

Abstract
BACKGROUND AND PURPOSE
The passage of drugs across the blood–brain barrier (BBB) limits the efficacy of chemotherapy in brain tumours. For instance, the anticancer drug doxorubicin, which is effective against glioblastoma in vitro, has poor efficacy in vivo, because it is extruded by P-glycoprotein (Pgp/ABCB1), multidrug resistance-related proteins and breast cancer resistance protein (BCRP/ABCG2) in BBB cells. The aim of this study was to convert poorly permeant drugs like doxorubicin into drugs able to cross the BBB.
EXPERIMENTAL APPROACH
Experiments were performed on primary human cerebral microvascular endothelial hCMEC/D3 cells, alone and co-cultured with human brain and epithelial tumour cells.
KEY RESULTS
Statins reduced the efflux activity of Pgp/ABCB1 and BCRP/ABCG2 in hCMEC/D3 cells by increasing the synthesis of NO, which elicits the nitration of critical tyrosine residues on these transporters. Statins also increased the number of low-density lipoprotein (LDL) receptors exposed on the surface of BBB cells, as well as on tumour cells like human glioblastoma. We showed that the association of statins plus drug-loaded nanoparticles engineered as LDLs was effective as a vehicle for non-permeant drugs like doxorubicin to cross the BBB, allowing its delivery into primary and metastatic brain tumour cells and to achieve significant anti-tumour cytotoxicity.
CONCLUSIONS AND IMPLICATIONS
We suggest that our ‘Trojan horse’ approach, based on the administration of statins plus a LDL receptor-targeted liposomal drug, might have potential applications in the pharmacological therapy of different brain diseases for which the BBB represents an obstacle.
Keywords: blood–brain barrier, statins, ATP-binding cassette transporters, doxorubicin, nitric oxide, low-density lipoproteins receptor, liposomes, central nervous system tumours
Introduction
Primary CNS tumours, like glioblastoma, and CNS metastasis of solid tumours are poorly responsive to chemotherapy, as a consequence of the intrinsic tumour resistance and of the low penetration of many anticancer drugs across the blood–brain barrier (BBB). Both tumour cells and BBB endothelial cells are rich in ATP-binding cassette (ABC) transporters, like P-glycoprotein (Pgp/ABCB1), multidrug resistance-related proteins (MRPs/ABCCs) and breast cancer resistance protein (BCRP/ABCG2) that extrude drugs from the luminal side of endothelial cells back into the blood and out of the tumour cells (Declèves et al., 2006; Mercer et al., 2009; Robey et al., 2010). Doxorubicin, for instance, is a very effective anticancer drug against glioblastoma cells in vitro, but since it is a substrate of Pgp/ABCB1, MRP1/ABCC1 and BCRP/ABCG2, its delivery across the BBB is hampered and it is ineffective clinically at treating this tumour.
The effects of a pegylated liposomal doxorubicin (Doxil or Caelyx) have recently been investigated in glioblastoma, but the results obtained are unconvincing (Glas et al., 2007; Beier et al., 2009; Ananda et al., 2011). As alternative strategy, liposome-encapsulated drugs that have shown selective intratumour drug delivery, lower side effects and particular efficacy against aggressive and chemoresistant tumours (Jabr-Milane et al., 2008; Riganti et al., 2011), have been proposed as vehicles able to cross the BBB (through a diffusion process or a receptor-mediated endocytosis) and deliver their cargo into the CNS (Deelen and Loscher, 2007). Certain types of drug-containing nanoparticles adsorb apoB-100 and apo-E proteins from human low-density lipoprotein (LDLs; Kim et al., 2007), are recognized by the LDL receptor and achieve an efficient drug delivery into brain parenchyma (Michaelis et al., 2006; Nikanjam et al., 2007) because both neurons and glial cells express LDL receptors (Ambruosi et al., 2006).
In addition to increasing the delivery across the BBB, an ‘ideal’ chemotherapy for primary and metastatic tumours of CNS must simultaneously overcome the ABC transporters-dependent drug resistance of tumour cells. To meet these two requisites, in the present study we have investigated a nanoparticle-based approach – that is, the association of cholesterol-lowering drugs like statin plus a LDL receptor-targeting liposomal doxorubicin (termed ‘apo-Lipodox’; Kopecka et al., 2011).
Previous works from our group has shown that statins have chemosensitizing properties in epithelial tumours (Riganti et al., 2005; Riganti et al., 2006; Kopecka et al., 2011). Moreover statins are reported to exert cytotoxic effects against CNS tumour cells (Bababeygy et al., 2009; Yanae et al., 2011). To our knowledge, no data exist on the effects of statins on the metabolism and permeability of BBB cells. In this study, we investigated whether the association of statin with LDL receptor-targeted liposome-encapsulated doxorubicin simultaneously increases the drug permeability across the BBB and the cytotoxicity against CNS tumour cells, which would meet the two requisites of an optimal chemotherapy for brain tumours.
Methods
Chemicals
Plasticware for cell cultures was from Falcon (Becton Dickinson, Franklin Lakes, NJ, USA). Mevastatin, simvastatin, rhodamine 123 and Hoechst 33342 were purchased from Calbiochem (San Diego, CA, USA). Electrophoresis reagents were obtained from Bio-Rad Laboratories (Hercules, CA, USA); the protein content of cell monolayers and lysates was assessed with the BCA kit from Sigma Chemical Co (St. Louis, MO, USA). When not otherwise specified, all the other reagents were purchased from Sigma Chemical Co.
Cell lines
The hCMEC/D3 cells, a human brain microvascular endothelial cell line, were cultured as previously described (Weksler et al., 2005). Human glioblastoma U87-MG cells were cultured in DMEM medium, human neuroblastoma SJKNP cells and human breast cancer MDA-MB-231 cells were grown in RPMI-1640 medium, human lung cancer A549 cells were grown in Ham's F12 medium. These cell lines (purchased from American Type Cells Collection) were maintained with 1% v v-1 penicillin-streptomycin and 10% v v-1 FBS, at 37°C and 5% CO2 and were preliminarily characterized for the amount of Pgp, MRP1, BCRP and LDL receptor and for drug-resistance parameters (intracellular doxorubicin accumulation, extracellular release of lactate dehydrogenase, LDH) as reported below.
Cholesterol and geranylgeranyl pyrophosphate de novo synthesis
Cells grown to confluence in 35-mm diameter Petri dishes, incubated as reported in the Results section, were labelled for 24 h with 1 µCi·mL−1[3H]-acetate (3600 mCi·mmol−1; Amersham Bioscience, Piscataway, NJ, USA), then washed and subjected to lipid extraction with the methanol/hexane method (Kopecka et al., 2011). Samples were resuspended in 30 µL chloroform and separated by TLC, using 1:1 v v-1 ether/hexane as the mobile phase. Solutions of 10 mg·mL−1 cholesterol and 10 mg·mL−1 geranylgeranyl pyrophosphate (GGPP) were used as standards. After exposure for 1 h to an iodine-saturated atmosphere, the migrated spots were cut out and their radioactivity was measured by liquid scintillation, using a Tri-Carb Liquid Scintillation Analyzer (PerkinElmer, Waltham, MA, USA). Cholesterol and GGPP synthesis were expressed as pmol [3H]-cholesterol per 106 cells or [3H]-GGPP per 106 cells, according to the corresponding calibration curve.
Spectrophotometric measurement of membrane cholesterol
Cells were rinsed with 0.5 mL PBS, sonicated on ice with two bursts of 10 s and centrifuged at 100 000×g for 1 h at 4°C. The pellets (cell membrane fractions) were resuspended in 0.25 mL PBS and the cholesterol concentration was measured with an enzymatic colorimetric assay kit (OSR6516, Olympus System Reagent, Olympus Europe Holding GmbH, Hamburg, Germany), following the manufacturer's instructions. The absorbance was measured at 540/600 nm by an Olympus Analyzers spectrophotometer (Olympus Europe Holding GmbH). β-Methyl-cyclodextrin (10 mmol·L−1 for 3 h) was used as a cholesterol chelator (Kopecka et al., 2011). A 50 µL aliquot was used to determine the protein content with the BCA kit. The results are expressed in µg cholesterol·mg−1 membrane proteins, according to a previously prepared titration curve.
RhoA and RhoA kinase activity
To evaluate RhoA activity, the GTP-bound fraction, taken as an index of monomeric G-proteins activation (Laufs and Liao, 2000), was measured using the G-LISA™ RhoA Activation Assay Biochem Kit (Cytoskeleton Inc, Denver, CO, USA), according to the manufacturer's instructions. Absorbance was read at 450 nm, using a Packard EL340 microplate reader (Bio-Tek Instruments, Winooski, VT, USA). For each set of experiments, a titration curve was prepared, using serial dilution of the Rho-GTP positive control of the kit. Data are expressed as absorbance units·mg−1 cell proteins. RhoA kinase activity was measured using the CycLex Rho Kinase Assay Kit (CycLex Co, Nagano, Japan), following the manufacturer's instructions. The titration curve was prepared with serial dilutions of recombinant RhoA kinase (Rock2, MBL Inc, Woburn, MA, USA). Data are expressed as absorbance units·mg−1 cell proteins.
NF-kB activity
Nuclear proteins were extracted using the Nuclear Extract Kit (Active Motif, Rixensart, Belgium) and quantified. The activity of NF-kB was assessed by the TransAM™ Flexi NFκB Family kit (Active Motif), by adding 1 pmol of the biotinylated probe containing the NF-kB consensus site 5′-GGGACTTTCC-3′ to 10 µg of nuclear extract proteins. The absorbance at 450 nm was measured with a Packard EL340 microplate reader (Bio-Tek Instruments). For each set of experiments, a blank was prepared with bi-distilled water, and its absorbance was subtracted from that obtained in the presence of nuclear extracts. Data are expressed as absorbance units·mg−1 cell proteins.
NOS activity and nitrite measurement
Cells were detached by trypsin/EDTA, resuspended in 0.3 mL of assay buffer (20 mmol·L−1 HEPES, 0.5 mmol·L−1 EDTA, and 1 mmol·L−1 dithiothreitol DTT; pH 7.2) and sonicated. NOS (EC 1.14.13.49) activity was measured in 100 µg of cell lysates with the Ultrasensitive Colorimetric Assay for Nitric Oxide Synthase kit (Oxford Biomedical Research, Oxford, MI, USA). Results are expressed as nmol nitrite·min−1·mg−1 cell proteins. Nitrite production was measured by adding 0.15 mL of cell culture medium to 0.15 mL of Griess reagent in a 96-well plate. After a 10 min incubation at 37°C in the dark, the absorbance was detected at 540 nm with a Packard EL340 microplate reader. A blank was prepared for each experiment in the absence of cells, and its absorbance was subtracted from that obtained in the presence of cells. Nitrite concentration was expressed as nmol nitrite·mg−1 cell protein.
Western blot analysis
Cells were rinsed with lysis buffer (50 mmol·L−1 Tris, 10 mmol·L−1 EDTA, 1% v v−1 Triton-X100), supplemented with the protease inhibitor cocktail set III (80 µmol·L−1 aprotinin, 5 mmol·L−1 bestatin, 1.5 mmol·L−1 leupeptin, 1 mmol·L−1 pepstatin; Calbiochem), 2 mmol·L−1 phenylmethylsulfonyl fluoride (PMSF) and 1 mmol·L−1 sodium orthovanadate, then sonicated and centrifuged at 13 000×g for 10 min at 4°C. Extracts of protein, 20 µg, were subjected to SDS-PAGE and probed with the following antibodies: anti-phospho-Ser(176/180)-IKKα/β (Cell Signaling Technology Inc, Danvers, MA, USA), anti-IKKα/β (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), anti-IkB-α (Santa Cruz Biotechnology Inc.), anti-neuronal NOS (nNOS/NOS I, Transduction Laboratories, Lexington, KY, USA), anti-inducible NOS (iNOS/NOS II, Transduction Laboratories), anti-endothelial NOS (eNOS/NOS III, Transduction Laboratories), anti-phospho-(Ser 1177) eNOS (Cell Signaling Technology Inc), anti-Pgp/ABCB1 (Santa Cruz Biotechnology Inc.), anti-MRP1/ABCC1 (Abcam, Cambridge, MA, USA), anti-BCRP/ABCG2 (Santa Cruz Biotechnology Inc.), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Santa Cruz Biotechnology Inc.) This procedure was followed by exposure to a peroxidase-conjugated secondary antibody (Bio-Rad). The membranes were washed with Tris-buffered saline (TBS)-Tween 0.1% v v-1, and proteins were detected by enhanced chemiluminescence (PerkinElmer).
To assess the presence of nitrated proteins, the whole cell extract was immunoprecipitated with a rabbit polyclonal anti-nitrotyrosine antibody (Millipore, Billerica, MA, USA), using the PureProteome Protein A and Protein G Magnetic Beads (Millipore). Immunoprecipitated proteins were separated by SDS-PAGE and probed with anti-Pgp/ABCB1, anti-MRP1/ABCC1 or anti-BCRP/ABCG2 antibody. Whole cells lysates, 30 µg, were probed with the same antibodies before the immunoprecipitation step to check the total amount of Pgp, MRP1 and BCRP.
ABC transporters activity
To measure the ATPase activity of Pgp/ABCB1, MRP1/ABCC1 and BCRP/ABCG2, cells were lysed in buffer A (50 mmol·L−1 HEPES, 750 mmol·L−1 KCl, 200 mmol·L−1 sucrose, 10 mmol·L−1 NaHCO3; pH 7.4), supplemented with protease inhibitor cocktail set III, centrifuged at 13 000×g for 5 min, then at 100 000×g for 1 h at 4°C. The pellet was resuspended in 1 mL buffer B (20 mmol·L−1 HEPES, 160 mmol·L−1 KCl, 1 mmol·L−1 MgCl2, 1 mmol·L−1 CaCl2, 0.5% v v−1 Triton X-100; pH 7.4). To obtain membrane fractions enriched in Pgp/ABCB1, MRP1/ABCC1 and BCRP/ABCG2, 100 µg of membrane proteins were immunoprecipitated overnight with the specific primary antibodies, then washed twice with 1 mL buffer B, supplemented with 2 mmol·L−1 DTT; 50 µg of each sample were mixed with 2 mmol·L−1 ATP, 2.5 mmol·L−1 phosphoenolpyruvate, 7.5 U pyruvate kinase and 8.0 U LDH to check ATPase activity, as previously described (Doublier et al., 2008). The reaction was started by adding 0.25 mmol·L−1 NADH and was followed for 10 min, measuring the absorbance at 340 nm with a Packard EL340 microplate reader. The reaction kinetics was linear throughout the time of measurement. The NADH oxidation rate (expressed as µmol NADH oxidized·min−1·mg−1 cell proteins) of each sample was subtracted from the oxidation obtained in the absence of immunoprecipitated proteins. The ATP hydrolysis rate was calculated stoichiometrically and ATPase activity was expressed as µmol ATP hydrolysed·min−1·mg−1 cell proteins.
The efflux of rhodamine 123, taken as an index of Pgp/ABCB1 and MRP1/ABCC1 activity, and the intracellular accumulation of Hoechst 33342, taken as an index of BCRP/ABCG2 activity, were measured as reported previously (Riganti et al., 2011).
LDL receptor expression
Total RNA was extracted and reverse transcribed using the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). RT-PCR was carried out using IQ™ SYBR Green Supermix (Bio-Rad), according to the manufacturer's instructions. The same cDNA preparation was used for the quantification of LDL receptor and GAPDH, used as a housekeeping gene. The sequences of LDL receptor primers were 5′-TGAACTGGTGAGAGGACCAC-3′, 5′-TGTTCTTAAGCCGCCAGTTGTT-3′; the sequences of the GAPDH primers were 5′-TGGTCACCAGGGCTGCTT-3′, 5′-AGCTTCCCGTTCTCAGCCTT-3′. The relative quantification of each sample was obtained by comparing the LDL receptor PCR product with the GAPDH PCR product, with the Bio-Rad Software Gene Expression Quantitation (Bio-Rad).
For flow cytometry detection of surface LDL receptors, cells were washed with PBS, detached with Cell Dissociation Solution (Sigma) and resuspended at 5 × 105 cells·mL−1 in 1 mL RPMI medium containing 5% v v-1 FBS. Samples were washed with 0.25% w v-1 BSA-PBS, incubated with the primary antibody for LDL receptors (Abcam) for 45 min at 4°C, then washed twice and incubated with secondary FITC-conjugated antibody for 30 min at 4°C. After washing and fixation with paraformaldehyde 2% w v-1, the number of LDL receptors expressed on the surface was detected on 100 000 cells by a FACSCalibur system (Becton Dickinson), using the Cell Quest software (Becton Dickinson). Control experiments included incubation of cells with non-immune isotypic antibody, followed by the appropriate labelled secondary antibody.
Synthesis of LDL receptor-targeted liposome-encapsulated doxorubicin
LDL receptor-targeted liposome-encapsulated doxorubicin was prepared using anionic pegylated liposomes (COATSOME EL-01-PA, NOF Corporation, Tokyo, Japan) of the following composition: 1,2 distearoyl-sn-3-glycero-3-phosphoethanolamine conjugated with polyethylene glycol (PEG), cholesterol, 1,2 dipalmitoyl-sn-glycero-3-phosphocoline, 1,2 dipalmitoyl-sn-glycero-3-phosphoglycerol, at a molar ratio 4.2:11.4:15.2:11.4. For each preparation, 30 mg desiccated liposomes were incubated with 1.5 mmol·L−1 doxorubicin in sterile aqueous solution, according to the manufacturer's instructions. The residual non-encapsulated drug was removed by gel filtration in a Sephadex G-50 (Amersham Bioscience) column. The amount of encapsulated doxorubicin was quantified by diluting 50 µL of liposomal suspension in 0.5 mL of 1:1 v v-1 ethanol/HCl 0.3 N, sonicating the liposomes and measuring the fluorescence emitted by the drug with a LS-5 spectrofluorimeter (PerkinElmer). Excitation and emission wavelengths were 475 nm and 553 nm respectively. The encapsulation efficiency was calculated as described (Wong et al., 2004). The liposomes with an encapsulation efficiency higher than 85% (termed ‘Lipodox’) were collected, stored at a doxorubicin concentration of 0.5 mmol·L−1 and used to produce apoB100-conjugated doxorubicin-loaded liposomes (termed ‘apo-Lipodox’). With this aim, the following recombinant peptide from human apoB100 was used: DWLKAFYDKVAEKLKEAFRLTRKRGLKLA (LDL receptor-binding site is underlined; GenScript, Piscataway, NJ, USA). An aqueous solution of the peptide, 2.5 µmol·L−1, was added to liposomal doxorubicin, vortexed and incubated for 30 min at room temperature, with mild agitation. The unbound peptide was removed by dialysis as described previously (Nikanjam et al., 2007). The amount of peptide attached to the liposomes was detected by the QuantiPro BCA Assay kit (Sigma).
To assess the role of PEG in the peptide attachment, anionic non-pegylated liposomes (COATSOME EL01A, NOF Corporation), with the following composition: 1,2 dipalmitoyl-sn-glycero-3-phosphocoline, cholesterol, 1,2 dipalmitoyl-sn-glycero-3-phosphoglycerol (molar ratio of 3:4:3), were used in control experiments. To investigate the effects of liposomal shell alone, empty anionic pegylated liposomes (COATSOME EL-01-PA series), conjugated or not with the LDL receptor-targeted peptide, were used.
The size of the liposomes was evaluated by dynamic light scattering: 10 µL of liposome suspension were diluted in 1 mL of 120 mmol·L−1 NaCl solution and analysed with an ALV-NIBS dynamic light scattering instrument (Langen, Germany) provided with a Ne–He laser and an ALV-5000 multiple tau digital correlator. The scattered light intensity was recorded for 30 s on suspensions at 37°C. The hydrodynamic radius of liposomes was evaluated by using both the cumulant method and the CONTIN algorithm (Provencher, 1982). The mean radius of each liposomal preparation is presented in Table 1.
Table 1.
Dynamic light scatter analysis of liposomes
| Liposomesa | Acronymb | Doxorubicin | PEG | Peptide | Radius (mean ± SD) | P.I.c |
|---|---|---|---|---|---|---|
| COATSOME EL01A | – | Yes | No | Yes | 70.05 nm ± 22.45 nm | 0.187 |
| COATSOME EL-01-PA | Lipodox | Yes | Yes | No | 70.63 nm ± 28.55 nm | 0.183 |
| COATSOME EL-01-PA | apo-Lipodox | Yes | Yes | Yes | 72.05 nm ± 23.14 nm | 0.180 |
| COATSOME EL-01-PA | EL | No | Yes | No | 69.57 nm ± 27.35 nm | 0.171 |
| COATSOME EL-01-PA | apoEL | No | Yes | Yes | 71.77 nm ± 18.71 nm | 0.177 |
Commercial name (NOF Corporation).
Acronyms used in text and figures.
Polydispersity Index.
The morphological analysis of apo-Lipodox was performed on diluted samples (5 mg·mL−1 liposomes in 120 mmol·L−1 NaCl solution), using a Philips CM10 transmission electron microscope (TEM; Philips Amsterdam, The Netherlands), at an acceleration voltage of 80 kV. For each sample, a minimum of three microscopic fields was examined.
Permeability coefficient across the BBB cells
The permeability to inulin, taken as a parameter of the integrity of the tight junctions (Monnaert et al., 2004), was measured on hCMEC/D3 cells seeded at 50 000·cm−2 and grown for 7 days up to confluence in 6-multiwell Transwell devices (0.4 µm diameter pores-size, Corning Life Sciences, Chorges, France). Cells were incubated with or without mevastatin or simvastatin (0.1 µmol·L−1 for 24 h) or β-methyl-ciclodextrin (10 mmol·L−1 for 3 h), then the culture medium was replaced in the upper and lower chambers and 2 µCi·mL−1[14C]-inulin (10 mCi·mmol−1; PerkinElmer) was added to the upper chamber of Transwell. After 3 h, the medium in the lower chamber was collected and the amount of [14C]-inulin was measured using a Tri-Carb Liquid Scintillation Analyzer (PerkinElmer). Radioactivity was converted to nmol inulin·cm−2, using a calibration curve prepared previously.
For doxorubicin permeability, hCMEC/D3 cells seeded as reported above, were incubated with or without mevastatin or simvastatin (0.1 µmol·L−1 for 24 o 48 h). After this period, the culture medium was replaced in the upper and lower chambers and 5 µmol·L−1 doxorubicin or apo-Lipodox were added in the upper chamber of Transwell for 3 h, then the medium in lower chamber was collected and the amount of doxorubicin was measured fluorimetrically, using a LS-5 spectrofluorimeter (PerkinElmer). Excitation and emission wavelengths were 475 nm and 553 nm, respectively. Fluorescence was converted in nmol doxorubicin·cm−2, using a calibration curve previously set.
The permeability coefficients were calculated as described previously (Siflinger-Birnboim et al., 1987).
Intratumour drug accumulation and toxicity in co-culture models
The hCMEC/D3 cells (50 000·cm−2) were grown for 7 days up to confluence in 6-multiwell Transwell devices (0.4 µm diameter pores-size), whereas in the lower chamber, 500 000 cells (U87-MG, SJKNP, A549 or MDA-MB-231 cells) were seeded at day 4. At day 7, 0.1 µmol·L−1 simvastatin was added to the upper chamber medium for 48 h, when indicated. During the final 24 h, 5 µmol·L−1 doxorubicin or apo-Lipodox were added into the upper chamber of Transwell, then the inserts were removed and the extracellular medium of cells in the lower chamber was checked for the LDH activity, taken as an index of cytotoxicity and necrosis (Kopecka et al., 2011). Cells in the lower chamber were detached, and divided into two aliquots of 250 000 cells each: the first aliquot was lysed in 0.5 mL ethanol/HCl 0.3 N (1:1 v v-1) and analysed for the intracellular content of doxorubicin, as reported above. Results are expressed as nmol doxorubicin·mg−1 cell proteins. The second aliquot was lysed in 0.5 mL of caspase lysis buffer (20 mmol·L−1 HEPES/KOH, 10 mmol·L−1 KCl, 1.5 mmol·L−1 MgCl2, 1 mmol·L−1 EGTA, 1 mmol·L−1 EDTA, 1 mmol·L−1 DTT, 1 mmol·L−1 PMSF, 10 µg·mL−1 leupeptin; pH 7.5). Then 20 µg cell lysates were incubated for 1 h at 37°C with 20 µmol·L−1 of the fluorogenic substrate of caspase 3, DEVD-7-amino-4-methylcumarine (DEVD-AMC), in 0.25 mL caspase assay buffer (25 mmol·L−1 HEPES, 0.1% w v-1 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate CHAPS, 10% w v-1 sucrose, 10 mmol·L−1 DTT, 0.01% w v-1 egg albumin; pH 7.5). The reaction was stopped by adding 0.75 mL ice-cold 0.1% w v-1 trichloroacetic acid and the fluorescence of the AMC fragment released by active caspase-3 was read using a LS-5 spectrofluorimeter (PerkinElmer). Excitation and emission wavelengths were 380 nm and 460 nm respectively. Fluorescence was converted in pmol·µg−1 cell protein using a calibration curve prepared previously with standard solutions of AMC.
For fluorescence microscope analysis, tumour cells in the lower chamber were seeded on sterile glass coverslips and treated as reported above. At the end of the incubation time, samples were rinsed with PBS, fixed with 4% w v-1 paraformaldehyde for 15 min, washed three times with PBS and incubated with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) for 3 min at room temperature in the dark. Cells were washed three times with PBS and once with water, then the slides were mounted with 4 µL of Gel Mount Aqueous Mounting and examined with a Leica DC100 fluorescence microscope (Leica Microsystems GmbH, Wetzlar, Germany). For each experimental point, a minimum of five microscopic fields were examined.
Statistical analysis
All data in the text and figures are presented as means ± SD. The results were analysed by a one-way ANOVA. A P < 0.05 was considered significant.
Results
Statins induce the synthesis of NO in human primary BBB cells by a RhoA/RhoA kinase/NF-kB-dependent mechanism
As inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase enzyme, statins decrease the synthesis of cholesterol and isoprenoids like farnesyl pyrophosphate and GGPP in mammalian cells (Liao and Laufs, 2005). We observed in solid tumours that a decrease in GGPP reduces the activity of small GTPase of the RhoA family, activates the NF-kB transcription factor and up-regulates the inducible NO synthase (iNOS/NOS II) gene (Riganti et al., 2006; Riganti et al., 2008). The human brain microvascular endothelial hCMEC/D3 cells were sensitive to both mevastatin and simvastatin, two lipophilic statins that reduced the endogenous synthesis of cholesterol and GGPP in a dose- and time-dependent manner (Figure 1A). The lowest concentration that significantly decreased cholesterol and GGPP level after 24 h was 0.1 µmol·L−1 and this was chosen for all the subsequent experiments. The cholesterol chelator β-methyl-cyclodextrin significantly depleted the cell membrane of cholesterol (Supporting information Figure S1A) and in parallel disrupted the integrity of the tight junctions in hCMEC/D3 cells, as demonstrated by the increased permeability coefficient of inulin (Supporting information Figure S1B). In the experimental conditions used in this study, mevastatin and simvastatin reduced the amount of cholesterol in the cell membrane by a lesser extent than β-methyl-cyclodextrin (Supporting information Figure S1A) and did not affect the permeability to inulin (Supporting information Figure S1B).
Figure 1.
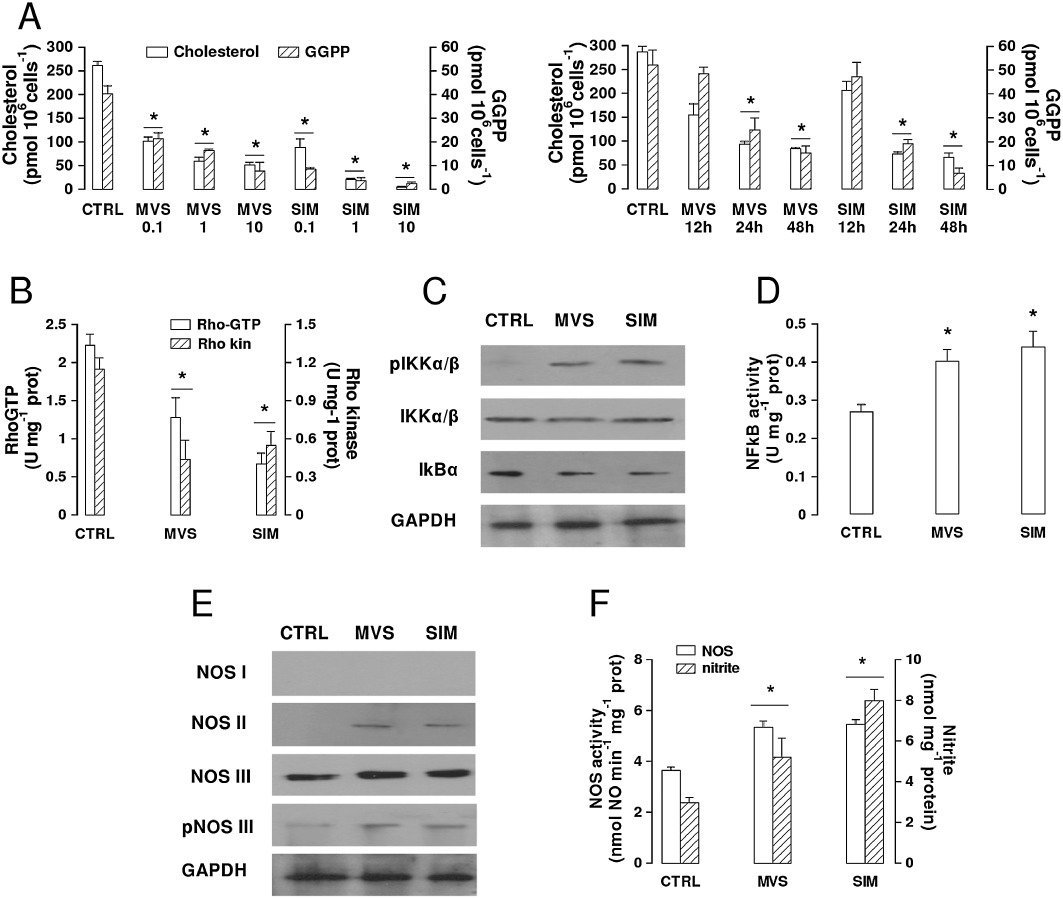
Effects of statins on cholesterol and GGPP synthesis, RhoA/RhoA kinase activity, NF-kB pathway and NO synthesis in BBB cells. (A) Cholesterol and GGPP synthesis. hCMEC/D3 cells were incubated in the absence (CTRL) or presence of different concentrations of 0.1, 1, 10 µmol·L−1 mevastatin (MVS) and simvastatin (SIM) for 24 h (left panel), or in the presence of 0.1 µmol·L−1statin for 12, 24 or 48 h (right panel). In the subsequent 24 h, the cells were labelled with [3H]-acetate, then cholesterol (open columns) and GGPP (hatched columns) synthesis was measured as reported under Methods. Data are presented as means ± SD (n= 3). Versus CTRL: *P < 0.05. For the subsequent experiments, 0.1 µmol·L−1MVS or SIM for 24 h was used. (B) RhoA/RhoA kinase activity. Samples were subjected to ELISA assays to measure the amount of RhoA-GTP (open bars) and the activity of RhoA kinase (hatched bars). The experiments were performed in duplicate, as described in the Methods section. Data are presented as means ± SD (n= 3). Versus CTRL: *P < 0.05. (C) Western blot detection of phospho-Ser(176/180)-IKK α/β, IKK α/β and IkB-α protein in extracts from hCMEC/D3 cells. The expression of GAPDH was used to check the equal protein loading. The figure is representative of three experiments with superimposable results. (D) NF-kB activity. The activity of NF-kB was detected in the nuclear extracts measuring the DNA-binding capacity of NF-kB on its target sequence (see Methods). Measurements were performed in duplicate and data are presented as means ± SD (n= 3). Versus CTRL: *P < 0.05. (E) Western blot detection of NOS isoforms (nNOS/NOS I; iNOS/NOS II; eNOS/NOS III) and of phospho-(Ser 1177)eNOS/NOS III protein in extracts from hCMEC/D3 cells. The expression of GAPDH was used to check the equal protein loading. The figure is representative of three experiments with superimposable results. (F) NO synthesis. NOS activity in cell lysates and nitrite accumulation in the extracellular medium were measured by use of spectrophotometric assays, as reported in Methods. Data are presented as means ± SD (n= 3). Versus CTRL: *P < 0.05.
In keeping with the decrease in GGPP, the activity of the geranylgeranylated protein RhoA, measured as the amount of active GTP-bound protein, was reduced by both statins, resulting in a lower activation of the downstream effector RhoA kinase (Figure 1B). In parallel, the amount of IKKα/β complex phosphorylated on serine 176/180, which was nearly undetectable in untreated hCMEC/D3 cells, was increased by mevastatin and simvastatin, without significant changes in the expression of IKKα/β protein (Figure 1C). Such an increase was accompanied by a decrease in the inhibitory protein Ik-Bα (Figure 1C) and by the activation of NF-kB, measured as the ability of the transcription factor translocated into the nucleus to bind its specific target sequence (Figure 1D).
Differently from the constitutive NOS isoforms (neuronal NOS/NOS I and endothelial NOS/NOS III), the inducible iNOS/NOS II is usually absent in non-stimulated cells and is up-regulated by different stimuli, like cytokines and bacterial lipopolysaccharide that induce the activation of NF-kB (Pautz et al., 2010). Statins increased the expression of NOS II; this was undetectable in untreated hCMEC/D3 cells (Figure 1E). The statins did not induce NOS I and did not change the expression of NOS III, but a slight increase in the amount of the active phospho-(Ser1177)-NOS III was detected following statins treatment (Figure 1E). As a consequence of these changes, the global enzymatic activity of NOS in cell lysates and the synthesis of NO, measured as the amount of the stable NO-derivative nitrite in culture supernatant, were significantly increased in hCMEC/D3 cells treated with mevastatin or simvastatin (Figure 1F).
Statins reduce the activity of Pgp and BCRP and increase the permeability of doxorubicin across the BBB cells by a NO-dependent mechanism
hCMEC/D3 cells constitutively express several ABC transporters (Carl et al., 2010), like Pgp/ABCB1, MRP1/ABCC1, BCRP/ABCG2 (Figure 2A). The immunoprecipitation of membrane extracts with an anti-nitrotyrosine antibody, followed by the Western blot detection with the specific primary antibodies, revealed that mevastatin and simvastatin promoted a nitration on tyrosine of Pgp/ABCB1 and BCRP/ABCG2, similar to the one produced by the NO donor sodium nitroprusside (Figure 2A). Both these transporters also showed a low level of nitration in untreated cells, whereas no nitration was detectable on MRP1/ABCC1 in each experimental condition. In parallel the ATPase activity of Pgp/ABCB1 and BCRP/ABCG2, but not that MRP1/ABCC1, was decreased in the presence of the statins (Figure 2B), suggesting that the nitration probably impairs the catalytic cycle of the pumps. Indeed, the intracellular retention of rhodamine 123 (Figure 2C) that is inversely related to the activity of Pgp/ABCB1, and the intracellular retention of Hoechst 33342 (Figure 2D) that is inversely related to the activity of BCRP/ABCG2, were both increased by the statins. Although the tyrosine nitration and the decrease in ATPase activity in Pgp/ABCB1 and BCRP/ABCG2 were already detectable after 24 h of incubation with mevastatin or simvastatin (Figure 2A and B) and were not lost after 48 h (data not shown), the effects of these changes on the activity of Pgp/ABCB1 and BCRP/ABCG2 were stronger after 48 h (Figure 2C and D).
Figure 2.
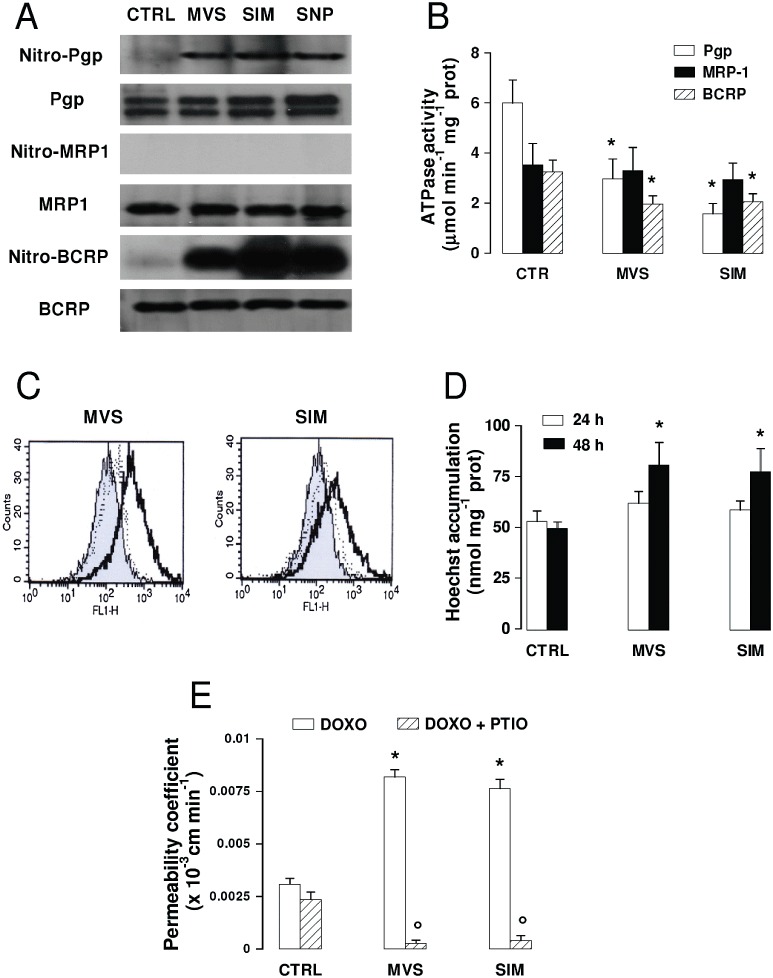
Effects of statins on ABC transporters activity and doxorubicin permeability across BBB cells. The hCMEC/D3 cells were incubated in the absence (CTRL) or presence of 0.1 µmol·L−1mevastatin (MVS) or simvastatin (SIM) for 24 h or 48 h, then subjected to the following investigations. (A) Nitration of ABC transporters. After a 24 h incubation, cells were lysed and the whole cell extracts were immunoprecipitated with an anti-nitrotyrosine polyclonal antibody. The immunoprecipitated proteins were subjected to Western blotting, using an anti-Pgp, an anti-MRP1 or an anti-BCRP antibody (see Methods). The NO donor sodium nitroprusside (100 µmol·L−1 for 24 h, SNP) was used as a positive control of nitration. The figure is representative of three experiments with similar results. (B) ATPase activity was measured spectrophotometrically after immunoprecipitation of Pgp, MRP1, BCRP from membrane fractions, as described in the Methods. Measurements were performed in duplicate and data are presented as means ± SD (n= 3). Versus CTRL: *P < 0.05. (C) Rhodamine assay. Cells were incubated for 20 min at 37°C with the fluorescent Pgp substrate rhodamine 123. The intracellular fluorescence was assessed by flow cytometry analysis in untreated cells (grey peak) and in cells treated with statins for 24 h (dotted line) or 48 h (continuous line). The figures shown here are representative of three similar experiments performed in duplicate. (D) Hoechst 33342 assay. Cells were incubated for 15 min at 37°C with Hoechst 33342, lysed and analysed fluorimetrically for the intracellular content of the dye. Measurements were performed in duplicate and data are presented as mean ± SD (n= 3). Versus CTRL: *P < 0.05. (E) Transport of doxorubicin across BBB monolayer. hCMEC/D3 cells were grown up to the confluence in Transwell insert, in fresh medium or in the presence of statins for 48 h, alone or co-incubated with the NO scavenger PTIO (100 µmol·L−1). 5 µmol·L−1doxorubicin was then added in the upper chamber. After 3 h, the medium was recovered by the lower chamber and the amount of doxorubicin was measured fluorimetrically. Measurements were performed in duplicate and data are presented as means ± SD (n= 4). Versus CTRL: *P < 0.05; versus condition without PTIO: °P < 0.001.
As doxorubicin is transported by Pgp/ABCB1, MRP1/ABCC1 and BCRP/ABCG2, its ability to cross the BBB cells is poor in vivo; also in hCMEC/D3 cells, the permeability coefficient was low (Figure 2E). However, the treatment with statins for 48 h significantly increased the permeability of the drug. The presence of the NO scavenger 2-phenyl-4,4,5,5,tetramethylimidazoline-1-oxyl 3-oxide (PTIO) prevented the increase in permeability to doxorubicin (Figure 2E), suggesting that NO levels play a crucial role in regulating the activity of Pgp/ABCB1 and BCRP/ABCG2 in hCMEC/D3 cells.
The association of statins plus a LDL receptor-targeted liposome-encapsulated doxorubicin further increases the drug transport across BBB cells
By lowering the de novo synthesis of cholesterol, statins force mammalian cells to expose the LDL receptor on their surface (Liao and Laufs, 2005). In hCMEC/D3 cells treated with mevastatin and simvastatin, the mRNA level for LDL receptors was significantly increased after 24 h (Figure 3A), a time point at which the synthesis of cholesterol was decreased (Figure 1A). The amount of LDL receptor protein on the cell surface was further increased by statins after 48 h (Figure 3B).
Figure 3.
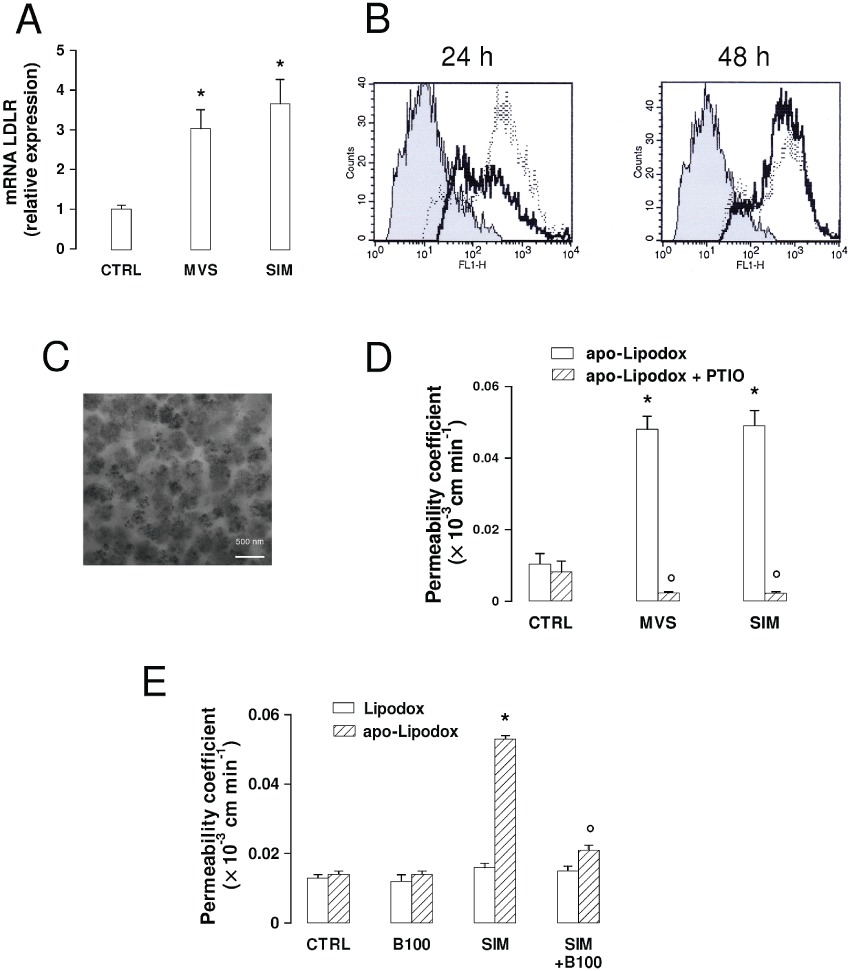
Effects of statins on the expression of LDL receptors and on the permeability of LDL receptor-targeted liposomal doxorubicin across the BBB. The hCMEC/D3 cells were incubated in the absence (CTRL) or presence of 0.1 µmol·L−1mevastatin (MVS) or simvastatin (SIM) for 24 h (A, B), 48 h (B, D and E), then subjected to the following investigations. (A) RT-PCR of LDL receptors. Total RNA was extracted, reverse transcribed and amplified by RT-PCR, as indicated in Methods. Measurements were performed in triplicate and data are presented as means ± SD (n= 3). Versus CTRL: *P < 0.005. (B) Flow cytometry analysis of surface LDL receptors in untreated cells (grey peak) or treated with MVS (continuous line) or SIM (dotted line) for 48 h. The figures shown here are representative of 3 similar experiments, performed in triplicate. (C) Apo-Lipodox imaging by TEM. The micrographs (28 500× magnification) are representative of three similar experiments. (D) Transport of apo-Lipodox across the BBB monolayer. The hCMEC/D3 cells were grown to the confluence in Transwell insert, in fresh medium or in the presence of statins or of the NO scavenger PTIO (100 µmol·L−1 for 48 h), then 5 µmol·L−1 apo-Lipodox was added in the upper chamber. After 3 h, the amount of the drug recovered by the lower chamber medium was measured fluorimetrically. Measurements were performed in duplicate and data are presented as means ± SD (n= 4). Versus CTRL: *P < 0.005; versus condition without PTIO: °P < 0.001. (E) Transport of Lipodox and apo-Lipodox across the BBB monolayer. The hCMEC/D3 cells were grown up to the confluence in Transwell insert, in fresh medium or in the presence of SIM, then 5 µmol·L−1 Lipodox or apo-Lipodox was added in the upper chamber. When indicated, the free LDL receptor-targeted apoB100 peptide (the same that was conjugated with apo-Lipodox), was co-incubated with the liposomes (100 µmol·L−1; B100). After 3 h, the amount of doxorubicin recovered by the lower chamber medium was measured fluorimetrically. Measurements were performed in duplicate and data are presented as means ± SD (n= 3). Versus CTRL: *P < 0.005; versus SIM: °P < 0.05.
We have previously shown that anionic pegylated liposomes loaded with doxorubicin and conjugated with a synthetic peptide containing the LDL receptor-binding site from human apo-B100 (the so-called ‘apo-Lipodox’) was internalized efficiently via a LDL receptor-mediated endocytosis and was less extruded by Pgp/ABCB1 in solid tumour cells (Kopecka et al., 2011). We thus applied apo-Lipodox that appeared as round-shaped particles with electron-dense areas due to the doxorubicin packed inside the liposomal shell (Figure 3C), to hCMEC/D3 cells untreated or treated with mevastatin and simvastatin for 48 h. In control cells, the permeability coefficient of apo-Lipodox was higher than that of free doxorubicin (see Figures 2E and 3D; comparing these data the significance of apo-Lipodox vs. doxorubicin was P < 0.005). The transport of apo-Lipodox across the BBB monolayer was even higher than that of free doxorubicin in cells exposed to statins (Figures 2E and 3D; significance of apo-Lipodox vs. doxorubicin in statin-treated cells: P < 0.001). Again, the addition of PTIO dramatically decreased the permeability of apo-Lipodox, suggesting that the NO levels – and the consequent status of nitration on ABC transporters – are also critical for the transport of apo-Lipodox in hCMEC/D3 cells.
In addition to the increased intracellular retention due to the inhibition of ABC transporters, we also investigated whether an increased uptake by LDL receptor-mediated endocytosis was critical in determining the greater permeability of apo-Lipodox. Apo-Lipodox exhibited a mean concentration of LDL receptor-targeted peptide of 20 ± 1.6 µg·µL−1, corresponding to about 3 × 1012 peptides·µL−1 (n= 3).
The permeabilities of Lipodox (liposomal doxorubicin without the LDL receptor-targeted peptide) and apo-Lipodox were similar in untreated hCMEC/D3 cells and were unaffected by an excess of free LDL receptor-targeted peptide under basal conditions (Figure 3E). However, apo-Lipodox crossed the BBB monolayer at a significantly higher rate when cells where pre-incubated with simvastatin, which increased the amount of LDL receptors (Figure 3A and B); this transport was dramatically decreased by the competing free LDL receptor-targeted peptide (Figure 3E). In contrast, Lipodox permeability was the same in the presence of simvastatin or of the competing peptide (Figure 3E), suggesting that the uptake of non-targeted liposomes was not dependent on the LDL receptor-mediated endocytosis.
To assess whether the presence of PEG influences the attachment of the LDL receptor-targeted peptide to the liposomal shell and/or the binding to the LDL receptor, parallel experiments were performed with anionic non-pegylated liposomes. These particles did not significantly differ in their radius (Table 1) or in the amount of peptide attached (22 ± 1.9 µg·µL−1; n= 3). The permeability of pegylated and non-pegylated apo-Lipodox was superimposable in both untreated and simvastatin-treated hCMEC/D3 cells (Supporting information Figure S2), suggesting that the presence of PEG did not interfere with the attachment of LDL receptor-targeted peptide or with the liposome uptake, either by simple endocytosis or LDL receptor-mediated endocytosis.
Empty liposomes may affect different intracellular pathways modulating the activity of Pgp/ABCB1 in epithelial cells (Riganti et al., 2011). To clarify whether empty liposomes affected the activation of NF-kB, the synthesis of nitrite, the activity of NOS enzyme, the induction of necrosis or apoptosis, we measured these parameters (Supporting information Figure S3A–C) in hCMEC/D3 cells, exposed to the same empty anionic pegylated liposomes used for apo-Lipodox, conjugated or not with the LDL receptor-targeted peptide. None of the liposomal formulations modified the above parameters compared to the untreated cells. Furthermore, when we co-incubated free doxorubicin with empty liposomes, with or without the LDL receptor-targeted peptide, in hCMEC/D3 cells pretreated or not with simvastatin (0.1 µmol·L−1 for 48 h), we did not detect a different permeability compared with free doxorubicin alone (Supporting information Figure S3D). We thus excluded a permeabilizing effect exerted by the liposomal shell itself.
The association of statins plus LDL receptor-targeted liposome-encapsulated doxorubicin is effective at delivering doxorubicin into tumour cells co-cultured with BBB cells
The abundance of ABC transporters on primary CNS tumours, on cerebral metastasis of epithelial solid tumours and on BBB cells determines a poor response to chemotherapy. To clarify if the association of statins and apo-Lipodox can overcome such resistance, we produced co-culture models by growing hCMEC/D3 cells on Transwell insert and tumour cells – that is, human glioblastoma multiforme U87-MG cells, human neuroblastoma SJKNP cells, human invasive breast cancer MD-MBA-231 cells and human non-small cells lung cancer A549 cells – in the lower chamber.
A preliminary characterization of these cell lines showed that they all expressed at various levels Pgp/ABCB1, MRP1/ABCC1 and BCRP/ABCG2 (Figure 4A), and that simvastatin did not modify the amount of these transporters (Figure 4A) but increased the surface LDL receptors (Figure 4B). Whereas the intracellular content of free doxorubicin was low in all the cell lines (giving a fluorescence similar to the autofluorescence of untreated cells, Figure 4C) and not sufficient to elicit cytotoxicity (Figure 4C), apo-Lipodox was accumulated to higher extent, exerting significant cell damage, as revealed by the release of LDH in the culture supernatant. The addition of simvastatin to free doxorubicin enhanced the drug retention and the LDH release compared to doxorubicin alone. For all the cell lines, the maximal increase in these parameters was achieved by the combination of simvastatin plus apo-Lipodox (Figure 4C).
Figure 4.
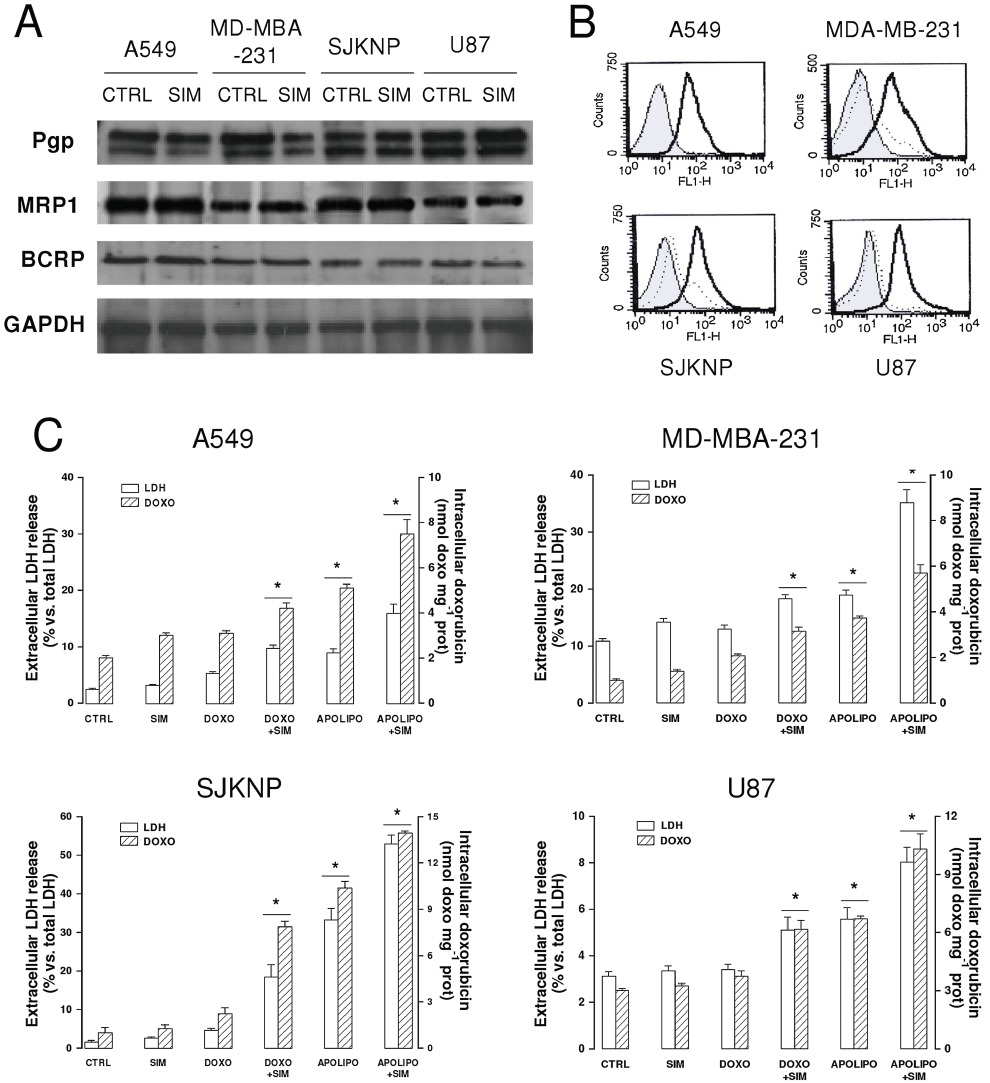
Characterization of different CNS- and non-CNS-derived tumour cells for ABC transporters expression, surface LDL receptors and resistance to doxorubicin. Human glioblastoma U87-MG cells, human neuroblastoma SJKNP cells, human breast cancer MDA-MB-231, human lung cancer A549 cells were grown in the absence (CTRL) or presence of simvastatin (0.1 µmol·L−1for 48 h; SIM). When indicated, 5 µmol·L−1doxorubicin (DOXO) or apo-Lipodox (APOLIPO) were added in the last 24 h. (A) Western blot analysis of ABC transporters. The expression of Pgp, MRP1 and BCRP on whole cells extracts was detected by Western blotting. The expression of GAPDH was used to check the equal protein loading. The figure is representative of three experiments with superimposable results. (B) Flow cytometry analysis of surface LDL receptors in untreated cells (dotted line) or SIM-treated cells (continuous line). Negative controls, with non-immune isotypic antibodies, are represented by the grey peak. The figures shown here are representative of two similar experiments, each performed in duplicate. (C) Intracellular doxorubicin accumulation and toxicity. Culture supernatant was checked for the extracellular activity of LDH; cells were detached and lysed to quantify the intracellular amount of doxorubicin, as described in Methods. The hatched columns in the conditions ‘CTRL’ and ‘SIM’ refer to the cell autofluorescence, measured in the absence of doxorubicin or apo-Lipodox administration. Measurements were performed in duplicate and data are presented as means ± SD (n= 3). Versus CTRL: *P < 0.05.
When added to the upper chamber of the Transwell containing confluent hCMEC/D3 cells on the insert and U87-MG in the lower chamber, doxorubicin poorly entered glioblastoma cells, as shown by the low red fluorescence in U87-MG cells recovered by the lower chamber (Figure 5A); the drug delivery into tumour cells was improved by pretreating BBB cells with simvastatin for 48 h followed by doxorubicin or apo-Lipodox in the last 24 h. The strongest fluorescent signal was achieved by the association of simvastatin (for 48 h) plus apo-Lipodox (in the last 24 h; Figure 5A).
Figure 5.
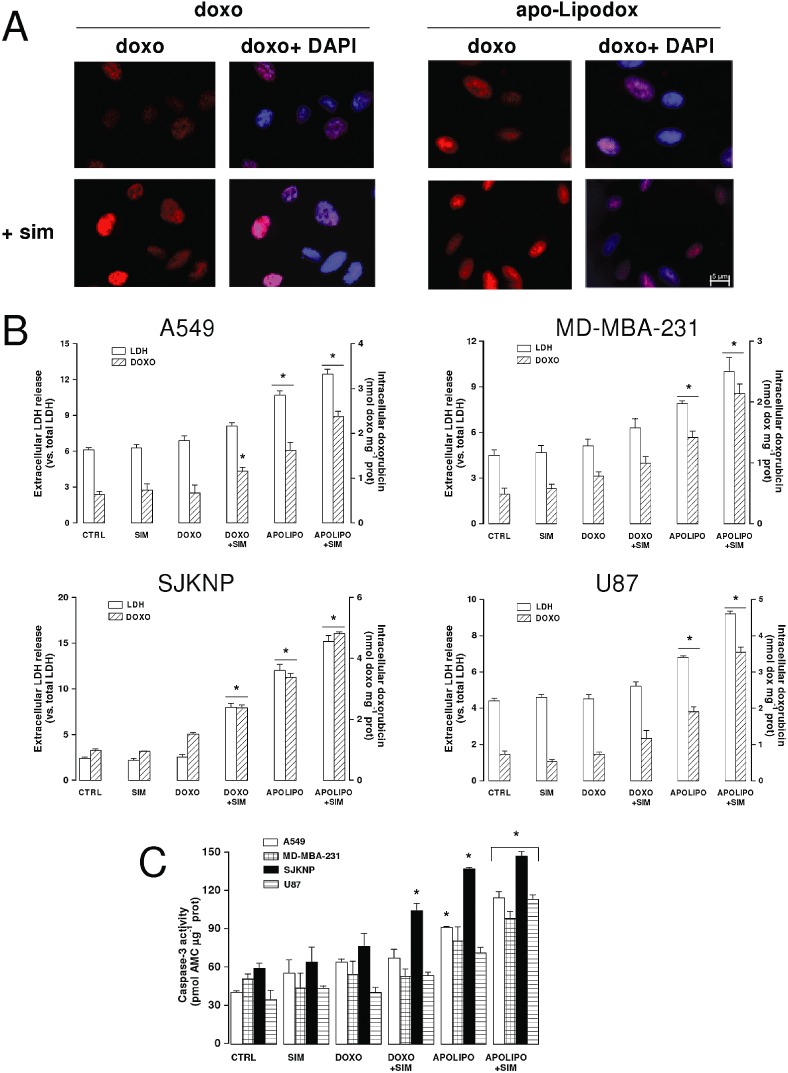
Drug delivery and antitumour efficacy of statins plus LDL receptor-targeted liposomal doxorubicin in co-culture models. The hCMEC/D3 cells were grown for 7 days up to confluence in Transwell inserts, whereas U87-MG, SJKNP, A549 or MDA-MB-231 cells were seeded at day 4 in the lower chamber. At day 0, supernatant in the upper chamber was replaced with fresh medium without (CTRL) or with simvastatin (0.1 µmol·L−1for 48 h; SIM). 5 µmol·L−1doxorubicin (DOXO) or apo-Lipodox (APOLIPO) were added in the upper chamber of Transwell in the last 24 h, then the following investigations were performed. (A) Microscope analysis of doxorubicin accumulation. U87-MG cells were seeded on sterile glass coverslips, treated as reported above, then analysed by fluorescence microscopy to detect the intracellular accumulation of doxorubicin. The cells were also counterstained with the nuclear fluorescent probe DAPI. The micrographs are representative of three experiments with similar results. (B) Culture supernatant of tumour cells was checked for the extracellular activity of LDH, cells were detached and lysed to quantify the intracellular amount of doxorubicin, as described in Methods. The hatched columns of the conditions ‘CTRL’ and ‘SIM’ refer to the cell autofluorescence, measured in the absence of doxorubicin or apo-Lipodox administration. Measurements were performed in duplicate and data are presented as means ± SD (n= 4). Versus CTRL: *P < 0.05. (C) Apoptosis induction. The activation of caspase-3 in tumour cells lysates was measured fluorimetrically as described in the Methods section. Measurements were performed in duplicate and data are presented as means ± SD (n= 3). Versus CTRL: *P < 0.05.
Quantitative fluorimetric analysis of intratumour doxorubicin confirmed that the drug content was significantly higher when simvastatin was combined with apo-Lipodox rather than with doxorubicin in all the cell lines co-cultured with hCMEC/D3 cells (Figure 5B). In keeping with these results, doxorubicin alone did not exert any relevant cytotoxic effect in tumour cells, in terms of LDH release (Figure 5B) and caspase-3 activation (Figure 5C). The association of free doxorubicin plus statin or the use of apo-Lipodox, two experimental conditions that increased the intracellular amount of doxorubicin in the cell lines without a BBB monolayer (Figure 4C), were significantly less effective in the presence of BBB monolayer (Figure 5B), although apo-Lipodox increased the release of LDH in all the cell lines and induced the activation of caspase-3 in A549 and SJKNP cells (Figure 5C). Also in BBB-tumour cells co-cultures, the association of simvastatin plus apo-Lipodox was the most effective at achieving a significant delivery of doxorubicin (Figure 5B) and inducing cell death by both necrosis (Figure 5B) and apoptosis (Figure 5C). Free doxorubicin co-incubated with empty liposomes, with or without the LDL receptor-targeted peptide, did not accumulate within U87-MG cells more than doxorubicin alone, either in the absence or in the presence of simvastatin (Supporting information Figure S4A). Similarly, the death of glioblastoma cells, measured as release of LDH and activation of caspase-3, was not increased by the addition of free doxorubicin to empty liposomes (Supporting information Figure S4B).
Discussion and conclusions
The therapeutic management of CNS tumours, currently based on surgery, radiotherapy and chemotherapy, is not completely safe and compatible with an acceptable quality of life. Chemotherapy is the first choice in disseminated tumours, such as invasive glioblastoma, high-risk medulloblastoma or multiple metastasis, but the percentage of success remains low. New targeted therapies, anti-angiogenic therapies or gene therapies show a real benefit only in limited groups of patients with known specific molecular defects (Sathornsumetee et al., 2007; Rossi et al., 2008). Thereby, the development of new pharmacological therapies for CNS tumours is still needed.
The low drug delivery across BBB and the low drug accumulation within the tumour are among the main factors that decrease the efficacy of chemotherapy in primary and metastatic brain tumours. In addition, having no fenestrations and many tight junctions, the brain microvascular endothelium has high levels of drug efflux pumps of the ABC transporter family (Declèves et al., 2006; Mercer et al., 2009; Robey et al., 2010). The latter have also been identified in CNS tumours (Nakagawa et al., 2009), where they contribute to the frequently observed chemoresistance. Therefore, an effective pharmacological therapy for CNS malignancies should overcome the extrusion of the drugs from both endothelial and tumour cells. In this perspective, we suggest a new combination approach, based on the association of statins plus LDL receptor-targeting liposomal drug, starting from two considerations: (i) both statins and liposomal drugs have chemosensitizer properties in solid tumours; (ii) liposomal drugs are delivered across the BBB more easily than free drugs.
We have previously observed that in cancer cells, statins increase the synthesis of NO, which inhibits ABC transporters efflux (Riganti et al., 2005; Riganti et al., 2006; Riganti et al., 2008), and decrease the amount of membrane cholesterol, which also lowers the activity of Pgp/ABCB1 (Kopecka et al., 2011).
In the past few years, statins have been proposed as drugs with potential benefits in cerebrovascular and neurodegenerative diseases, but to our knowledge, whether they have effects on the transport functions of brain microvascular endothelium has not been investigated. We found here that in human BBB cells, the lipophilic statins mevastatin and simvastatin decreased the synthesis of cholesterol and isoprenoids and reduced the amount of cholesterol in plasma membrane. This event has been previously linked to the loss of integrity of tight junctions and to the increased paracellular leakage of drugs across the BBB monolayer (Monnaert et al., 2004). In our work, we observed that a strong decrease in membrane cholesterol, caused by β-methyl-cyclodextrin, actually impaired the integrity of tight junctions in hCMEC/D3 cells, but the smaller variations of membrane cholesterol produced by statins did not. According to these data, it seems unlikely that statins – at least at the concentrations used in this study – increase the paracellular transport across BBB. At low (micromolar) concentrations, that is, in keeping with the Ki of statins for 3-hydroxy-3-methylglutaryl coenzyme A reductase (Liao and Laufs, 2005), mevastatin and simvastatin lowered the synthesis of isoprenoids in hCMEC/D3 cells and decreased the activity of RhoGTPase. In parallel, they increased the synthesis of NO, by up-regulating the IKK/NF-kB-mediated expression of iNOS/NOS II and by enhancing the phosphorylation on serine 1177 of eNOS/NOS III. The effects exerted by RhoA on NF-kB and NO synthesis are highly variable and depend on cell type (Nakata et al., 2007; Ahn et al., 2008; 32: Ye et al., 2008). Lovastatin has been reported to inhibit RhoA, stimulate the activity of IKK-α and NF-kB and enhance the expression of iNOS in rat glioma cells (33: Rattan et al., 2003); we hypothesize that such a mechanism occurs also in our model. It has been also shown that RhoA kinase reduces the stability of eNOS mRNA and prevents the Akt-dependent phosphorylation of eNOS (Rikitake and Liao, 2005). In accord with these observations, we detected an increased phosphorylation of eNOS in BBB cells treated with statins that inhibited RhoA kinase. This event, together with the up-regulation of iNOS isoform, led to a significant increase in NO levels and this suggests that this is the mechanism by which statins reduce the vascular tone and increase cerebral flow, as inhibitors of Rho kinase produce similar effects (Rikitake et al., 2005).
A high blood flow usually ensures better delivery of anticancer drugs within tumour areas and better tumour oxygenation enhances the cytotoxicity of many anticancer drugs including doxorubicin. In addition to these theoretical benefits, the increased production of NO elicited by the statins reduced the activity of at least two proteins – Pgp/ABCB1 and BCRP/ABCG2 – that are involved in the apical extrusion of drugs in hCMEC/D3 cells (Tai et al., 2009) and in vivo (Declèves et al., 2006; Mercer et al., 2009; Robey et al., 2010).
Activators of endogenous NO synthesis, such as lipopolysaccharide, TNF-α and endothelin-1, as well as the NO donor SNP, are known to affect Pgp/ABCB1 in rat brain capillaries, producing a decrease in the transporter expression and activity after a 3 h incubation (Bauer et al., 2007; Hartz et al., 2007) followed by an increase at 6 h (Bauer et al., 2007). Since the expression of Pgp/ABCB1 is subjected to rapid changes in BBB cells (Bauer et al., 2007; Hawkins et al., 2010), we cannot exclude the possibility that the NO synthesis induced by statins produces a decrease in Pgp/ABCB1 expression at earlier time points (e.g. less than 6 h), followed by a return to a ‘steady state’ at 24 h. Furthermore, the effects of NO are highly dependent on the species studied and on the amount and rate of NO released (Huerta et al., 2008). This variability may explain some apparently contrasting findings, that is, a high concentration of exogenous NO for 6 h increased the expression of Pgp/ABCB1 in rat endothelial cells (Bauer et al., 2007), whereas weak inducers of NOS II like statins did not produce any change of Pgp/ABCB1 expression in human hCMEC/D3 cells after 24 h (Figure 2A). Such low levels of endogenous NO, however, were sufficient to elicit a detectable nitration of the transporter, which decreased its activity. The reduction was even more pronounced after 48 h, indicating that statins induce a persistent inhibition of Pgp/ABCB1 activity after a longer time. As we considered the prolonged half-life of Pgp/ABCB1 in low-serum mediums (Zhang and Ling, 2000), the standard culture condition for hCMEC/D3 cells, we expected the reduction in activity of the ABC pumps to be higher at 48 h than at 24 h, although a clear nitration of the pump was already detectable after 24 h. The reversal of statins' effects by the NO scavenger PTIO suggests that the persistent inhibition of Pgp/ABCB1 and BCRP/ABCG2 activity was due to a continuous synthesis of NO by hCMEC/D3 cells.
Since in other models of BBB, the rate of drug efflux was NO independent (Salkeni et al., 2009), we cannot exclude a priori that statins also reduce the activity of ABC transporters by other mechanisms, for example, by decreasing the Pgp/ABCB1 expression and glycosylation (Sieczkowski et al., 2010) or by reducing the amount of cholesterol in the plasma membrane, an event that impairs the activity of Pgp/ABCB1 (Troost et al., 2004; Kopecka et al., 2011). Whatever the pleiotropic effects of statins on ABC transporters are, the final result of treating BBB cells with statins was a dramatic increase in the permeability of doxorubicin, a substrate of both Pgp/ABCB1 and BCRP/ABCG2.
In all mammalian cells, statins also up-regulate the transcription and surface exposure of LDL receptors, offering a flexible method of designing specific, nanoparticle-based targeted therapies. We have previously validated the efficacy of an engineered LDL receptor-targeted liposome-encapsulated doxorubicin (‘apo-Lipodox’) in solid tumours with high expression of LDL receptors (Kopecka et al., 2011). Besides being taken up more by LDL receptor-driven endocytosis (Kopecka et al., 2011), liposomal particles alter the lipid composition of the plasma membrane microdomains (i.e. lipid rafts) where Pgp/ABCB1 is active, interfere with ATP hydrolysis and substrate binding, and elicit a lower extrusion of doxorubicin in Pgp-overexpressing cells (Riganti et al., 2011). In untreated hCMEC/D3 cells, the permeability for apo-Lipodox was significantly greater than that of free doxorubicin, probably as a consequence of the decreased efflux via Pgp/ABCB1 and of the increased uptake. Since both the non-LDL receptor-targeted Lipodox and the LDL receptor-targeted apo-Lipodox had a higher uptake than free doxorubicin in the absence of statins, it is likely that liposomes have a facilitated entry within BBB cells by simple endocytosis. The uptake of apo-Lipodox was unaffected by an excess of the free LDL receptor-binding apoB100 peptide under basal conditions, suggesting that the LDL receptors present on the surface of untreated hCMEC/D3 cells were too few to play a critical role in the liposome uptake. In contrast, the LDL receptor became a helpful tool to increase the endocytosis of targeted liposomes when its levels increased on the cells' surface, that is, after exposure to statins. Simvastatin, which forced BBB cells to expose more LDL receptors and simultaneously decreased the activity of Pgp/ABCB1, indeed enhanced the apo-Lipodox uptake and reduced the apical efflux of doxorubicin, increasing the trans-cellular transport of the drug. Empty liposomal shells, which inhibited the activity of Pgp/ABCB1 in chemoresistant tumours (Riganti et al., 2011), did not exert any permeabilizing activity on the delivery of doxorubicin across the BBB, suggesting that the effects of the lipidic envelope alone may be tissue-dependent.
We show here that simvastatin enhanced the number of LDL receptors exposed and induced chemosensitization in different human tumour (glioblastoma, neuroblastoma, non-small cell lung cancer, breast cancer) cells with constitutive expression of Pgp/ABCB1, MRP1/ABCC1 and BCRP/ABCG2 and different degrees of resistance to doxorubicin. Pretreatment of the tumour cells with simvastatin followed by the apo-Lipodox showed the greatest cytotoxic efficacy not only in primary CNS tumours like glioblastoma, but also in epithelial tumours, such as lung and breast cancers cells, whose metastasis within the CNS are often unresponsive to therapy. In co-culture models, the application of free doxorubicin on the luminal side of hCMEC/D3 monolayer did not yield any strong drug delivery into the tumour cells growing under the BBB. Apo-Lipodox alone or the combination of simvastatin plus free doxorubicin increased drug delivery and toxicity. However, the extent of this increase was variable, depending on the different levels and activity of ABC transporters and LDL receptors, and on the existence of other mechanisms of resistance characterizing each cell line. The presence of specific tumour cells on the basal side of hCMEC/D3 cells may also affect the permeability of the BBB differently, determining different rate and kinetics of drug delivery. Only the pretreatment of the co-cultures with simvastatin followed by apo-Lipodox elicited a clear increase in doxorubicin delivery and toxicity, consisting of both necrotic and apoptotic death, in all the models investigated, including the highly chemoresistant glioblastoma.
A direct anti-tumour activity of statins against glioblastoma cells has been reported previously (Bababeygy et al., 2009; Yanae et al., 2011): for instance, in rat C6 glioma cells, 5 µmol·L−1 mevastatin and simvastatin induced apoptosis by decreasing the intracellular availability of GGPP and the activation of ERK1/2 and Akt (Yanae et al., 2011), suggesting that the effect could be mediated by a decrease in the activity of a geranylgeranylated protein. We did not detect direct cytotoxic effects of statins in co-cultured glioblastoma cells, perhaps because we used simvastatin at lower dose. It is noteworthy, however, that statins, at concentrations that are found in the blood of patients receiving anti-cholesterolemic therapy, enhanced the efficacy of a chemotherapeutic drug like doxorubicin against glioblastoma cells.
Phase I/II trials with pegylated liposomal doxorubicin have shown that the drug has very few side effects and is well tolerated, but does not offer a significant advantage in terms of overall survival or progression-free survival in comparison to the standard protocols based on radio-chemotherapy (Glas et al., 2007; Beier et al., 2009; Ananda et al., 2011). For this reason, chemical modifications and improvements of drug-loaded nanoparticles are under intensive investigations (Michaelis et al., 2006; Nikanjam et al., 2007; Guo et al., 2011; Wohlfart et al., 2011). The approach proposed in our work differs from the other studies on CNS-penetrating nanoparticles because it is more ‘physiological’: it takes advantage of the usual metabolic effects of therapeutic doses of statins – the increase in LDL receptors exposed and the synthesis of NO – and from the properties of liposomes, that induce permeability across the BBB and efficacy against drug-resistant tumours. We are currently testing this association in animal models bearing primary and metastatic CNS tumours, in order to determine the pharmacokinetic profile, the optimal administration regimen, the immunogenicity of apoLipodox, the antitumour effect and the presence of side effects. Testing our strategy in animals will add further important details about the molecular mechanisms of apo-Lipodox, inferred by these in vitro experiments, in order to validate the feasibility and efficacy of the association proposed.
The problem of poor permeability across the BBB is not limited to anticancer drugs, but also affects the delivery of agents used in epilepsy and neurodegenerative diseases. In a general perspective, our ‘Trojan horse’ approach, based on the administration of statins followed by a LDL receptor-targeting liposomal drug, might have potential applications in the pharmacological therapy of different CNS diseases.
Acknowledgments
We thank Mr Costanzo Costamagna, Department of Genetics, Biology and Biochemistry, University of Turin, for the technical assistance and Mr Silvano Panero, Department of Vegetal Biology, University of Turin, for the help with the TEM analysis. We are grateful to Dr Marco Minella and Prof Claudio Minero, Department of Analytical Chemistry, University of Turin, for having provided the dynamic light scattering facility and for the valuable advices in the interpretation of the data. We are indebted with Prof Davide Schiffer, Neuro-Bio-Oncology Center, Vercelli, Italy, for the helpful discussion and the critical revision of the manuscript.
This work has been supported by grants from Compagnia di San Paolo, Italy (Neuroscience Program; grant 2008.1136) and Italian Association for Cancer Research (AIRC; MFAG 11475) to CR. M L P-D is a recipient of an ERACOL Erasmus Mundus fellowship.
Glossary
- ABC
ATP-binding cassette
- AMC
amino-4-methylcumarine
- BBB
blood–brain barrier
- BCRP/ABCG2
breast cancer resistance protein
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GGPP
geranylgeranyl pyrophosphate
- LDL
low-density lipoprotein
- MRPs/ABCCs
multidrug resistance-related proteins
- PEG
polyethylene glycol
- Pgp/ABCB1
P-glycoprotein
- PTIO
phenyl-4,4,5,5,tetramethylimidazoline-1-oxyl 3-oxide
- TBS
Tris-buffered saline
Conflict of in interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effects of statins on membrane cholesterol and tight junctions integrity in hCMEC/D3 cells. (A) Cells were incubated in the absence (CTRL) or presence of 0.1 μmol·L−1 mevastatin (MVS) or simvastatin (SIM) for 24 h or 10 mmol·L−1 β-methyl cyclodextrin (MCD) for 3 h, then lysed and subjected to cell membranes isolation. The cholesterol content in membrane extracts was measured spectrophotometrically as described in the Methods section. Measurements were performed in duplicate and data are presented as means ± SD (n = 3). Vs CTRL: *P< 0.01. (B) Cells were seeded on Transwell insert, grown up to confluence for 7 days and incubated as reported in A; 2μCi·mL−1[14C]-inulin was then added in the upper chamber. After 3 h the medium was recovered by the lower chamber and the amount of [14C]-inulin was measured by liquid scintillation. Measurements were performed in duplicate and data are presented as means ± SD (n= 4). Vs CTRL: *P < 0.05.
Figure S2 Effects of pegylation on the permeability of LDL receptor-targeted liposomal doxorubicin across the BBB. Cells were grown up to the confluence for 7 days in Transwell insert, then incubated in the absence (CTRL) or presence of 0.1 μmol·L−1 simvastatin (SIM) for 48h. 5 μmol·L−1 apo-Lipodox obtained from anionic non-pegylated liposomes (COATSOME EL01A series; −PEG) or from anionic pegylated liposomes (COATSOME EL-01-PA series; + PEG) was added in the upper chamber and after 3 h the amount ofthe drug recovered by the lower chamber medium was measured fluorimetrically. Measurements were performed in duplicate and data are presented as means ± SD (n = 3). Vs CTRL: *P < 0.005.
Figure S3 Effects of empty liposomes on NF-kB activity, NO synthesis, cytotoxicity and permeability in hCMEC/D3 cells. The hCMEC/D3 cells were incubated in the absence (CTRL) or presence of 5 μmol·L−1 empty anionic pegylated liposomes (EL) or empty anionic pegylated LDL receptor-targeted conjugated-liposomes (apoEL) for 24 h, then subjected to the following investigations. (A) NF-kB activity. The activity of NF-kB was detected in the nuclear extracts measuring the DNA-binding capacity of NF-kB on its target sequence (see Methods). Measurements were performed in duplicate and data are presented as means ± SD (n = 3). (B) NO synthesis. NOS activity in cell lysates (open bars) and nitrite accumulationin the extracellular medium (hatched bars) were measured with spectrophotometric assays, as reported in the Methods section. Data are presented as means ± SD (n = 3). (C) Cytotoxicity. Culture supernatant of cells was checked for the extracellular activity of LDH (open bars), cells were detached and lysed to measure the activity of caspase-3 (hatched bars), as described in the Methods section. Measurements were performed in duplicate and data are presented as means ± SD (n =3). (D) Transport of doxorubicin across BBB monolayer in the presence of empty liposomes. The hCMEC/D3 cells were grown up tothe confluence for 7 days in Transwell insert, in fresh medium orin the presence of simvastatin (5 μmol·L−1 for 48 h; SIM); then 5 μmol·L−1 doxorubicin (DOXO), alone or co-incubated with 5 μmol·L−1 empty anionic pegylated liposomes (EL) or empty anionic pegylated LDL receptor-targeted conjugated-liposomes (apoEL), were added in the upper chamber. After 3 h the amount of the drug recovered by the lower chamber medium was measured fluorimetrically. Measurements were performed in duplicate and data are presented as means± SD (n = 3). Vs the corresponding condition without SIM: * P < 0.005.
Figure S4 Effects of empty liposomes on the delivery and cytotoxicity of free doxorubicin in co-culture models. The hCMEC/D3 cells were grown for 7 days up to confluence in Transwell inserts, whereas U87-MG cells were seeded at day 4 in the lower chamber. At day 0, supernatant in the upper chamber was replaced with fresh medium without (CTRL) or with simvastatin (0.1μmol·L−1 for 48 h; SIM). 5μmol·L−1 doxorubicin (DOXO), alone or co-incubated with 5 μmol·L−1 empty anionic pegylated liposomes (EL) or empty anionic pegylated LDL receptor-targeted conjugated-liposomes (apoEL), were added in the upper chamber of Transwell in the last 24 h, then the following investigations were performed. (A) U87-MG cells were lysed in ethanol/HCl and the intracellular amount of doxorubicin was measured fluorimetrically (see Methods section). Measurements were performed in duplicate and data are presented as means ± SD (n = 3). (B) The culture supernatant of tumor cells was checked for the extracellular activity of LDH (open bars), cells were detached and lysed to measure the activity of caspase-3 (hatched bars), as described in the Methods section. Measurements were performed in duplicate and data are presented as means± SD (n = 3).
References
- Ahn KS, Sethi G, Chaturvedi MM, Aggarwal BB. Simvastatin, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, suppresses osteoclastogenesis induced by receptor activator of nuclear factor-kB ligand through modulation of NF-kB pathway. Int J Cancer. 2008;123:1733–1740. doi: 10.1002/ijc.23745. [DOI] [PubMed] [Google Scholar]
- Ambruosi A, Khalansky AS, Yamamoto H, Gelperina SE, Begley DJ, Kreuter J. Biodistribution of polysorbate 80-coated doxorubicin-loaded [14C]-poly(butyl cyanoacrylate) nanoparticles after intravenous administration to glioblastoma-bearing rats. J Drug Target. 2006;14:97–105. doi: 10.1080/10611860600636135. [DOI] [PubMed] [Google Scholar]
- Ananda S, Nowak AK, Cher L, Dowling A, Brown C, Simes J, et al. Phase 2 trial of temozolomide and pegylated liposomal doxorubicin in the treatment of patients with glioblastoma multiforme following concurrent radiotherapy and chemotherapy. J Clin Neurosci. 2011;18:1444–1448. doi: 10.1016/j.jocn.2011.02.026. [DOI] [PubMed] [Google Scholar]
- Bababeygy SR, Polevaya NV, Youssef S, Sun A, Xiong A, Prugpichailers T, et al. HMG-CoA reductase inhibition causes increased necrosis and apoptosis in an in vivo mouse glioblastoma multiforme model. Anticancer Res. 2009;29:4901–4908. [PubMed] [Google Scholar]
- Bauer B, Hartz AMS, Miller DS. Tumor necrosis factor α and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol Pharmacol. 2007;71:667–675. doi: 10.1124/mol.106.029512. [DOI] [PubMed] [Google Scholar]
- Beier CP, Schmid C, Gorlia T, Kleinletzenberger C, Beier D, Grauer O, et al. RNOP-09: pegylated liposomal doxorubicine and prolonged temozolomide in addition to radiotherapy in newly diagnosed glioblastoma-a phase II study. BMC Cancer. 2009;9:308–318. doi: 10.1186/1471-2407-9-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl SM, Lindley DJ, Couraud PO, Weksler BB, Romero I, Mowery SA, et al. ABC and SLC transporter expression and pot substrate characterization across the human CMEC/D3 blood-brain barrier cell line. Mol Pharm. 2010;7:1057–1068. doi: 10.1021/mp900178j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declèves X, Amiel A, Delattre JY, Scherrmann JM. Role of ABC transporters in the chemoresistance of human gliomas. Curr Cancer Drug Targets. 2006;6:433–445. doi: 10.2174/156800906777723930. [DOI] [PubMed] [Google Scholar]
- Deelen JF, Loscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res. 2007;13:1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]
- Doublier S, Riganti C, Voena C, Costamagna C, Aldieri E, Pescarmona G, et al. RhoA silencing reverts the resistance to doxorubicin in human colon cancer cells. Mol Cancer Res. 2008;6:1607–1620. doi: 10.1158/1541-7786.MCR-08-0251. [DOI] [PubMed] [Google Scholar]
- Glas M, Koch H, Hirschmann B, Jauch T, Steinbrecher A, Herrlinger U, et al. Pegylated liposomal doxorubicin in recurrent malignant glioma: analysis of a case series. Oncology. 2007;72:302–307. doi: 10.1159/000113052. [DOI] [PubMed] [Google Scholar]
- Guo L, Fan L, Pang Z, Ren J, Ren Y, Li J, et al. TRAIL and doxorubicin combination enhances anti-glioblastoma effect based on passive tumor targeting of liposomes. J Control Release. 2011;154:93–102. doi: 10.1016/j.jconrel.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Hartz AMS, Bauer B, Fricker G, Miller DS. Rapid modulation of P-glycoprotein-mediated transport at the blood-brain barrier by tumor necrosis factor-α and lipopolysaccharide. Mol Pharmacol. 2007;69:462–470. doi: 10.1124/mol.105.017954. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Sykes DB, Miller DS. Rapid, reversible modulation of blood-brain barrier P-glycoprotein transport activity by vascular endothelial growth factor. J Neurosci. 2010;30:1417–1425. doi: 10.1523/JNEUROSCI.5103-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta S, Chilka S, Bonavida B. Nitric oxide donors: novel cancer therapeutics. Int J Oncol. 2008;33:909–927. [PubMed] [Google Scholar]
- Jabr-Milane LS, van Vlerken LE, Yadav S, Amiji MM. Multi-functional nanocarriers to overcome tumour drug resistance. Cancer Treat Rev. 2008;34:592–602. doi: 10.1016/j.ctrv.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Andrieux K, Gil S, Taverna M, Chacun H, Desmaele D, et al. Translocation of poly(ethylene glycol-co-hexadecyl)cyanoacrylate nanoparticles into rat brain endothelial cells: role of apolipoproteins in receptor-mediated endocytosis. Biomacromolecules. 2007;8:793–799. doi: 10.1021/bm060711a. [DOI] [PubMed] [Google Scholar]
- Kopecka J, Campia I, Olivero P, Pescarmona G, Ghigo D, Bosia A, et al. A LDL-masked liposomal-doxorubicin reverses drug resistance in human cancer cells. J Control Release. 2011;149:196–205. doi: 10.1016/j.jconrel.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Laufs U, Liao JK. Targeting Rho in cardiovascular disease. Circ Res. 2000;87:526–528. doi: 10.1161/01.res.87.7.526. [DOI] [PubMed] [Google Scholar]
- Liao K, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer RW, Tyler MA, Ulasov IV, Maciej S. Targeted therapies for malignant glioma. BioDrugs. 2009;23:25–35. doi: 10.2165/00063030-200923010-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis K, Hoffmann MM, Dreis S, Herbert E, Alyautdin RN, Michaelis M, et al. Covalent linkage of apolipoprotein E to albumin nanoparticles strongly enhances drug transport into the brain. J Pharmacol Exp Ther. 2006;317:1246–1253. doi: 10.1124/jpet.105.097139. [DOI] [PubMed] [Google Scholar]
- Monnaert V, Betbeder D, Fenart L, Bricout H, Lenfant AM, Landry C, et al. Effects of γ- and hydroxypropyl- γ -cyclodextrins on the transport of doxorubicin across an in vitro model of blood-brain barrier. J Pharmacol Exp Ther. 2004;311:1115–1120. doi: 10.1124/jpet.104.071845. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Ido K, Sakuma T, Takeuchi H, Sato K, Kubota T. Prognostic significance of the immunohistochemical expression of O6-methylguanine-DNA methyltransferase, P-glycoprotein, and multidrug resistance protein-1 in glioblastomas. Neuropathology. 2009;29:379–388. doi: 10.1111/j.1440-1789.2008.00983.x. [DOI] [PubMed] [Google Scholar]
- Nakata S, Tsutsui M, Shimokawa H, Yamashita T, Tanimoto A, Tasaki H, et al. Statin treatment upregulates vascular neuronal nitric oxide synthase through Akt/NF-kB pathway. Arterioscler Thromb Vasc Biol. 2007;27:92–98. doi: 10.1161/01.ATV.0000251615.61858.33. [DOI] [PubMed] [Google Scholar]
- Nikanjam M, Blakely EA, Bjornstad KA, Shu X, Budinger TF, Forte TM. Synthetic nano-low density lipoprotein as targeted drug delivery vehicle for glioblastoma multiforme. Int J Pharm. 2007;328:86–94. doi: 10.1016/j.ijpharm.2006.07.046. [DOI] [PubMed] [Google Scholar]
- Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide. 2010;23:75–93. doi: 10.1016/j.niox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Provencher SW. A constrained regularization method for inverting data represented by linear algebraic or integral equations. Comput Phys Commun. 1982;27:213–227. [Google Scholar]
- Rattan R, Giri S, Singh AK, Singh I. RhoA negatively regulates cytokine-mediated inducible nitric oxide synthase expression in brain derived transformed cell lines: negative regulation of IKKa. Free Radic Biol Med. 2003;39:1037–1050. doi: 10.1016/s0891-5849(03)00459-3. [DOI] [PubMed] [Google Scholar]
- Riganti C, Miraglia E, Viarisio D, Costamagna C, Pescarmona G, Ghigo D, et al. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res. 2005;65:516–525. [PubMed] [Google Scholar]
- Riganti C, Orecchia S, Pescarmona G, Betta PG, Ghigo D, Bosia A. Statins revert doxorubicin resistance via nitric oxide in malignant mesothelioma. Int J Cancer. 2006;119:17–27. doi: 10.1002/ijc.21832. [DOI] [PubMed] [Google Scholar]
- Riganti C, Doublier S, Costamagna C, Aldieri E, Pescarmona G, Ghigo D, et al. Activation of nuclear factor-kappa B pathway by simvastatin and RhoA silencing increases doxorubicin cytotoxicity in human colon cancer HT29 cells. Mol Pharmacol. 2008;74:476–484. doi: 10.1124/mol.108.045286. [DOI] [PubMed] [Google Scholar]
- Riganti C, Voena C, Kopecka J, Corsetto P, Montorfano G, Enrico E, et al. Liposome-encapsulated doxorubicin reverses drug-resistance by inhibiting P-glycoprotein in human cancer cells. Mol Pharm. 2011;8:683–700. doi: 10.1021/mp2001389. [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res. 2005;97:1232–1235. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, et al. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. 2005;36:2251–2257. doi: 10.1161/01.STR.0000181077.84981.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Massey PR, Amiri-Kordestani L, Bates SE. ABC transporters: unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med Chem. 2010;10:625–633. doi: 10.2174/187152010794473957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Caracciolo V, Russo G, Reiss K, Giordano A. Medulloblastoma: from molecular pathology to therapy. Clin Cancer Res. 2008;14:971–976. doi: 10.1158/1078-0432.CCR-07-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salkeni MA, Lynch JL, Otamis-Price T, Banks WA. Lipopolysaccharide impairs blood–brain barrier P-glycoprotein function in mice through prostaglandin- and nitric oxide-independent pathways. J Neuroimmune Pharmacol. 2009;4:276–282. doi: 10.1007/s11481-008-9138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110:13–24. doi: 10.1002/cncr.22741. [DOI] [PubMed] [Google Scholar]
- Sieczkowski E, Lehner C, Ambros PF, Hohenegger M. Double impact on pglycoprotein by statins enhances doxorubicin cytotoxicity in human neuroblastoma cells. Int J Cancer. 2010;126:2025–2035. doi: 10.1002/ijc.24885. [DOI] [PubMed] [Google Scholar]
- Siflinger-Birnboim A, Del Vecchio PJ, Cooper JA, Blumenstock FA, Shepard JM, Malik AB. Molecular sieving characteristics of the cultured endothelial monolayer. J Cell Physiol. 1987;132:111–117. doi: 10.1002/jcp.1041320115. [DOI] [PubMed] [Google Scholar]
- Tai LM, Loughlin AJ, Male DK, Romero IA. P-glycoprotein and breast cancer resistance protein restrict apical-to-basolateral permeability of human brain endothelium to amyloid-β. J Cereb Blood Flow Metab. 2009;29:1079–1083. doi: 10.1038/jcbfm.2009.42. [DOI] [PubMed] [Google Scholar]
- Troost J, Lindenmaier H, Haefeli WE, Weiss J. Modulation of cellular cholesterol alters P-glycoprotein activity in multidrug-resistant cells. Mol Pharmacol. 2004;66:1329–1332. doi: 10.1124/mol.104.002329. [DOI] [PubMed] [Google Scholar]
- Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- Wohlfart S, Khalansky AS, Gelperina S, Begley D, Kreuter J. Kinetics of transport of doxorubicin bound to nanoparticles across the blood–brain barrier. J Control Release. 2011;154:103–107. doi: 10.1016/j.jconrel.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Wong HL, Bendayan R, Rauth AM, Wu XY. Development of solid lipid nanoparticles containing ionically complexed chemotherapeutic drugs and chemosensitizers. J Pharm Sci. 2004;93:1993–2008. doi: 10.1002/jps.20100. [DOI] [PubMed] [Google Scholar]
- Yanae M, Tsubaki M, Satou T, Itoh T, Imano M, Yamazoe Y, et al. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J Exp Clin Cancer Res. 2011;30:74–82. doi: 10.1186/1756-9966-30-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Martinez JD, Perez-Polo RJ, Lin Y, Uretsky BF, Birnbaum Y. The role of eNOS, iNOS, and NF-kB in upregulation and activation of cyclooxygenase-2 and infarct size reduction by atorvastatin. Am J Physiol Heart Circ Physiol. 2008;295:H343–H351. doi: 10.1152/ajpheart.01350.2007. [DOI] [PubMed] [Google Scholar]
- Zhang W, Ling V. Cell-cycle–dependent turnover of P-glycoprotein in multidrug-resistant cells. J Cell Physiol. 2000;184:17–26. doi: 10.1002/(SICI)1097-4652(200007)184:1<17::AID-JCP2>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.