

Abstract
Cetuximab and panitumumab, two antibodies targeting the extracellular domain of the epidermal growth factor receptor (EGFR), are of major clinical importance particularly in the treatment of metastatic colorectal cancer. As patients may acquire resistance-mediating mutations within the extracellular EGFR domain, functional dissection of the exact binding sites of EGFR targeting antibodies may help predict treatment responses. We therefore assessed the epitope recognition of panitumumab by screening phage-displayed random cyclic 7mer and linear 12mer peptide libraries on this antibody. Phage screenings revealed two strong, potentially epitope-mimicking consensus motifs targeted by panitumumab. A computational approach was used to map the sequences back to the potential epitope region on domain III of EGFR. The presumed epitope regions (386)WPEXRT(391) and a biochemically similar though discontinuous region P349-F352-D355 on a neighboring loop of domain III could be confirmed as part of the functionally relevant binding site of panitumumab by site-directed mutational analysis. To more accurately differentiate the panitumumab epitope from the previously characterized cetuximab epitope, binding studies were performed on a broad range of additional mutants. Taken together, this analysis revealed two large, partially overlapping functional epitopes consisting of 17 critical amino acid positions. Four of these positions were selectively targeted by cetuximab (I467, S468, Q408, and H409), whereas another four were selectively recognized by panitumumab (W386, E388, R390, and T391). In view of the clinical significance of extracellular domain mutations, our data may help guide treatment decisions in selected patients receiving EGFR-targeted therapies.
Introduction
The epidermal growth factor receptor (EGFR) is a major target in oncology, and monoclonal EGFR antibodies as well as small molecule tyrosine kinase inhibitors are used as standard treatment for patients with a variety of solid tumors [1,2]. The most important antibodies targeting the extracellular domain of the EGFR are the chimeric IgG1 mouse/human antibody cetuximab [3,4] and the fully human IgG2 antibody panitumumab [5]. On binding to EGFR, the antibodies compete with epidermal growth factor (EGF) binding, inhibit downstream pathway signaling, and therefore block proliferation of tumor cells [6]. While cetuximab has been approved for the treatment of colorectal cancer [3,7–9] as well as for head and neck cancer [10,11], panitumumab has only been approved for its use in colorectal cancer, so far [12,13]. Yet, recent preliminary data suggest a role for panitumumab in the treatment of patients with human papilloma virus-negative head and neck cancer [14] and the drug is under investigation for the treatment of malignant gliomas [15]. In metastatic colorectal cancer, both antibodies are considered equally effective. Nonetheless, primary resistance to these targeted agents has been extensively documented to be mediated by mutations in downstream signaling molecules [16,17]. Of these, KRAS is the only biomarker currently used in daily practice to select patients with metastatic colorectal cancer for anti-EGFR-targeted treatment. Other biomarkers such as BRAF, PIK3CA, PTEN, or NRAS are promising but so far lack enough evidence to be used in the clinics.
The conformational epitope recognized by cetuximab covers a large surface on domain III of the EGFR [18,19], whereas the exact binding site of panitumumab remains unclear. Previous studies suggest that the panitumumab epitope is in close proximity to the cetuximab epitope or may even partially overlap with the latter [20,21]. However, there is clear evidence that both epitopes are not identical. This notion may be supported by the description of effective treatment with panitumumab in patients after progression under cetuximab [22,23]. Most convincing data, however, come from a clinical study showing that a patient with colorectal cancer who acquired a point mutation under treatment with cetuximab, leading to the substitution of serine by arginine in position 468 of the extracellular EGFR domain (denominated 492 by Montagut et al. [24]), developed resistance to treatment with this antibody, whereas panitumumab was still effective in this patient. On the molecular level, this corresponded to an abrogation of cetuximab binding to the mutated EGFR, while panitumumab binding remained unaffected. Although extracellular domain mutations may only account for a small subset of clinically relevant resistance mechanisms to EGFR-targeted therapies in different tumors, characterization of the binding site of panitumumab could help predict the response to this targeted therapy in selected patients with resistance-mediating mutations [25].
We therefore explored the epitope recognition of panitumumab by screening random phage display peptide libraries that provide a powerful technical platform for epitope mapping of antibodies [26–30]. Phage display screenings on panitumumab identified a discontinuous epitope that overlapped with the large conformational cetuximab epitope. Our findings could subsequently be confirmed by mutational analysis and may help guide treatment decisions in selected patients with domain III EGFR mutations.
Materials and Methods
Phage Display Library Screening
The cyclic 7mer and linear 12mer phage libraries were purchased from New England Biolabs (Frankfurt, Germany). Three consecutive screening rounds on panitumumab (Amgen, Thousand Oaks, CA) were performed with both libraries. Each round started with a two-fold negative selection on polyclonal IgG (Octapharma, Lachen, Switzerland), followed by positive selection on panitumumab. Unbound phage were removed by washing with PBS-Tween 20. Bound phage were eluted with 0.2 M glycine (pH 2.2) and neutralized with 1 M Tris-HCl (pH9.0). Eluted phage were amplified in Escherichia coli strain K12 ER2738 (New England Biolabs) and purified by polyethylene glycol (PEG) precipitation. After three selection rounds, single-phage clones were amplified and tested for selective binding to panitumumab versus IgG or cetuximab (Bristol-Myers Squibb, New York, NY) by enzyme linked immunosorbent assay (ELISA). Therefore, phage were added to immobilized panitumumab and detected by an anti-M13, HRP-conjugated monoclonal antibody (GE Healthcare, Munich, Germany). ABTS solution (Roche Diagnostics, Grenzach-Wyhlen, Germany) was used as substrate and the absorption was measured at 405 nm using a Sunrise microplate reader (Tecan, Mannedorf, Switzerland). Selectively binding phage were sequenced (GATC, Constance, Germany). Phage displaying the random peptide YMTPPLSSQQKS were used as control.
Computational Mapping of Epitope-mimicking Consensus Motifs
The MIMOX algorithm was used to search for similarities between the phage-displayed consensus motifs and accessible amino acids on the three-dimensional structure of the EGFR surface [31]. The algorithm is freely available as a web-based tool (http://immunet.cn/mimox/).
Blocking Assay with Glutathione S-Transferase Fusion Protein
The oligonucleotide encoding the phage-derived peptide IYPPLLRTS-QAM was amplified from phage DNA by polymerase chain reaction, digested with BamHI and EcoRI and ligated into pGEX-2TK (GE Healthcare, Buckinghamshire, England). Glutathione S-transferase (GST) fusion proteins were expressed in E. coli ER2655 (Invitrogen, Camarillo, CA), purified following GE Healthcare's instructions and used for a phage competition assay as previously described [26,28].
Generation of Human EGFR Mutants
The coding sequence for the human wild-type EGFR was amplified from cDNA of A431 cells (ATCC, Middlesex, United Kingdom) and cloned into pBluescript II KS(+) (Fermentas, St Leon-Rot, Germany). For expression in CHO cells (ATCC, Wesel, Germany), cDNAs coding for human wild-type EGFR or EGFRvIII (a kind gift from Dr Hrvoje Miletic) were cloned into pcDNA 3.1+ (Invitrogen). EGFR mutants with point mutations were generated from cDNA constructs using the QuikChange XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) as described [32] using individually designed oligonucleotides. Successful introduction of point mutations was verified by sequence analysis (Seqlab, Göttingen, Germany).
Flow Cytometry
CHO cells (ATCC, Middlesex, United Kingdom) were maintained in RPMI medium and NIH 3T3 cells (ATCC, Middlesex, United Kingdom) in DMEM medium, both containing 10% FBS and 1% penicillin/streptomycin. Cell lines were genotypically and phenotypically tested to confirm identity by the supplier; 5 x 105 CHO or NIH 3T3 cells were transfected with 15 µg of wild-type or mutant EGFR vector using polyethylenimine (jet PEI by Polyplus Transfection, Illkirch, France) according to the manufacturer's protocol. After 48 hours, cells were either stained with a polyclonal goat anti-human EGFR antibody (R&D Systems, Minneapolis, MN) or with panitumumab or cetuximab, respectively. Secondary antibodies were fluorescein isothiocyanate-labeled rabbit anti-human (Sigma-Aldrich, St Gallen, Switzerland) or polyclonal rabbit anti-goat antibodies (Dako Cytomation, Copenhagen, Denmark). Cells were analyzed on a FACS Calibur (BD Biosciences, Franklin Lakes, NJ). Data analysis was performed using the FlowJo software 7.6.5 (Tree Star, Ashland, OR).
Results
Identification of Epitope-mimicking Peptides by Phage Library Screenings on Panitumumab
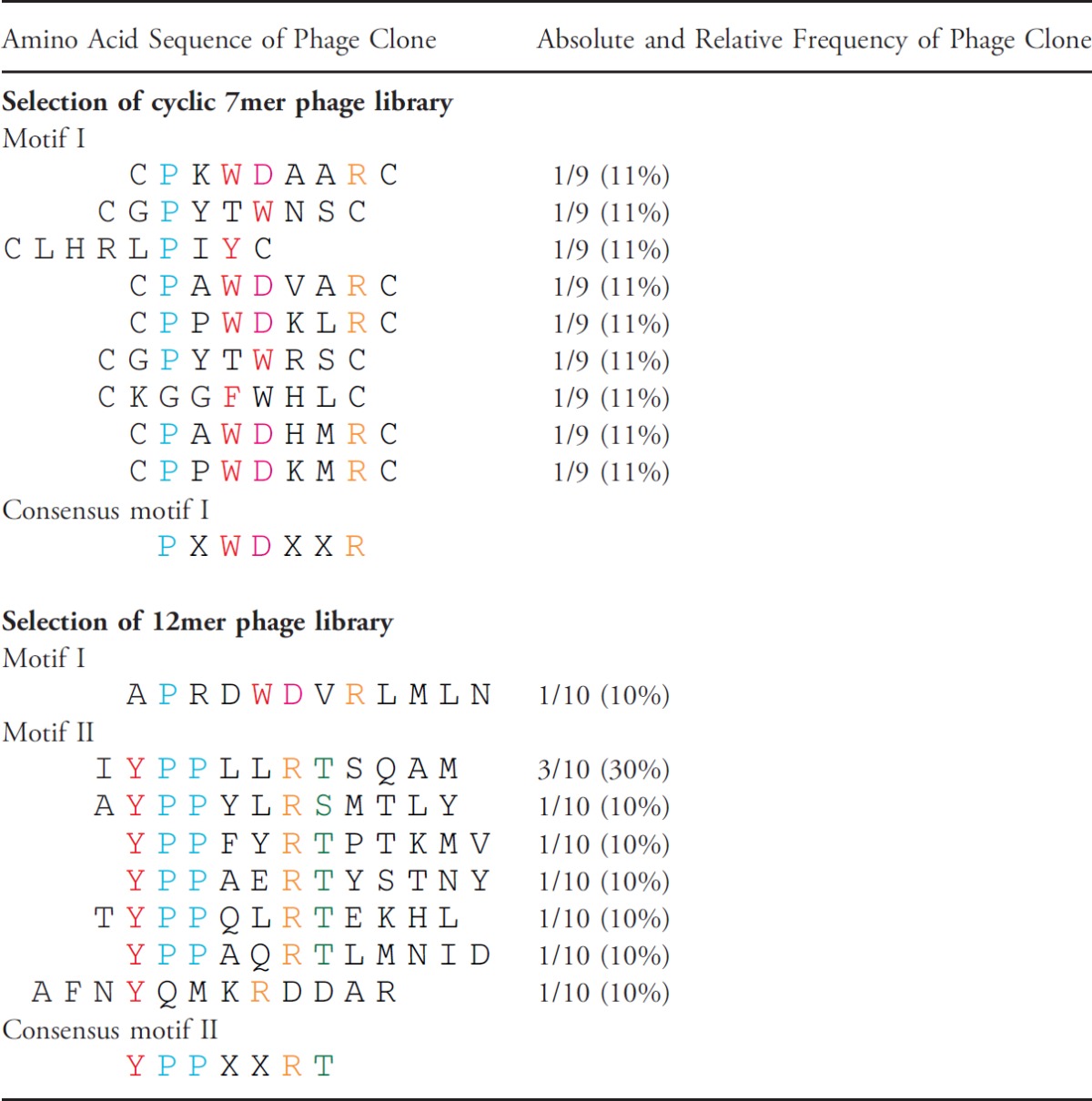
To map the epitope targeted by panitumumab, we screened a cyclic 7mer (CX7C) and a linear 12mer (X12) random peptide phage display library on this antibody (C, cysteine; X, any amino acid). With both libraries specifically binding phage could be enriched over several selection rounds after negative selection on polyclonal IgG. Figure 1A shows the binding of selected phage pools to panitumumab and control IgG after each selection round for 7mer phage (left panel) and 12mer phage (right panel). After the third selection round, single-phage clones were amplified and their binding was evaluated by ELISA. All selected phage, but not random control phage, bound specifically to panitumumab as demonstrated in Figure 1B (7mer phage on the left and 12mer on the right). Sequencing of their inserts revealed two distinct consensus motifs (Table 1). While consensus motif I (PXWDXXR) appeared in both selections, consensus motif II (YPPXXRT) was only displayed by phage from the 12mer library. To validate that phage binding to panitumumab is mediated by the cognate peptide displayed on the phage surface, we performed competition assays using a representative phage peptide as fusion protein to block the phage-panitumumab interaction. The GST-conjugated peptide IYPPLLRTSQAM inhibited binding of phage IYPPLLRTSQAM to panitumumab in a dose-dependent manner, whereas GST alone had no effect on phage binding (Figure 1C).
Figure 1.

Selection of epitope mimics on panitumumab. (A) Panitumumab binding 7mer (left panel) and 12mer (right panel) phage were enriched over three selection rounds. Binding of recovered phage pools to panitumumab and control IgG was monitored after each selection round by measuring bacterial infection. TU, transducing units. (B) Single-phage clones displaying 7mer (left panel) or 12mer (right panel) peptides bind specifically to panitumumab but not to control IgG; 1 x 108 TU of phage displaying the selected peptides as well as random control phage were incubated on immobilized panitumumab or control IgG. Phage binding was measured by ELISA. Data are means from triplicate experiments ± SEM. (C) GST-IYPPLLRTSQAM blocks binding of phage IYPPLLRTSQAM to panitumumab; 1 x 108 TU of phage IYPPLLRTSQAM were incubated on immobilized panitumumab in the presence or absence of various concentrations of GST-IYPPLLRTSQAM or GST alone. Bound phage were quantified as described in B. Data are means from triplicate experiments ± SEM. (D) Representative phage selected on panitumumab are not recognized by cetuximab. Phage binding was measured by ELISA as described in B. Data are means from triplicate experiments ± SEM.
Table 1.
Phage-Displayed Peptide Sequences Selected on Panitumumab*.
|
Sequences are displayed using a single-letter amino acid code.
The Epitope-mimicking Phage Selected on Panitumumab Are Not Recognized by Cetuximab
As cetuximab binds to a large conformational epitope on domain III of EGFR that may overlap with the panitumumab epitope, we asked whether phage clones mimicking the panitumumab epitope were recognized by cetuximab as well. The binding of one representative phage clone of each consensus motif was tested on panitumumab, cetuximab, and control IgG (Figure 1D). Both panitumumab-binding phage clones did not show any binding to cetuximab, indicating that neither of the phage-displayed peptides mimics the cetuximab epitope. We therefore concluded that the epitopes targeted by panitumumab and cetuximab are not identical. In fact, this lack of cross-reactivity was expected as entirely unrelated peptides had previously been described to structurally mimic the cetuximab epitope [33,34]. However, these data do not exclude a substantial overlap of both epitopes.
Computational Mapping of Panitumumab Epitope-mimicking Consensus Motifs to EGFR
The MIMOX algorithm was used to search for similarities between the consensus motifs and accessible amino acids on the three dimensional EGFR surface. Whereas the YPP(XX)RT consensus motif did not yield any results by MIMOX analysis, the P(X)WD(XX)R motif gave one single hit with high accessibility (198.72 Å2). This consensus motif was mapped back to the following region: W386-P387-E388-R390. Interestingly, the identified region belonged to domain III of the receptor, which—according to previous findings—is the domain targeted by panitumumab [20]. Figure W1 shows the localization of the potential epitope region on domain III of EGFR. Although both distinct consensus motifs could well structurally mimic the same linear epitope, we also had to consider a discontinuous epitope with critical amino acids on more than one loop. When manually aligning both consensus motifs with surface-exposed amino acids on neighboring loops, further potential homologies were found in spatial proximity to the previously identified region: P349-F352-D355. The computational and manual alignments of the epitope-mimicking peptides were therefore compatible with a discontinuous epitope targeted by panitumumab.
Mutational Analysis Dissects Functional Panitumumab and Cetuximab Epitopes on EGFR
To corroborate our findings, we assessed the contribution of each amino acid to the presumed epitope by mutation to alanine. In the context of a human EGFR construct for eukaryotic expression, we mutated the amino acids identified by our phage display screening (W386A/P387A/E388A, W386A, P387A, E388A, R390A/T391A, R390A, T391A, P349A/F352A, P349A, F352A, D355A) as schematically displayed in Figure W2. Moreover, we generated eight control mutants in different parts of domain III, some of them adjacent to the epitope regions in solvent-accessible surface areas (R353A, F357A, P362A, V417A, S418A, N420A) and some of them in a distant part of domain III (G458A/T459A/S460A, K463A/T464A; Figure W2). Primers used for the mutagenesis reaction are shown in Table 2. All constructs were transfected into EGFR-negative CHO cells. Polyclonal EGFR control antibody (used as a probe to assess proper presentation of EGFR on the surface of transfected CHO cells) as well as panitumumab and cetuximab binding to transfected CHO cells was then studied by flow cytometry. Transfection efficiency of the EGFR constructs ranged around 40% of viable cells. As a lot of mutants exhibited a slightly reduced binding of the monoclonal antibodies cetuximab and panitumumab compared to binding of the polyclonal EGFR control antibody and compared to wild-type EGFR, we had to specify arbitrary thresholds in binding reduction defining functionally relevant amino acid positions. A >50% binding reduction on mutation compared to the binding of polyclonal EGFR antibody was considered to indicate functionally relevant amino acid positions (although this threshold was arbitrary and not evaluated clinically). Positions with reproducibly at least 30% binding reduction were considered less essential for antibody binding. Positions that resulted in <30% binding reduction on mutation were not considered as part of the functional epitope.
Table 2.
Denomination and Sequences of Mutagenesis Primers*.
| Primer Denomination | Primer Sequence |
| S418A | CAGTTTTCTCTTGCAGTCGTCGCCCTGAACATAACATCC |
| G458A/T459A/S450A | GGAAAAAACTGTTTGCGGCCGCCGGTCAGAAAACC |
| R353A | CCGGTGGCATTTGCGGGTGACTCCTTCACAC |
| F357A | GGGGTGACTCCGCCACACATACTCCTCC |
| S468A | GGTCAGAAAACCAAAATTATAGCCAACAGAGGTGAAAACAGC |
| N420A | GCAGTCGTCAGCCTGGCCATAACATCCTTGG |
| V417A | CAGTTTTCTCTTGCAGTCGCCAGCCTGAACATAACATCC |
| K463A/T464A | GGGACCTCCGGTCAGGCAGCCAAAATTATAAGC |
| I438A | GGAGATAAGTGATGGAGATGTGGCAATTTCAGGAAAC |
| P362A | CCTTCACACATACTCCTGCTCTGGATCCACAGG |
| F412A | GCAACATGGTCAGGCTTCTCTTGCAGTCGTCAGCC |
| Q408M/H409E | CGCGGCAGGACCAAGATGCAGGGTCAGTTTTCTCTTGC |
| P349A | GATCTCCACATCCTGGCGGTGGCATTTAGGG |
| K443A | GGAGATGTGATAATTTCAGGAAACGCAAATTTGTGCTATGC |
| P387A | GCTGATTCAGGCTTGGGCTGAAAACAGGACGG |
| K465E | GGGACCTCCGGTCAGAAAACCGAAATTATAAGCAACAGAGG |
| D355A | GTGGCATTTAGGGGTGCCTCCTTCACACATAC |
| P349A/F352A | CTCCACATCCTGGCGGTGGCAGCTAGGGGTGAC |
| F352A | CTGCCGGTGGCAGCTAGGGGTGACTCCTTC |
| S468I | GGTCAGAAAACCAAAATTATAATCAACAGAGGTGAAAACAGC |
| Q408A/H409A | CGCGGCAGGACCAAGGCAGCTGGTCAGTTTTCTCTTGC |
| I467M | GGGACCTCCGGTCAGAAAACCAAAATTATGAGCAACAGAGG |
| S468R | GGTCAGAAAACCAAAATTATAAGAAACAGAGGTGAAAACAGC |
| R390A/T391A | GCTTGGCCTGAAAACGCGGCGGACCTCCATGCC |
| R390A | CTTGGCCTGAAAACGCGACGGACCTCCATGC |
| T391A | CCTGAAAACAGGGCGGACCTCCATGCCTTTG |
| W386A/P387A/E388A | GCTGATTCAGGCTGGGGCTGCAAACAGGACGGACC |
| W386A | GCTGATTCAGGCTGGGCCTGAAAACAGGACG |
| E388A | GATTCAGGCTTGGCCTGCAAACAGGACGGACC |
All primers are designed in a 5′-3′ orientation. Only forward primers are shown; corresponding reverse primer sequences are complementary reverse. Base exchanges are underlined.
All groups of amino acid positions identified by our phage display analysis—W386A/P387A/E388A, R390A/T391A, P349A/F352A and D355A—were confirmed to be critical for panitumumab binding by mutational analysis although R390A/T391A to a lesser extent [exemplary fluorescence-activated cell sorting (FACS) plots are shown in Figure 2A]. All of these mutants also exhibited reduced cetuximab binding, except for R390A/T391A. When we analyzed all single positions separately, all of these mutants displayed reduced panitumumab binding; only P349 and T391 proved to be less critical for this interaction (Figure 2B). Some of these single residues also proved to be of significant relevance for cetuximab binding, especially F352, D355, and P387 (Figure 2B). Neither the control mutations adjacent to the presumed epitope regions nor the distant ones had an impact on panitumumab or cetuximab binding (Figure 2B). Only control mutation P362, which is in close proximity to the critical residues, showed a minor binding reduction of about 40% for both antibodies (Figure 2B). EGFRvIII, a clinically relevant EGFR mutant exhibiting a deletion within domains I and II of EGFR [35], was used as a control. This deletion mutant showed preserved binding of cetuximab and panitumumab, as expected (Figure 2B).
Figure 2.


Mutational analysis of EGFR reveals functionally critical amino acid positions of the conformational epitopes of panitumumab and cetuximab. (A) Exemplary flow cytometry analysis of mutants W386A/P387A/E388A, P349A/F352A, D355A, and R390A/T391A. EGFR-negative CHO cells were transfected with wild-type EGFR or mutations thereof. Binding of panitumumab, cetuximab, or a control polyclonal EGFR antibody was assessed by FACS analysis 48 hours after transfection. FSC, forward scatter. (B) Evaluation of 30 mutants reveals the extent of the functional panitumumab and cetuximab epitopes. Flow cytometry experiments on transfected CHO cells were performed as in A. Data are shown as relative values compared to EGFR-positive cells (percentage of EGFR-positive cells set to 1) and are means of duplicate experiments ± SEM.
In addition to the phage display.inspired mutational analysis, we generated a number of mutants presumably critical for cetuximab (Q408A/H409A, Q408M/H409E, K443A, K465E, I467M, S468R, S468I, S468A) [18,19,24,36] or panitumumab (F412A, I438A) [21] binding to more accurately differentiate between both epitopes. As expected, Q408 and H409, previously described as functional core positions within the cetuximab epitope, differentially affected cetuximab and panitumumab binding on mutation to alanine. The same applied for I467 on mutation to methionine. Interestingly, K443A and K465E resulted in a critical binding reduction of both antibodies, thereby adding two additional positions to the functional panitumumab epitope, which had not been suggested by the phage display analysis. Likewise, mutants F412A and I438A showed a somewhat reduced binding of both antibodies, although the inhibition ranged only between 30%and 40%. Mutations of position S468 (denominated 492 by Montagut et al. [24]) showed divergent results, depending on the amino acid used for replacement. While mutation to arginine completely abrogated cetuximab binding, as described [24], mutation to isoleucine showed only a weak binding reduction and an alanine replacement did not affect the binding of cetuximab at all. In addition, panitumumab binding was affected in the S468R mutant, although to a significantly lesser extent compared to cetuximab. As the interaction of panitumumab with this mutant had been described to be comparable to wild-type EGFR [24], we wished to further corroborate our discordant finding. Our sequence analysis of the S468R mutant did not show any additional mutations or deletions within the whole EGFR gene. In this way, we excluded that genetic aberrations other than the intentionally introduced point mutation of the target molecule accounted for reduced panitumumab binding. Moreover, we transfected the S468R mutant DNA into EGFR-negative NIH 3T3 cells that had been used for previously published experiments [24]. These additional transfection experiments resulted in exactly the same antibody binding profiles of all three S468 mutants as the transfections into CHO cells. Likewise, in these experiments, panitumumab binding to S468R was clearly reduced compared to binding of the polyclonal EGFR antibody and panitumumab binding to wild-type EGFR, thus confirming our findings (Figure W3). Figure 2B shows the binding profiles of all tested mutants.
Finally, we summarized the data generated by phage display and mutational analysis in a three-dimensional model of the two overlapping functional epitopes (Figure 3A). Four positions differentially interfering with the binding of panitumumab on mutation are depicted in red (W386, E388, R390, T391). In mutant T391A, we observed only very slight differences in cetuximab and panitumumab binding, but as these differences were amplified in combination with mutant R390A we considered this position as selectively targeted by panitumumab. Another four positions differentially affecting cetuximab binding on mutation are depicted in blue (I467, S468, Q408, H409). Nine residues critical for the binding of both antibodies are shown in light (>30% binding reduction) and dark (>50% binding reduction) gray. Both conformational epitopes overlapped with amino acids functionally characterized as critical for EGF binding (Figure 3B). This finding illustrates the underlying mechanism of competitive EGF inhibition by panitumumab and cetuximab.
Figure 3.

Three-dimensional model of the functional panitumumab and cetuximab epitopes. (A) Functional dissection of panitumumab and cetuximab epitopes. Amino acid positions critical for panitumumab binding are shown in red and for cetuximab in blue. Residues critical for the binding of both antibodies are shown in shades of gray (light gray, >30% binding reduction; dark gray, >50% binding reduction). (B) Overlap of panitumumab and cetuximab epitopes with EGF binding site. The left panel shows the panitumumab epitope in red and the EGF binding site in yellow. The overlapping amino acids are depicted in orange. In the right panel, the cetuximab epitope is depicted in blue, the EGF binding site is depicted in yellow, and the overlapping amino acid positions are shown in green (pani., panitumumab; cetuxi., cetuximab).
Discussion
Targeting the extracellular domain of EGFR with the monoclonal antibodies cetuximab and panitumumab is a common treatment option in patients with solid tumors. Resistance has been found to be conferred by mutations in downstream signaling molecules such as KRAS [9,17,37]. However, very recently acquired extracellular domain mutations have been shown to mediate resistance to targeted agents as well [24]. Although little is known so far about the general incidence of such mutations in different tumors, these new findings put the epitopes of EGFR targeting antibodies into focus as the antibody binding sites may help predict responses to therapy in selected patients.
We therefore mapped the binding site of panitumumab by phage display library screenings and confirmed our findings by mutational analysis. In this way, we defined a functional epitope targeted by panitumumab, which substantially overlapped with the previously identified cetuximab epitope on domain III of EGFR. As our mutational analysis comprised a total of 30 mutants covering almost all surface accessible amino acid positions in the critical part of EGFR domain III, this analysis may provide a comprehensive picture of both functional epitopes.
Our data need to be discussed in the context of previously published work. So far, there has been only limited knowledge about the epitope targeted by panitumumab. In line with our findings, however, the cross-blocking experiments by Dechant et al. [20] suggested that panitumumab and cetuximab bind to EGFR in close spatial proximity. Freeman et al. [21] describe several amino acid positions as critical for panitumumab binding: P349, D355, F412, and I438. While our phage display analysis also suggested that P349 and D355 may be part of the panitumumab epitope and our alanine mutational analysis clearly confirmed these two residues as critical positions, the mutation of F412 and I438 to alanine only resulted in a 30% to 40% abrogation of panitumumab as well as cetuximab binding to mutated EGFR in our hands. We therefore considered positions F412 and I438 less critical for the binding of both antibodies. Because D355 is involved in binding of the EGF to its receptor, the identification of D355 as part of the panitumumab epitope is especially meaningful as it may provide the molecular basis for the mechanism of action of this antibody. Thus, the experimentally observed competition of panitumumab with EGF [6] can be explained by an overlap of binding sites.
Our work also adds some new aspects to the functional definition of the previously identified cetuximab epitope. This epitope has been studied intensively by several groups both in a structural and in a functional way. The crystal structure of the EGFR-cetuximab complex revealed that the epitope covers a large surface on domain III of the EGFR [18]. Moreover, some functionally critical residues, such as Q384, Q408, H409, K443, K465, I467, and S468, have been identified by different groups [18,19,24,36]. By selective alanine scanning, we identified some more residues in two regions of EGFR domain III (F352, D355, P387), which are apparently involved in cetuximab binding as well, as their mutation results in an abrogation (>50%) of antibody binding to EGFR. Positions P349, P362, W386, E388, R390, F412, and I438 seem to be of some minor importance for the EGFR-cetuximab interaction as their mutation results in a binding reduction of 30% to 50%.
In most of our mutants, alanine replacement was used as it represents a standard technical tool for mapping of functional epitopes [38]. This amino acid is usually chosen as it does not impose conformational changes and extreme electrostatic or steric effects. In some specific cases, we generated mutants with alternative replacement mutations, if these had been well characterized previously. In this context, we noticed that some residues are not significantly impacted by mutation to alanine compared to substitutions with other amino acids. Whereas the clinically relevant mutation of serine in position 468 to arginine results in a complete abrogation of cetuximab binding, the substitution of the same residue with isoleucine only results in a partial binding inhibition and the replacement with alanine does not significantly alter the binding of cetuximab at all [24,36]. Vice versa, in position D355, the mutation to alanine results in a loss of cetuximab binding in our hands, whereas the substitution with threonine [18] does not produce this effect. Likewise, our data indicated that the mutation of positions S418 and R353 to alanine does not impair antibody binding although these residues are part of the structurally defined binding site of cetuximab [18]. These findings nicely demonstrate the complexity of epitope definition, taking into account the structural as well as the functional dimension of the binding site, which are known to show imperfect congruence [39].
From a clinical perspective, this complexity may impede the prediction of resistance to antibody-based EGFR targeting. Residues defined as critical for cetuximab or panitumumab binding by an alanine scanning approach may well be mutated in vivo to other amino acids without functional consequences. Therefore, even if the binding site has been functionally defined in vitro by mutational analysis, every unrecognized domain III mutation occurring in an individual tumor sample would have to be tested in vitro for functional consequences to more reliably predict the response to therapy. Likewise, we currently lack a biologically meaningful in vitro binding threshold, allowing us to estimate if a given mutation will result in a clinically relevant abrogation of antibody binding in vivo. While a complete binding abrogation in vitro (as found for cetuximab binding to S468R) will most likely result in clinical ineffectiveness of the antibody (at least in the tumor subclone harboring the mutation), it remains unclear what clinical consequences will arise from mutations inducing only partial impairment of antibody binding. In this context, it is quite interesting to note that in our hands panitumumab binding to the S468R mutant was reduced to 30%, although the antibody has been described to be active in tumors harboring this mutation [24].
Taken together, we functionally defined the conformational epitope targeted by panitumumab within domain III of the EGFR, thereby differentiating it from the cetuximab epitope. These data are of clinical relevance as they may help predict which patients with EGFR domain III mutations may benefit from cetuximab or panitumumab treatment and may therefore guide treatment decisions in selected patients.
Supplementary Material
Acknowledgments
We thank Hrvoje Miletic (Bergen, Norway) for providing a vector encoding the EGFR deletion variant III.
Footnotes
This work was supported by the Wilhelm Sander-Stiftung (grant 2009.035.1 to M.B.). The authors declare no financial conflict of interest.
This article refers to supplementary materials, which are designated by Figures W1 to W3 and are available online at www.neoplasia.com.
References
- 1.Mendelsohn J. The epidermal growth factor receptor as a target for cancer therapy. Endocr Relat Cancer. 2001;8:3–9. doi: 10.1677/erc.0.0080003. [DOI] [PubMed] [Google Scholar]
- 2.Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:15–31. doi: 10.1517/14728222.2011.648617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 4.Robert F, Ezekiel MP, Spencer SA, Meredith RF, Bonner JA, Khazaeli MB, Saleh MN, Carey D, LoBuglio AF, Wheeler RH, et al. Phase I study of anti-epidermal growth factor receptor antibody cetuximab in combination with radiation therapy in patients with advanced head and neck cancer. J Clin Oncol. 2001;19:3234–3243. doi: 10.1200/JCO.2001.19.13.3234. [DOI] [PubMed] [Google Scholar]
- 5.Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol Hematol. 2001;38:17–23. doi: 10.1016/s1040-8428(00)00134-7. [DOI] [PubMed] [Google Scholar]
- 6.Gill GN, Kawamoto T, Cochet C, Le A, Sato JD, Masui H, McLeod C, Mendelsohn J. Monoclonal anti-epidermal growth factor receptor antibodies which are inhibitors of epidermal growth factor binding and antagonists of epidermal growth factor binding and antagonists of epidermal growth factor-stimulated tyrosine protein kinase activity. J Biol Chem. 1984;259:7755–7760. [PubMed] [Google Scholar]
- 7.Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–2048. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 8.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 9.Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pinter T, Lim R, Bodoky G, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 10.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116–1127. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 11.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 12.Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 13.Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, Andre T, Chan E, Lordick F, Punt CJ, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28:4706–4713. doi: 10.1200/JCO.2009.27.6055. [DOI] [PubMed] [Google Scholar]
- 14.Gilbert DC, Simcock R, Schache A, Shaw R. Epidermal growth factor receptor and the changing face of oropharyngeal cancer. J Clin Oncol. 2012;30:890–891. doi: 10.1200/JCO.2011.40.4848. [DOI] [PubMed] [Google Scholar]
- 15.Berezowska S, Schlegel J. Targeting ErbB receptors in high-grade glioma. Curr Pharm Des. 2011;17:2468–2487. doi: 10.2174/138161211797249233. [DOI] [PubMed] [Google Scholar]
- 16.Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28:1254–1261. doi: 10.1200/JCO.2009.24.6116. [DOI] [PubMed] [Google Scholar]
- 17.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 18.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Chao G, Cochran JR, Wittrup KD. Fine epitope mapping of anti-epidermal growth factor receptor antibodies through random mutagenesis and yeast surface display. J Mol Biol. 2004;342:539–550. doi: 10.1016/j.jmb.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 20.Dechant M, Weisner W, Berger S, Peipp M, Beyer T, Schneider-Merck T, Lammerts van Bueren JJ, Bleeker WK, Parren PW, van de Winkel JG, et al. Complement-dependent tumor cell lysis triggered by combinations of epidermal growth factor receptor antibodies. Cancer Res. 2008;68:4998–5003. doi: 10.1158/0008-5472.CAN-07-6226. [DOI] [PubMed] [Google Scholar]
- 21.Freeman D, Sun J, Bass R, Jung K, Ogbagabriel S, Elliot G, Randinsky R. Panitumumab and cetuximab epitope mapping and in vitro activity; Posterior session presented at the ASCO-Gastrointestinal Cancers Symposium; Orlando, FL. 2008. [Google Scholar]
- 22.Saif MW, Kaley K, Chu E, Copur MS. Safety and efficacy of panitumumab therapy after progression with cetuximab: experience at two institutions. Clin Colorectal Cancer. 2010;9:315–318. doi: 10.3816/CCC.2010.n.046. [DOI] [PubMed] [Google Scholar]
- 23.Wadlow RC, Hezel AF, Abrams TA, Blaszkowsky LS, Fuchs CS, Kulke MH, Kwak EL, Meyerhardt JA, Ryan DP, Szymonifka J, et al. Panitumumab in patients with KRAS wild-type colorectal cancer after progression on cetuximab. Oncologist. 2012;17:e14–e35. doi: 10.1634/theoncologist.2011-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–223. doi: 10.1038/nm.2609. [DOI] [PubMed] [Google Scholar]
- 25.Bardelli A, Janne PA. The road to resistance: EGFR mutation and cetuximab. Nat Med. 2012;18:199–200. doi: 10.1038/nm.2646. [DOI] [PubMed] [Google Scholar]
- 26.Binder M, Otto F, Mertelsmann R, Veelken H, Trepel M. The epitope recognized by rituximab. Blood. 2006;108:1975–1978. doi: 10.1182/blood-2006-04-014639. [DOI] [PubMed] [Google Scholar]
- 27.Binder M, Vogtle FN, Michelfelder S, Muller F, Illerhaus G, Sundararajan S, Mertelsmann R, Trepel M. Identification of their epitope reveals the structural basis for the mechanism of action of the immunosuppressive antibodies basiliximab and daclizumab. Cancer Res. 2007;67:3518–3523. doi: 10.1158/0008-5472.CAN-06-3919. [DOI] [PubMed] [Google Scholar]
- 28.Binder M, Muller F, Jackst A, Lechenne B, Pantic M, Bacher U, Zu Eulenburg C, Veelken H, Mertelsmann R, Pasqualini R, et al. B-cell receptor epitope recognition correlates with the clinical course of chronic lymphocytic leukemia. Cancer. 2011;117:1891–1900. doi: 10.1002/cncr.25755. [DOI] [PubMed] [Google Scholar]
- 29.Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 30.Vidal CI, Mintz PJ, Lu K, Ellis LM, Manenti L, Giavazzi R, Gershenson DM, Broaddus R, Liu J, Arap W, et al. An HSP90-mimic peptide revealed by fingerprinting the pool of antibodies from ovarian cancer patients. Oncogene. 2004;23:8859–8867. doi: 10.1038/sj.onc.1208082. [DOI] [PubMed] [Google Scholar]
- 31.Huang J, Gutteridge A, Honda W, Kanehisa M. MIMOX: a web tool for phage display based epitope mapping. BMC Bioinformatics. 2006;7:451. doi: 10.1186/1471-2105-7-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller OJ, Kaul F, Weitzman MD, Pasqualini R, Arap W, Kleinschmidt JA, Trepel M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol. 2003);21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 33.Hartmann C, Muller N, Blaukat A, Koch J, Benhar I, Wels WS. Peptide mimotopes recognized by antibodies cetuximab and matuzumab induce a functionally equivalent anti-EGFR immune response. Oncogene. 2010;29:4517–4527. doi: 10.1038/onc.2010.195. [DOI] [PubMed] [Google Scholar]
- 34.Riemer AB, Kurz H, Klinger M, Scheiner O, Zielinski CC, Jensen-Jarolim E. Vaccination with cetuximab mimotopes and biological properties of induced anti-epidermal growth factor receptor antibodies. J Natl Cancer Inst. 2005;97:1663–1670. doi: 10.1093/jnci/dji373. [DOI] [PubMed] [Google Scholar]
- 35.Schulte A, Gunther HS, Martens T, Zapf S, Riethdorf S, Wulfing C, Stoupiec M, Westphal M, Lamszus K. Glioblastoma stem-like cell lines with either maintenance or loss of high-level EGFR amplification, generated via modulation of ligand concentration. Clin Cancer Res. 2012;18:1901–1913. doi: 10.1158/1078-0432.CCR-11-3084. [DOI] [PubMed] [Google Scholar]
- 36.Li S, Kussie P, Ferguson KM. Structural basis for EGF receptor inhibition by the therapeutic antibody IMC-11F8. Structure. 2008;16:216–227. doi: 10.1016/j.str.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 37.Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 38.Cunningham BC, Wells JA. High-resolution epitope mapping of hGHreceptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 39.Cunningham BC, Wells JA. Comparison of a structural and a functional epitope. J Mol Biol. 1993;234:554–563. doi: 10.1006/jmbi.1993.1611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.