lincRNA and nearest protein-coding gene tended to be correlated, especially for the gene pairs in tandem and divergent orientations (Fig. 4D). Longer-range correlations were also observed for large clusters of genes within 200-kb regions (Supplemental Table S8). For instance, within a 200-kb region centered on the dauer-specific linc-3 gene, for which expression peaked in the dauer-entry stage (Fig. 5A,B), the protein-coding genes also tended to be expressed in a dauer-specific manner, peaking after dauer entry (Fig. 5C). As a result, the expression correlations between linc-3 and each of the 59 genes in this larger region tended to be higher than that for 1000 cohorts of number-matched controls (mean r = 0.24, adjusted P < 4 × 10−7, one-sample t-test). This region included genes for serpentine receptors, which are seven trans-membrane G-coupled chemo-receptors thought to function in a dauer pheromone signaling pathway (Kim et al. 2009; McGrath et al. 2011). The functions of these coexpressed neighboring genes supported the idea that linc-3 might also play a role in dauer formation or maintenance. Although short- and longer-range correlations were observed between the expression of lincRNAs genes and neighboring protein-coding genes, the correlations did not significantly differ from those observed between two neighboring protein-coding genes (Fig. 4D). These results resembled those observed in vertebrates (Ulitsky et al. 2011) and suggested that, compared to protein-coding genes, the lincRNA genes were no more likely to be misannotated extensions of neighboring genes and no more likely to influence expression of their neighboring genes.
Figure 5.

Long-range expression correlations involving the dauer-specific linc-3. (A) Expression of genes located within a 200-kb region centered on linc-3. The RNA-seq tracks illustrate that linc-3 and many other genes in the region were expressed higher in dauer entry and dauer stages compared with dauer exit and L3 stages. (Inset) Gene structure of linc-3 and its very high expression during dauer entry, with a read maximum exceeding that of any other gene in the region. The gene models are color-coded based on the correlation between their expression and that of linc-3 (key). (B) The expression profile of linc-3 across 35 different developmental stages/conditions. (C) The expression profile of the 59 genes within 200 kb of the linc-3 gene, visualized by plotting the mean z scores for each stage/condition. The error bars indicate standard deviation.
Expression correlation of a lincRNA and complementary mRNAs
Five lincRNAs had a long region significantly similar to the sense strand of an mRNA (≥100 nt, E-value < 10−51) (Supplemental Table S9A), and one lincRNA had a long region significantly antisense to an mRNA (≥100 nt, E-value < 10−51) (Supplemental Table S9B). Although these six unique lincRNAs might either derive from pseudogenes of protein-coding genes or simply share a common repeat element (Supplemental Table S9A,B), they nonetheless represented only 3.5% of our annotated lincRNAs, a much lower fraction than observed for mRNAs with homology with other mRNAs (19%).
Examination of shorter regions of homology identified 31 lincRNAs aligning antisense to one or more mRNAs (E-value < 10−5), comprising 168 gene pairs (Supplemental Table S10A). This fraction (18.2%) was significantly higher than that observed for number- and length-matched mRNA sequences (10.3 ± 1.8%, comprising an average of 31 pairs for 100 cohorts of computational controls, P < 0.021, Fisher's exact test). However, when excluding lincRNAs (and mRNA controls) associated with repeat elements, only 16 aligned antisense to one or more mRNAs, and the fraction of lincRNAs with antisense matches (11.4%) was not significantly higher than that for the controls (11.6%, P = 0.43). These results indicated that the tendency to map antisense to short regions of mRNAs occurred through repeat elements, raising the question as to whether it occurred by chance or has functional implications. Even after controlling for repeats, the number of the antisense pairs (78) was twice as high for the lincRNAs as for mRNA controls (30 ± 17), largely because a short conserved region of linc-55 mapped antisense to 37 members of a large gene family encoding major sperm proteins and their hypothetical paralogs (Fig. 6). These 37- to 52-nt regions of complementarity did not trigger endo-siRNAs. Overall, there was not a strong tendency for the expression of linc-55 or that of other lincRNAs with short regions of antisense complementarity to be anti-correlated with expression of their complementary mRNAs (Supplemental Table S10B).
Figure 6.

A short conserved segment of linc-55 complementary to members of the major sperm protein (MSP) family. Conservation and alignment tracks show an ∼70-nt segment conserved in four additional sequenced species. This segment has extensive complementarity to 37 members of the major sperm protein family (E-value < 10−5), including some hypothetical genes (e.g., ZK1248.17).
Discussion
Methods for annotating lncRNAs are improving but are still far from perfect. As with lists from previous efforts in other species, our lists of C. elegans ancRNAs and lincRNAs contain some very confident annotations and others that are less confident, primarily because they are not as well supported in the RNA-seq and 3P-seq data sets. The lower expression of lincRNAs compared to mRNAs has been used as evidence that they represent transcriptional noise or lack biological significance (Birney et al. 2007; Clark et al. 2011). However, the lower expression level might be due in part to their tissue-, stage-, and condition-specific expression patterns. Although we identified hundreds of lncRNAs in C. elegans, we suspect that, with additional data, more lncRNAs will be confidently and accurately annotated in this species. These will include many genes that lacked exon-junction reads for one of their introns and thus were missed because the unannotated intron disrupted connectivity to a 3P-supported poly(A) site. In fact, even after considering lincRNAs and the available RNA-seq data, some clusters of endo-siRNAs and 8436 poly(A) sites (13.2%) identified using 3P-seq remain unassociated with known gene models. Other lncRNAs that remain unannotated include those with tandem overlap with protein-coding genes, as we excluded any candidates with even a single nucleotide of sense overlap because of the difficulties in distinguishing between authentic lincRNAs and alternative 5′ or 3′ extensions of known genes.
Other potential sources of false-negatives in our lncRNA data sets were the stringent criteria used to filter out potential protein-coding genes. Most notable was our use of RNAcode (Washietl et al. 2011), an algorithm that compares the rates of synonymous and nonsynonymous changes in whole-genome alignments to find evidence of conserved protein-coding potential. Because RNAcode can evaluate only sequences that are aligned to other genomes, any lncRNAs genes mistakenly flagged and removed by the algorithm would be conserved in other species and thus would be among those most attractive for experimental follow-up. When applying less stringent criteria (CPC < 0 and no consideration of RNAcode and polyribosome association), an additional 133 lincRNA and 102 ancRNA candidates were retained (Supplemental Table S11).
Another source of false negatives might have been our exclusion of annotated protein-coding genes, particularly the hypothetical protein-coding genes. With this in mind, we tested the coding potential of 19,907 RefSeq protein-coding genes. Eleven passed our criteria for annotation as potential lincRNAs, and three of these also had 3P-seq-supported poly(A) sites (Supplemental Table S12). Nine had been classified as hypothetical proteins, and the other two were fungus- and bacteria-response genes. None had evidence for trans-splicing.
Although more lncRNAs will undoubtedly be found, the identification of lincRNAs and ancRNAs in C. elegans, with initial characterization of their evolution, genomics, and expression, provides a starting point for the study of lincRNA biology in an invertebrate animal. For some of the lincRNAs, expression or sequence features already associate them with processes such as dauer formation, male identity, sperm formation, and interaction with sperm-specific mRNAs. The study of these and other newly identified lncRNAs in C. elegans, with its established tools for rapid molecular genetic analyses, can now contribute to the understanding of the fascinating biology and mechanisms of these enigmatic transcripts.
Methods
Data sources
C. elegans genome assembly ce6 was used throughout the study. For comparison to our de novo gene annotations and to analyze endo-siRNA clusters, NCBI RefSeq gene annotations (ce6, version Oct-3-2010) were used. To filter de novo transcripts overlapping with annotated genes, NCBI RefSeq gene annotations (ce6, version Oct-3-2010), Ensembl annotations (version 57), and WormBase annotations (WS231) were used. To find repeat loci, we used UCSC repeat-masking data (Jurka 2000). All public RNA-seq data, polyribosome data, and ribosome profiling data were obtained from NCBI SRA (SRA003622 and SRA049309) and NCBI GEO (GSE22410 and GSE19414). 3P-seq data were taken from NCBI GEO (GSE24924). Small-RNA data were from previous studies, supplemented with newly acquired 5′-monophosphate-independent sequencing of small RNAs from L4 and adult stages (Supplemental Table S5). Small-RNA sequencing was as described (Batista et al. 2008).
Analysis of start codon enrichment
The frequencies of the AUG start codon in the 30 nt downstream from trans-splicing sites of lincRNAs and mRNAs were compared to the background frequency observed within –500 to 100 nt of the trans-splicing sites. The P-values were estimated by the hypergeometric test and adjusted by the Bonferroni correction.
Expression correlation analysis
To measure expression correlation between mRNAs and lincRNAs and among lincRNAs, we used RPKM values across 35 different developmental stages/conditions. To measure expression correlation among ancRNAs, we used RPKM values across 10 different stages that have strand-specific RNA-seq data.
Sequence conservation analysis
For the comparisons, we excluded intronic regions and 3′ UTRs with sense overlap with an RNAcode region (P ≤ 0.01) because lincRNAs did not include RNAcode regions, and then randomly sampled 1000 exons and introns, and 500 3′ UTRs from genes. The introns were limited to those that did not overlap with any exons of alternative isoforms. For control exons, we considered intergenic regions, again excluding any region with sense overlap with an RNAcode region, and then randomly sampled 500 exon-length-matched regions. For each region, we calculated the mean phastCons score (Siepel et al. 2005), which was then adjusted by the fraction of residues aligned in multiple-genome alignments.
Additional bioinformatic analysis
To find sequence-similar lincRNAs and mRNAs and to find antisense-matching mRNAs, NCBI BLASTN was used with the parameter “blastn –e 0.001 –K 1” and E-value cutoff of 10−10 for lincRNA, 10−51 for mRNAs, and 10−5 for antisense-matching mRNAs.
Data access
The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) (Edgar et al. 2002) and are accessible through GEO Series accession number GSE36394 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36394).
Acknowledgments
We thank Wendy Johnston for technical support, the WI genome technology core for sequencing, David Garcia and Vikram Agarwal for helpful comments on the manuscript, Igor Ulitsky and Nick Burton for helpful discussions, and Paul Davis, Jonathan Hodgkin, and their WormBase colleagues for helpful discussions and inspection of our loci, which helped eliminate false positives from our final lists of lincRNA and ancRNA loci. This work was supported by a grant (GM067031) from the NIH. D.B. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.140475.112.
Freely available online through the Genome Research Open Access option.
References
- Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D 2003. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr Biol 13: 807–818 [DOI] [PubMed] [Google Scholar]
- Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, Chaves DA, Gu W, Vasale JJ, Duan S, et al. 2008. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol Cell 31: 67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal T, Gleason KS 2003. Caenorhabditis elegans operons: Form and function. Nat Rev Genet 4: 112–120 [DOI] [PubMed] [Google Scholar]
- Blumenthal T, Steward K 1997. RNA processing and gene structure. In C. elegans II (ed. DL Riddle et al.), pp. 117–145. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Borsani G, Tonlorenzi R, Simmler MC, Dandolo L, Arnaud D, Capra V, Grompe M, Pizzuti A, Muzny D, Lawrence C, et al. 1991. Characterization of a murine gene expressed from the inactive X chromosome. Nature 351: 325–329 [DOI] [PubMed] [Google Scholar]
- Brockdorff N, Ashworth A, Kay GF, McCabe VM, Norris DP, Cooper PJ, Swift S, Rastan S 1992. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 71: 515–526 [DOI] [PubMed] [Google Scholar]
- Brown CJ, Hendrich BD, Rupert JL, Lafreniere RG, Xing Y, Lawrence J, Willard HF 1992. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71: 527–542 [DOI] [PubMed] [Google Scholar]
- Burkhart KB, Guang S, Buckley BA, Wong L, Bochner AF, Kennedy S 2011. A pre-mRNA-associating factor links endogenous siRNAs to chromatin regulation. PLoS Genet 7: e1002249 doi: 10.1371/journal.pgen.1002249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL 2011. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. 2005. The transcriptional landscape of the mammalian genome. Science 309: 1559–1563 [DOI] [PubMed] [Google Scholar]
- Chen N, Stein LD 2006. Conservation and functional significance of gene topology in the genome of Caenorhabditis elegans. Genome Res 16: 606–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WH, de Meaux J, Lercher MJ 2010. Co-expression of neighbouring genes in Arabidopsis: Separating chromatin effects from direct interactions. BMC Genomics 11: 178 doi: 10.1186/1471-2164-11-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KV, Morillon A, et al. 2011. The reality of pervasive transcription. PLoS Biol 9: e1000625 doi: 10.1371/journal.pbio.1000625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claycomb JM, Batista PJ, Pang KM, Gu W, Vasale JJ, van Wolfswinkel JC, Chaves DA, Shirayama M, Mitani S, Ketting RF, et al. 2009. The Argonaute CSR-1 and its 22G-RNA cofactors are required for holocentric chromosome segregation. Cell 139: 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SE, Montgomery TA, Zhang C, Fahlgren N, Breen PC, Hwang A, Sullivan CM, Carrington JC, Ruvkun G 2011. The ERI-6/7 helicase acts at the first stage of an siRNA amplification pathway that targets recent gene duplications. PLoS Genet 7: e1002369 doi: 10.1371/journal.pgen.1002369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JA, Kugel JF 2006. Non-coding-RNA regulators of RNA polymerase II transcription. Nat Rev Mol Cell Biol 7: 612–616 [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Shirayama M, Conte D Jr, Vasale J, Batista PJ, Claycomb JM, Moresco JJ, Youngman EM, Keys J, Stoltz MJ, et al. 2009. Distinct argonaute-mediated 22G-RNA pathways direct genome surveillance in the C. elegans germline. Mol Cell 36: 231–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SG, Pak J, Guang S, Maniar JM, Kennedy S, Fire A 2012. Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat Genet 44: 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S 2010. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature 465: 1097–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. 2010. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464: 1071–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. 2009. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458: 223–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. 2010. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28: 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Reinke V, Green P, Hirst M, Marra MA, Waterston RH 2009. Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res 19: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M, Rinn JL 2010. Large non-coding RNAs: Missing links in cancer? Hum Mol Genet 19: R152–R161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. 2010. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142: 409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Friedman RC, Ruby JG, Bartel DP 2011. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 469: 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et al. 2003. MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22: 8031–8041 [DOI] [PubMed] [Google Scholar]
- Jia H, Osak M, Bogu GK, Stanton LW, Johnson R, Lipovich L 2010. Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 16: 1478–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J 2000. Repbase update: A database and an electronic journal of repetitive elements. Trends Genet 16: 418–420 [DOI] [PubMed] [Google Scholar]
- Kensche PR, Oti M, Dutilh BE, Huynen MA 2008. Conservation of divergent transcription in fungi. Trends Genet 24: 207–211 [DOI] [PubMed] [Google Scholar]
- Kim K, Sato K, Shibuya M, Zeiger DM, Butcher RA, Ragains JR, Clardy J, Touhara K, Sengupta P 2009. Two chemoreceptors mediate developmental effects of dauer pheromone in C. elegans. Science 326: 994–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465: 182–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Jensen LJ, von Mering C, Bork P 2004. Analysis of genomic context: Prediction of functional associations from conserved bidirectionally transcribed gene pairs. Nat Biotechnol 22: 911–917 [DOI] [PubMed] [Google Scholar]
- Koziol MJ, Rinn JL 2011. RNA traffic control of chromatin complexes. Curr Opin Genet Dev 20: 142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T 2002. Identification of tissue-specific microRNAs from mouse. Curr Biol 12: 735–739 [DOI] [PubMed] [Google Scholar]
- Lall S, Friedman CC, Jankowska-Anyszka M, Stepinski J, Darzynkiewicz E, Davis RE 2004. Contribution of trans-splicing, 5′-leader length, cap-poly(A) synergism, and initiation factors to nematode translation in an Ascaris suum embryo cell-free system. J Biol Chem 279: 45573–45585 [DOI] [PubMed] [Google Scholar]
- Lamm AT, Stadler MR, Zhang H, Gent JI, Fire AZ 2011. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res 21: 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, Kuroda MI 2011. X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471: 115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZJ, Yip KY, Wang G, Shou C, Hillier LW, Khurana E, Agarwal A, Auerbach R, Rozowsky J, Cheng C, et al. 2010. Prediction and characterization of noncoding RNAs in C. elegans by integrating conservation, secondary structure, and high-throughput sequencing and array data. Genome Res 21: 276–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangone M, Manoharan AP, Thierry-Mieg D, Thierry-Mieg J, Han T, Mackowiak SD, Mis E, Zegar C, Gutwein MR, Khivansara V, et al. 2010. The landscape of C. elegans 3′UTRs. Science 329: 432–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martianov I, Ramadass A, Serra Barros A, Chow N, Akoulitchev A 2007. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 445: 666–670 [DOI] [PubMed] [Google Scholar]
- McGrath PT, Xu Y, Ailion M, Garrison JL, Butcher RA, Bargmann CI 2011. Parallel evolution of domesticated Caenorhabditis species targets pheromone receptor genes. Nature 477: 321–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS 2009. Long non-coding RNAs: Insights into functions. Nat Rev Genet 10: 155–159 [DOI] [PubMed] [Google Scholar]
- Montgomery MK, Xu S, Fire A 1998. RNA as a target of double-stranded RNA-mediated genetic interference in Caenorhabditis elegans. Proc Natl Acad Sci 95: 15502–15507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata K, Kanai A, Saito R, Kondo S, Adachi J, Wilming LG, Hume DA, Hayashizaki Y, Tomita M 2003. Identification of putative noncoding RNAs among the RIKEN mouse full-length cDNA collection. Genome Res 13: 1301–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, et al. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563–573 [DOI] [PubMed] [Google Scholar]
- Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. 2010. Long noncoding RNAs with enhancer-like function in human cells. Cell 143: 46–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak J, Fire A 2007. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science 315: 241–244 [DOI] [PubMed] [Google Scholar]
- Pauli A, Rinn JL, Schier AF 2011a. Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet 12: 136–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, Levin JZ, Fan L, Sandelin A, Rinn JL, Regev A, et al. 2011b. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res 22: 577–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. 2007. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129: 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP 2006. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell 127: 1193–1207 [DOI] [PubMed] [Google Scholar]
- Sharan R, Maron-Katz A, Shamir R 2003. CLICK and EXPANDER: A system for clustering and visualizing gene expression data. Bioinformatics 19: 1787–1799 [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieth J, Brooke G, Kuersten S, Lea K, Blumenthal T 1993. Operons in C. elegans: Polycistronic mRNA precursors are processed by trans-splicing of SL2 to downstream coding regions. Cell 73: 521–532 [DOI] [PubMed] [Google Scholar]
- Stadler M, Fire A 2011. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA 17: 2063–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, et al. 2010. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39: 925–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY 2010. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329: 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Spitale RC, Chang HY 2011. Long intergenic noncoding RNAs: New links in cancer progression. Cancer Res 71: 3–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP 2011. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147: 1537–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R 2008. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454: 126–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GZ, Lercher MJ, Hurst LD 2011. Transcriptional coupling of neighboring genes and gene expression noise: Evidence that gene orientation and noncoding transcripts are modulators of noise. Genome Biol Evol 3: 320–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen J, Wei G, He H, Zhu X, Xiao T, Yuan J, Dong B, He S, Skogerbo G, et al. 2011. The Caenorhabditis elegans intermediate-size transcriptome shows high degree of stage-specific expression. Nucleic Acids Res 39: 5203–5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Findeiss S, Muller SA, Kalkhof S, von Bergen M, Hofacker IL, Stadler PF, Goldman N 2011. RNAcode: Robust discrimination of coding and noncoding regions in comparative sequence data. RNA 17: 578–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT 2008. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322: 750–756 [DOI] [PMC free article] [PubMed] [Google Scholar]