

Abstract
BACKGROUND AND PURPOSE
The accumulation of hypoxia-inducible factor-1α (HIF-1α) is under the influence of hydrogen sulfide (H2S), which regulates hypoxia responses. The regulation of HIF-1α accumulation by H2S has been shown, but the mechanisms for this effect are largely elusive and controversial. This study aimed at addressing the controversial mechanisms for and the functional importance of the interaction of H2S and HIF-1α protein.
EXPERIMENTAL APPROACH
HIF-1α protein levels and HIF-1α transcriptional activity were detected by Western blotting and luciferase assay. The mechanisms for H2S-regulated HIF-1α protein levels were determined using short interfering RNA transfection, co-immunoprecipitation and 7-methyl-GTP sepharose 4B pull-down assay. Angiogenic activity was evaluated using tube formation assay in EA.hy926 cells.
KEY RESULTS
The accumulation of HIF-1α protein under hypoxia (1% O2) or hypoxia-mimetic conditions was reversed by sodium hydrosulfide (NaHS). This effect of NaHS was not altered after blocking the ubiquitin-proteasomal pathway for HIF-1α degradation; however, blockade of protein translation with cycloheximide abolished the effect of NaHS on the half-life of HIF-1α protein. Knockdown of eukaryotic translation initiation factor 2α (eIF2α) suppressed the effect of NaHS on HIF-1α protein accumulation under hypoxia. NaHS inhibited the expression of VEGF under hypoxia. It also decreased in vitro capillary tube formation and cell proliferation of EA.hy926 cells under hypoxia, but stimulated the tube formation under normoxia.
CONCLUSIONS AND IMPLICATIONS
H2S suppresses HIF-1α translation by enhancing eIF2α phosphorylation under hypoxia. The interaction of H2S and HIF-1α inhibits the angiogenic activity of vascular endothelial cells under hypoxia through the down-regulation of VEGF.
Keywords: eukaryotic translation initiation factor 2α, hydrogen sulfide, hypoxia, hypoxia-inducible factor-1α
Introduction
Hydrogen sulfide (H2S) is a gasotransmitter, endogenously produced by cystathionine γ-lyase (CSE) and cystathionine β-synthase (Wang, 2003; 2010). Endogenous H2S participates in both physiological regulation and pathophysiological processes of different mammalian systems (Yang et al., 2008; Gil et al., 2011). The therapeutic value of exogenous H2S has been shown for dealing with different diseases (Esechie et al., 2009; Ekundi-Valentim et al., 2010; Pouokam and Diener, 2011).
A master regulator of cellular responses to changes in oxygen levels is hypoxia-inducible factor-1 (HIF-1), a transcription factor which activates more than 100 genes involved in angiogenesis, glucose metabolism, cell survival and metastasis (Semenza, 2006; Spagnuolo et al., 2011). The dominant form of HIF-1 is a heterodimer consisting of the inducibly regulated HIF-1α subunit and the constitutively expressed HIF-1β subunit (Wang et al., 1995; Yang et al., 2004). Under normoxia, the HIF-1α gene is continuously transcribed and translated, but the level of this protein is very low due to its rapid degradation via the ubiquitin-proteasomal pathway mediated by prolyl hydroxylase (Semenza, 2004). In contrast, hypoxia inhibits prolyl hydroxylase activity and consequently results in the accumulation of HIF-1α protein (Yee Koh et al., 2008). A wide range of stimuli and cytokines have been shown to affect HIF-1α protein stability, including two gasotransmitters, NO (Hagen et al., 2003; Metzen et al., 2003) and carbon monoxide (CO) (Choi et al., 2010). One of the two key control elements of HIF-1α gene translation is the PI3K-Akt-mammalian target of rapamycin (mTOR) pathway responsible for the initiation of cap-dependent HIF-1α gene translation (Holcik and Sonenberg, 2005; Spriggs et al., 2010). The other element is eukaryotic translation initiation factor 2α (eIF2α), a component of the eIF-2 ternary complex. eIF2α is responsible for the transformation of GDP to GTP, an essential step for translation commencement. Phosphorylation of eIF2α at Ser51 prevents the re-formation of the eIF-2 ternary complex and thus suppresses HIF-1α translation (Rocha, 2007; Yee Koh et al., 2008). Certain anti-tumour compounds decrease HIF-1α translation due to eIF2α phosphorylation (Jung et al., 2009; Zhang et al., 2010).
H2S has been shown to up-regulate HIF-1 protein levels and activity under normoxia in Caenorhabditis elegans or down-regulate HIF-1α protein levels and activity under hypoxia in cultured cells (Budde and Roth, 2009; Kai et al., 2012). The molecular mechanisms through which H2S mediates HIF-1α protein level are largely elusive and controversial. Elucidation of the mechanisms for H2S-induced alteration in HIF-1α protein level is critical for our understanding of the role and mode of action of H2S in hypoxia. The present study was undertaken to determine whether and how H2S regulates HIF-1α protein level under hypoxia and hypoxia-mimetic conditions in different cell lines. We also investigated the effect of H2S on HIF-1α translation and degradation and the relative importance of these two processes. HIF-1α transcriptional activity and the effect of H2S on angiogenic activity of endothelial cells were further tested.
Methods
Cell culture and reagents
HEK293T, Hep3B and EA.hy926 cells were obtained from ATCC (Manassas, VA, USA) and cultured in DMEM supplemented with 10% heat-inactivated FBS, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin at 37°C in a humidified atmosphere of 5% CO2 and 21% O2. For hypoxic treatment, the cells were incubated in a chamber (STEMCELL Technologies, Vancouver, BC, Canada) at 37°C with a gas mixture (1% O2, 5% CO2 and 94% N2). Sodium hydrosulfide (NaHS), cobalt chloride (CoCl2), desferrioxamine (DFX) and cycloheximide (CHX) were obtained from Sigma (Oakville, ON, Canada). Dimethyloxalyl glycine (DMOG) was purchased from Cayman (Burlington, ON, Canada). Rapamycin and MG132 were obtained from Cell Signaling (Beverly, MA, USA) and EMD (Philadelphia, PA, USA) respectively.
RNA extraction and real-time quantitative PCR
Total RNA was isolated from HEK293T cells using Trizol reagent (Sigma). First-strand cDNA was synthesized with SuperScript III First-Strand Synthesis System according to the protocol of the manufacturer (Invitrogen, Burlington, ON, Canada). Real-time quantitative PCR was performed as reported previously (Yang et al., 2007). Primers were designed for HIF-1α (forward) 5′-CTCAAAGTCGGACAGCCT- CA-3′, (reverse) 5′-CCCTGCAGTAGGTTTCTGCT-3′ (Metzen et al., 2003); actin (forward) 5′-GCACAGAGCCTCGCCTT-3′, (reverse) 5′-GTTGTCGACGACGAGCG-3′; and VEGF (forward) 5′-CCTTGCTGCTCTACCTCCAC-3′, (reverse) 5′-GCAGTAGCTGCGCTGATAGA-3′.
Western blotting
The cultured cells were harvested and solubilized at 4°C with a lysis buffer (40 mM Tris-HCl, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 0.5 mM Na3VO4, 20 mM NaF, pH 7.5) containing protease inhibitors (2 µg·mL−1 aprotinin, 2 µg·mL−1 leupeptim, 2 µg·mL−1 pepstatin A and 0.5 mM PMSF), incubated on ice for 20 min and centrifuged at 20817 g for 15 min at 4°C. Equal amounts of protein extracts were resolved on 10–15% SDS-polyacrylamide gel and transferred to nitrocellulose membranes (Pall Corporation, Pensacola, FL, USA). The membranes were probed with appropriate primary antibodies and detected using peroxidase-conjugated anti-mouse or anti-rabbit antibodies (1:5000) and visualized by ECL (GE Healthcare, Amersham, UK). Immunoblottings were performed with the following antibodies: mouse monoclonal anti-HIF-1α (BD Biosciences, Mississauga, ON, Canada); mouse monoclonal anti-ubiquitin (Invitrogen); rabbit polyclonal anti-eIF2α or anti-phospho-eIF2α, rabbit monoclonal anti-eIF4G, rabbit monoclonal anti-4E-BP1, mouse monoclonal anti-p53, mouse monoclonal anti-Cyclin D1 (Cell Signaling); rabbit monoclonal anti-CSE (Abnova, Taipei, Taiwan); mouse monoclonal anti-eIF4E, mouse monoclonal anti-p21 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and mouse monoclonal anti-β-actin (Sigma).
Co-immunoprecipitation assay
The cultured cells were harvested and lysed in a lysis buffer (25 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 2 mM EDTA, 2 mM EGTA, pH 7.6) containing protease inhibitors. Protein extracts of 1–2 mg were incubated with 2–3 µg anti-ubiquitin antibody for 4 h at 4°C followed by incubation with protein G (GE Healthcare) for 1 h at 4°C. The beads were washed three times with the lysis buffer, and the bound proteins were eluted by boiling for 5 min with 2 × SDS loading buffer and analysed by Western blotting.
Plasmids and DNA transfection
Plasmid haemagglutinin (HA)-HIF1α-pcDNA3 (#18949) (Kondo et al., 2002) and plasmid HA-Ubiquitin (#18712) (Kamitani et al., 1997) were purchased from Addgene (Cambridge, MA, USA). In brief, HEK293T cells were seeded onto 6-well plates (3 × 105 cells per well) and transfected with 1.6 µg plasmid using Lipofectamine™ 2000 Transfection Reagent (Invitrogen), according to manufacturer's specification.
Luciferase assay
HEK293T cells seeded onto 6-well plates were co-transfected with 600 ng plasmid HRE (hypoxia response element)-luciferase (#26731, Addgene) containing three hypoxia response elements from the Pgk-1 gene upstream of firefly luciferase (Emerling et al., 2008) and HA-HIF1α-pcDNA3 expressing vector (350 ng) or empty vector (EV) (350 ng) respectively. Fifty nanograms of pRL-TK vector (Promega, Madison, WI, USA) were co-transfected and served as an internal control. After 24 h of transfection, the cells were incubated with NaHS for 4 h under normoxic conditions and then analysed for luciferase activity using Dual-Luciferase® Reporter Assay System (Promega), followed by quantification with a Fluostar Luminometer (BMG Labtech, Offenburg, Germany).
Short interfering RNA (siRNA) transfection
Pre-designed eIF2α-targeted siRNA (eIF2α-siRNA) and control siRNA were purchased from Santa Cruz. Transfections were performed as previously reported (Teng et al., 2009). Briefly, HEK293T cells were seeded onto 6-well plates at a density of 1 × 105 cells per well and transfected using Lipofectamine™ 2000 Transfection Reagent. All transfections were performed according to manufacturer's instructions. The medium was replaced, 48 h after transfection, with normal medium containing NaHS under hypoxic condition for an additional 4 h.
7-Methyl-GTP Sepharose 4B pull-down assay
The cultured cells were lysed in the lysis buffer and centrifuged at 14000 rpm for 15 min at 4°C. The supernatants (500 µg total protein) were incubated with 25 µL of 7- methyl GTP Sepharose (GE Healthcare) on a rotator for 2 h at 4°C. Pelleted beads were washed three times with the lysis buffer and eluted by boiling for 5 min with 2 × SDS loading buffer and analysed by Western blotting.
Endothelial cell tube formation assay
EA.hy926 cells (2 × 104 cells per well) in DMEM supplemented with 0.1% heat-inactivated FBS were seeded in triplicates onto a 96-well plate coated with Matrigel Basement Membrane Matrix (BD Bioscience) and incubated for 8 h under normoxic or hypoxic conditions. Images of tube formation were taken under an IX71 inverted microscope (Olympus, Center Valley, PA, USA).
Cell viability assay
HEK293T and EA.hy926 cells were seeded at a density of 8 × 103 cells per well in a 96-well plate (Pei et al., 2011). After 24 h of incubation with NaHS, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution was added to each well and incubated further for 4 h. The MTT formazan was dissolved in 100 µL of DMSO. The plates were mixed for 30 min on a gyratory shaker, and absorbance was measured at 595 nm using a Fluostar Luminometer.
5-Bromo-2′-deoxyuridine (BrdU) cell proliferation assay
EA.hy926 cells were seeded at a density of 1.5 × 104 cells per well in a 96-well plate. After 24 h of incubation with NaHS, cell proliferation was assessed by measuring the incorporation of BrdU into DNA using BrdU Cell Proliferation Assay Kit (EMD, Philadelphia, PA, USA) according to the manufacturer's instructions. Quantification was performed using a Fluostar Luminometer at a wavelength of 450 nm with reference readings at a wavelength of 570 nm.
Oxygen partial pressure measurement
The cell culture medium was analysed for the partial pressures of O2 using an electronic blood-gas analyser (GEM Premier 3000, Instrumentation Laboratory, Bedford, MA, USA).
Statistical analysis
All data are expressed as means ± SEM of at least three independent experiments. Statistical analyses were made using Student's t-test, and P < 0.05 was considered statistically significant.
Results
H2S lowered HIF-1α protein levels in HEK293T cells challenged by hypoxia or hypoxia-mimetic agents
NaHS (an H2S donor) lowered hypoxia-elevated protein levels in HEK-293T cells in a dose-dependent manner and nearly eliminated the increase in HIF-1α at concentrations higher than 10 µM (Figure 1A). Overexpression of CSE in HEK293T cells reduced HIF-1α accumulation compared with vector transfected cells under hypoxia (Supporting Information Fig. S1).
Figure 1.
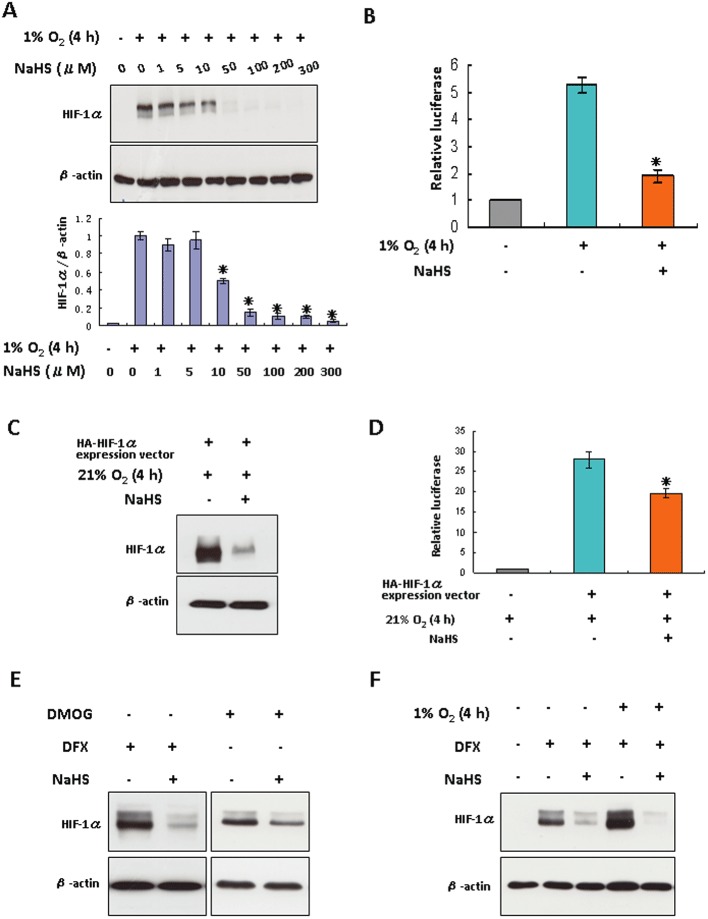
The effects of H2S on HIF-1α protein levels in HEK293T cells challenged by hypoxia or hypoxia-mimetic agents. (A) HEK293T cells were treated under hypoxia for 4 h in the presence or absence of increasing concentrations of NaHS. The lower panel shown is the densitometric quantification of HIF-1α protein normalized to β-actin, and presented as % of hypoxia-alone group. *P < 0.05 versus hypoxia-alone group, n= 5. (B) HEK293T cells were co-transfected with plasmid HRE-luciferase and pRL-TK vector as described in Methods. After 24 h of transfection, cells were exposed to hypoxia for 4 h with or without 100 µM NaHS. Luciferase activity was measured and normalized to control. Each column represents the mean ± SEM (n= 3). *P < 0.05 versus group without NaHS. (C) HEK293T cells were transfected with plasmid HA-HIF1α-pcDNA3 for 24 h, followed by the treatment of 100 µM NaHS for 4 h under normoxic conditions. (D) HEK293T cells were co-transfected with plasmid HRE-luciferase and pRL-TK vector, and either with plasmid HA-HIF1α-pcDNA3 or empty vector. After 24 h of transfection, cells were challenged with 100 µM NaHS under normoxia. Luciferase activity was measured and normalized to control. Each column represents the mean ± SEM (n= 3). *P < 0.05 versus group without NaHS. (E) HEK293T cells were treated with either 200 µM DFX or 1 mM DMOG for 2 h, followed by the addition of 100 µM NaHS for 4 h. (F) HEK293T cells were treated with 200 µM DFX in either normoxic or hypoxic conditions for 4 h, with or without 100 µM NaHS. In (A, C, E and F), total protein extracts (40 µg) were subjected to immunoblot assays with anti-HIF-1α or anti-β-actin antibodies.
To assess whether the suppressive effect of H2S on HIF-1α protein levels was associated with a decreased activity of HIF-1α, we transfected HEK293T cells with HRE-luciferase expression vector containing three hypoxia response elements from the Pgk-1 gene upstream of firefly luciferase(Emerling et al., 2008). As shown in Figure 1B, luciferase activity was significantly increased in response to hypoxia. The addition of NaHS substantially attenuated luciferase expression under hypoxia, indicating that H2S decreased HIF-1α transcriptional activity. We also detected the effect of NaHS on HIF-1α overexpressed HEK293T cells by using plasmid HA-HIF-1α-pcDNA3, which contains human wild-type HIF-1αcDNA sequences. Heterologous overexpression of HIF-1α gene in HEK293T cells led to an abundant expression of HIF-1α proteins under normoxic conditions. Application of NaHS for 4 h significantly lowered the HIF-1α protein level (Figure 1C) and HIF-1α activity (Figure 1D) compared with untreated cells.
Treatment of HEK293T cells with DFX (Figure 1E), DMOG (Figure 1E) or CoCl2 (Supporting Information Fig. S2) elevated HIF-1α protein levels. These agents have been shown to inhibit hydroxylation of HIF-1α, consequently promoting HIF-1α accumulation (Wang and Semenza, 1993; Keely et al., 2009; Wiley et al., 2010). In the presence of these hypoxia-mimetic agents, NaHS induced HIF-1α down-regulation. Even in the presence of hypoxia combined with DFX, NaHS still potently lowered HIF-1α protein level (Figure 1F). The oxygen partial pressure in the cell culture medium was significantly lower under hypoxic conditions than normoxic conditions (Supporting Information Fig. S3). With hypoxia-mimetic agents, however, the oxygen partial pressure in the cell culture medium was not altered compared with that in the absence of the agents under normoxic culture conditions. NaHS did not alter the oxygen partial pressure of the culture medium in either hypoxic or hypoxia-mimetic conditions (Supporting Information Fig. S3). Furthermore, NaHS down-regulated HIF-1α in cultured Hep3B cells (a human hepatoma cell line) or EA.hy926 cells (a human umbilical vein endothelial cell line) under both hypoxic and hypoxia-mimetic conditions (Supporting Information Fig. S4 & Figure 6A). Additionally, under normoxic conditions, NaHS treatment of HEK293T cells for either 1 or 4 h had no effect on HIF-1α protein level (Supporting Information Fig. S5). H2S did not affect HIF-1α expression at the transcription level as real-time quantitative PCR analysis did not reveal any decrease in HIF-1α mRNA level after 4 h of NaHS treatment under hypoxic conditions (Supporting Information Fig. S6).
Figure 6.
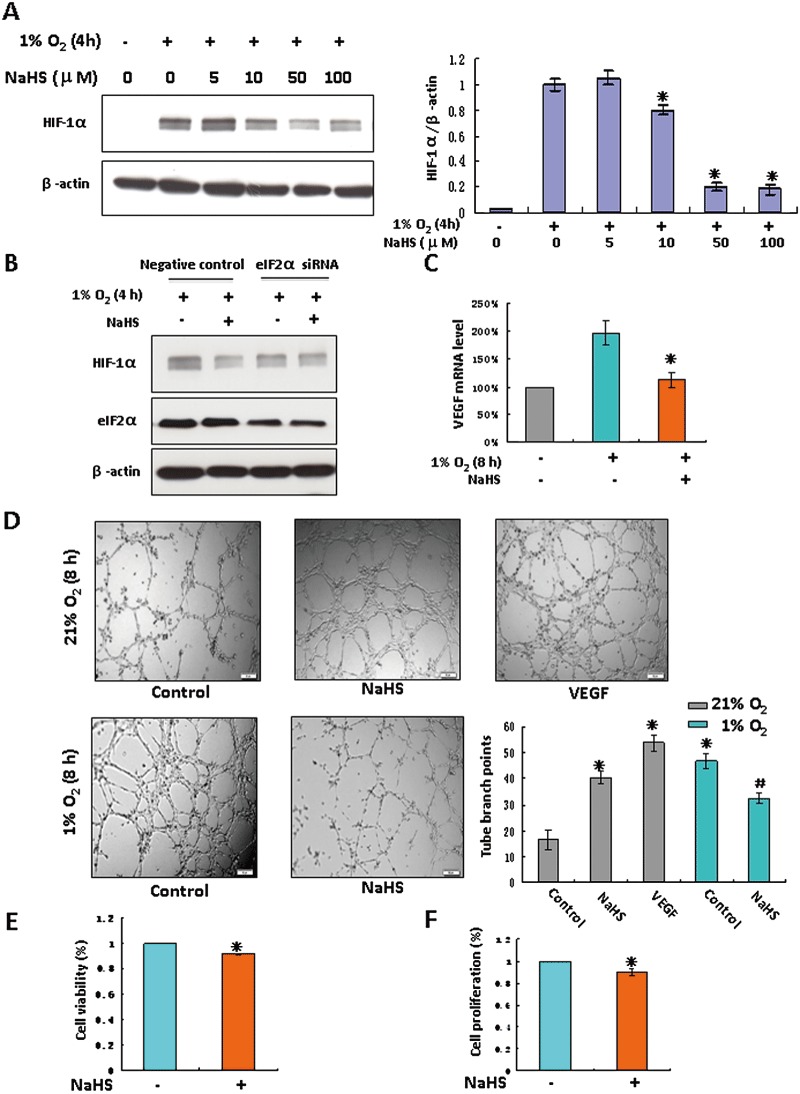
H2S inhibited in vitro angiogenic activity in EA.hy926 cells under hypoxia. (A) EA.hy926 cells were incubated in hypoxic conditions for 4 h in the presence of NaHS at indicated concentrations. The right panel shown is the densitometric quantification of HIF-1α protein normalized to β-actin, and presented as % of hypoxia alone group. *P < 0.05 versus hypoxia-alone group, n= 4. (B) EA.hy926 cells were transfected with a siRNA specific for eIF2α or a negative control siRNA. Transfected cells were exposed to hypoxia for 4 h in the presence or absence of 100 µM NaHS. Levels of HIF-1α and eIF2α were examined by immunoblotting and β-actin served as a loading control. (C) EA.hy926 cells were exposed to hypoxia for 8 h with or without 100 µM NaHS. Total RNA was isolated. VEGF mRNA was analysed by real-time quantitative PCR. Each column represents the mean ± SEM (n= 3). *P < 0.05 versus hypoxic group without NaHS. (D) EA.hy926 cells were seeded onto 96-well plates coated with Matrigel, followed by incubation with 100 µM NaHS for 8 h under normoxia or hypoxia. 50 ng·mL−1 VEGF was used as a positive control. Images of tube formation were taken under an inverted light microscope and tube branch points from each of four randomly chosen fields were quantified. Data represent the mean ± SEM, n= 3. *P < 0.05 versus control under normoxia. #P < 0.05 versus control under hypoxia. Cell viability (E) and cell proliferation (F) in EA.hy926 cells were detected under hypoxia in the presence of 100 µM NaHS for 24 h. *P < 0.05 versus hypoxia-alone group, n= 5.
The down-regulation of HIF-1α protein by NaHS was detected as early as 20 min after 4 h DFX treatment and manifested more as the NaHS treatment time was prolonged (Figure 2A). The NaHS-induced time-dependent suppression of HIF-1α protein levels was also obtained in HEK293T cells with overexpressed HIF-1α under normoxic conditions (Figure 2B). As shown in Figure 2C, NaHS treatment for 1–4 h decreased DFX-induced HIF-1α accumulation by appropriately 50%. This inhibitory effect subsided 8 h after NaHS treatment (Figure 2D).
Figure 2.
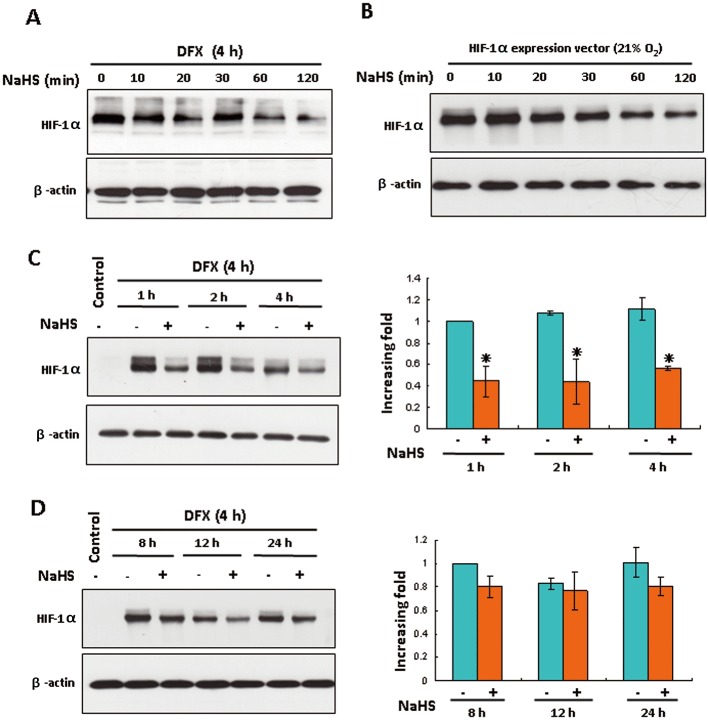
H2S reduced HIF-1α protein levels in HEK293T cells in a time-dependent manner. (A) HEK293T cells were treated with 200 µM DFX for 4 h followed by 100 µM NaHS treatment for the indicated time. The levels of HIF-1α and β-actin were analysed by Western blotting. (B) HEK293T cells transfected with plasmid HA-HIF1α-pcDNA3 were treated with or without 100 µM NaHS for the indicated time under normoxic conditions. The levels of HIF-1α and β-actin were analysed by Western blotting. (C and D) HEK293T cells were treated with 200 µM DFX for 4 h followed by 100 µM NaHS treatment for the indicated time. The right panel shown is the densitometric quantification of HIF-1α protein normalized to β-actin, and presented as % of cells at 1 h (C) or 8 h (D) without NaHS. In all panels, mean ± SEM is shown, *P < 0.05 versus group without NaHS at each time point. n= 4.
The inhibitory effect of H2S on HIF-1α protein level was independent of the ubiquitin-proteasomal degradation pathway
To investigate whether H2S-mediated repression of HIF-1α was due to increased HIF-1α degradation, we used the proteasome inhibitor MG132 to block the degradation of ubiquitinated HIF-1α. In the presence of MG132, NaHS down-regulated HIF-1α under either normoxia or hypoxia (Figure 3A, left). NaHS treatment of plasmid HA-HIF1α-pcDNA3 transfected HEK293T cells still lowered HIF-1α level after cells were pretreated with MG132 under normoxia (Figure 3A, right).
Figure 3.
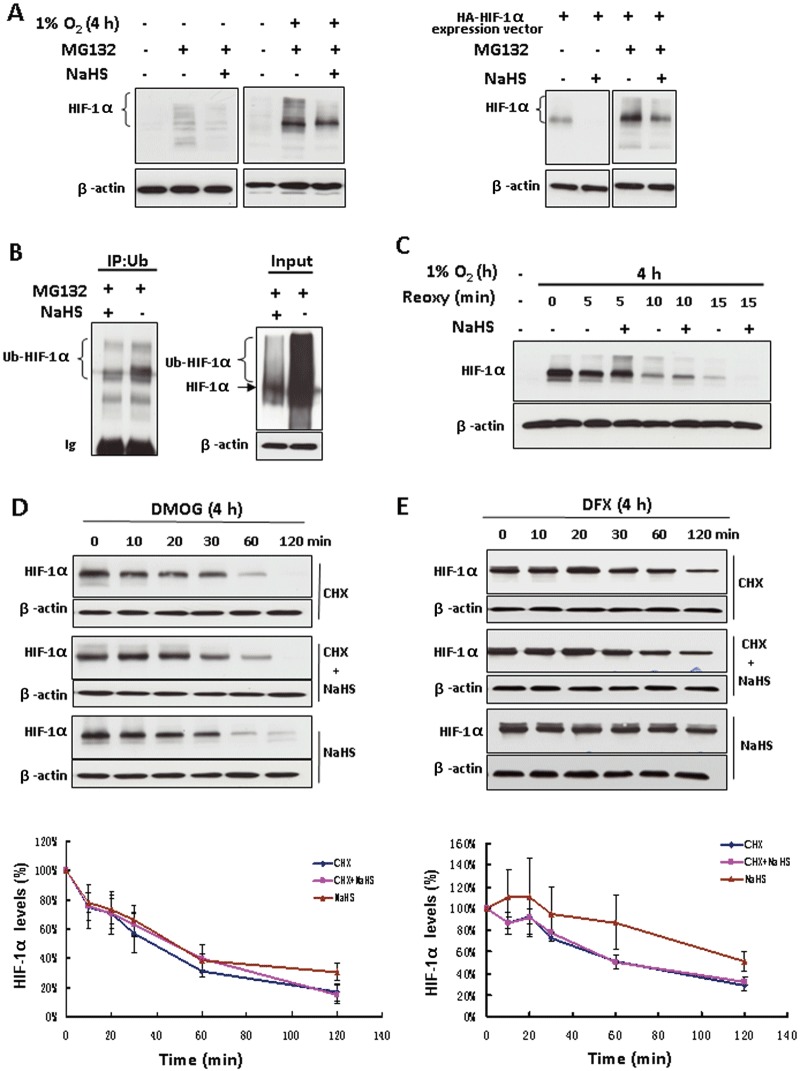
The inhibitory effect of H2S on HIF-1α protein level was independent of the ubiquitin-proteasomal degradation pathway. (A) Non-transfected HEK293T cells were treated with or without 100 µM NaHS in the presence of 20 µM MG132 for 4 h under either normoxia or hypoxia (left). Plasmid HA-HIF1α-pcDNA3 transfected HEK293T cells were exposed to the same treatments under normoxia (right). Total cell extracts (40 µg) were separated by SDS-PAGE and immunoblotted for HIF-1α protein; β-actin served as a loading control. (B) HEK293T cells co-transfected with plasmid HA-HIF1α-pcDNA3 and plasmid HA-ubiquitin were exposed to 20 µM MG132 with or without 100 µM NaHS for 4 h. Cell extracts were subjected to immunoprecipitation with antibody to ubiquitin (IP) followed by immunoblotting with anti-HIF-1α antibody. An aliquot of cell lysates that was reserved before IP was also analysed (input) by using anti-HIF-1α antibody. Ub, ubiquitin; Ub-HIF-1α, ubiquitinated HIF-1α. (C) HEK293T cells were treated with hypoxia for 4 h followed by re-oxygenation for the indicated time with or without 100 µM NaHS. Total cell extracts (40 µg) were prepared for immunoblotting. HEK293T cells were treated with either 1 mM DMOG (D) or 200 µM DFX (E) for 4 h, followed by the indicated treatments. The concentrations of CHX and NaHS are 25 and 100 µM respectively. Total cell extracts (40 µg) were prepared for immunoblotting with anti-HIF-1α antibody. The lower panels of (D) and (E) are the densitometric quantifications of HIF-1α normalized to β-actin, and presented as % of cells at 0 min. n= 3 for each group. Reoxy, re-oxygenation.
Ubiquitination of HIF-1α is a key step for ubiquitin-proteasome-dependent HIF-1α degradation. The interaction of H2S and ubiquitination of HIF-1α was studied by co-immunoprecipitation assay in HEK293T cells co-transfected with HIF-1α and ubiquitin expression vectors. After immunoprecipitation with antibodies against ubiquitin and then blotting with antibodies against HIF-1α, we detected that NaHS decreased the level of ubiquitinated HIF-1α after the degradation of ubiquitinated HIF-1α had been blocked with MG132 (Figure 3B, left). Consistent with the immunoprecipitation results, Western blot analysis on cell lysates with equal amounts of that used for immunoprecipitation (input) showed that NaHS decreased the levels of both un-ubiquitinated and ubiquitinated HIF-1α protein in the presence of MG132 (Figure 3B, right). We also did the same experiment but without MG132 treatment to further evaluate the ubiquitination of HIF-1α. In this case, NaHS inhibited HIF-1α accumulation, but did not significantly affect the ubiquitination of HIF-1α (Supporting Information Fig. S7). These results suggest that NaHS-induced HIF-1α down-regulation is not due to enhanced ubiquitination of HIF-1α.
As shown in Figure 3C, 4 h of exposure to hypoxia evoked massive HIF-1α accumulation in HEK293T cells. Five minutes of re-oxygenation caused a marked decrease in HIF-1α protein levels, and 15 min after re-oxygenation, HIF-1α proteins were only barely detectable. Re-oxygenation-induced HIF-1α reduction was not affected by NaHS.
Next we assessed the effect of NaHS on the rate of HIF-1α degradation in HEK293T cells. After pretreatment of cells with DMOG for 4 h to induce HIF-1α accumulation, CHX was added to block new HIF-1α translation. CHX alone or NaHS alone caused a gradual decrease in HIF-1α protein level. However, in the presence of CHX, the addition of NaHS did not cause significant changes in HIF-1α protein levels at each time point compared with that of CHX alone treated cells (Figure 3D). The half-life of HIF-1α was approximately 40–50 min in either CHX alone or NaHS combined with CHX treated cells. Similar results were obtained in the cells stimulated with DFX (Figure 3E). NaHS did not alter HIF-1α protein levels at any time point in the presence of CHX. The half-life of HIF-1α was approximately 60 min in either CHX alone or NaHS combined with CHX treated cells.
H2S-induced HIF-1α down-regulation was independent of the mTOR/4E-BP1 pathway
Under hypoxic conditions, H2S had no significant effect on the expression of p53, p21 and cyclin D1, all of which are short-lived proteins (Figure 4A). These data suggest that the inhibitory effect of H2S on HIF-1α expression is relatively specific to hypoxia.
Figure 4.
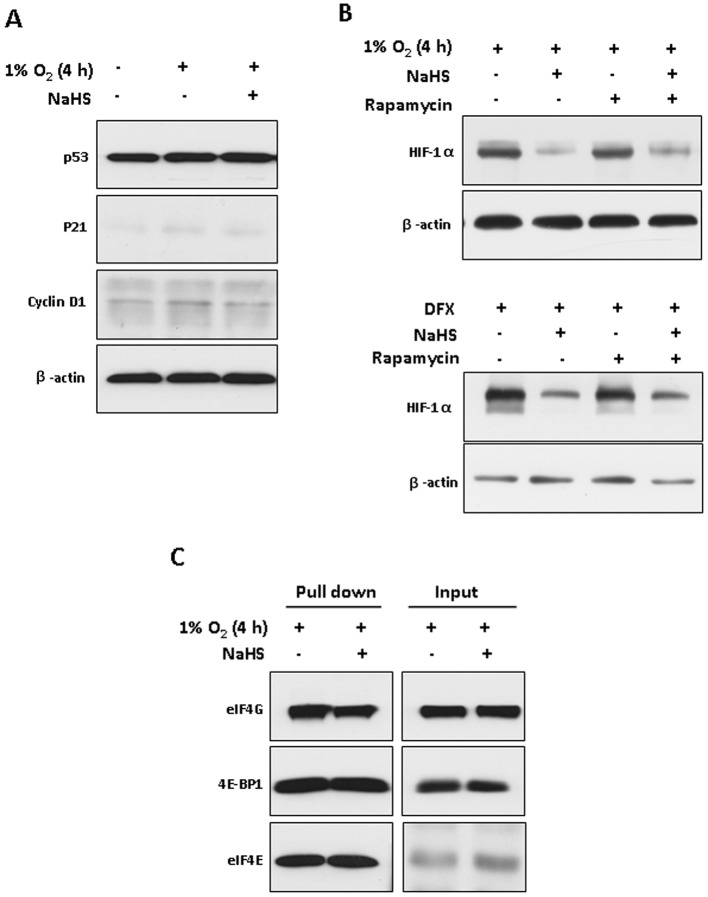
H2S-induced HIF-1α down-regulation was independent of the mTOR/4E-BP1 pathway. (A) Western blot analysis of p53, p21 and cyclin D1 in HEK293T cells treated under hypoxia for 4 h with or without 100 µM NaHS; β-actin served as a loading control. (B) HEK293T cells were pretreated with or without 100 nM rapamycin for 0.5 h followed by exposure to hypoxia (upper) or 200 µM DFX (lower) for 4 h in the presence or absence of 100 µM NaHS respectively. Western blot analyses of HIF-1α and β-actin are shown. (C) HEK293T cells were exposed to hypoxia for 4 h with or without 100 µM NaHS. Cell lysates (500 µg) were subjected to 7-methyl GTP Sepharose, followed by immunoblotting with antibodies for eIF4G, 4E-BP1 and eIF4E respectively. Equal amounts of proteins that were reserved before pull down were also analysed (input).
The PI3K-Akt-mTOR pathway has been implicated in the regulation of HIF-1α gene translation (Hudson et al., 2002). Activation of the PI3K-Akt-mTOR pathway phosphorylates the translational repressor 4E-binding protein 1 (4E-BP1). Phosphorylated 4E-BP1 dissociates from eIF4E, resulting in eIF4E binding to eIF4G, which is the limiting step in the assembly of the translation initiation complex eIF4F (Liu et al., 2006). Pretreatment with rapamycin, an inhibitor of mTOR, did not reverse H2S-induced reduction in HIF-1α protein in HEK293T cells exposed to either hypoxia or DFX treatment (Figure 4B), suggesting that mTOR may not be an effector of H2S for HIF-1α regulation. Next, we used 7-methyl GTP Sepharose pull-down assays to detect whether H2S modulates the interaction between 4E-BP1 and eIF4E (Chapuis et al., 2010). As shown in Figure 4C, H2S had no effect on the dissociation of 4E-BP1 from eIF4E in hypoxic conditions, as detected by pull-down assays. Equal amounts of eIF4G, 4E-BP1 and eIF4E in cell lysates for pull down were measured by Western blotting.
Phosphorylation of eIF2α was associated with H2S-induced repressing of HIF-1α translation
In normal oxygen levels, H2S reversibly phosphorylated eIF2α in HEK293T cells with the peak occurring at 30 min (Figure 5A). H2S treatment also induced a modest phosphorylation of eIF2α under hypoxic conditions (Figure 5B). eIF2α knockdown decreased the basal level of HIF-1α proteins by appropriately 30% in hypoxic conditions compared with the siRNA negative control (Figure 5C). The eIF2α-depleted cells were highly resistant to H2S, displaying only a 34% decrease in HIF-1α expression compared with the 83% decrease in cells transfected with the control siRNA, indicating that eIF2α is involved in the H2S-induced repression of HIF-1α in hypoxic conditions. However, application of the hypoxia-mimetic compound DFX, resulted in a similar H2S-induced reduction of HIF-1α in either negative control or eIF2α knockdown cells, 55 and 48% respectively (Figure 5D). In short, these results show that H2S-induced eIF2α phosphorylation contributed to HIF-1α translational repression under hypoxic but not hypoxia-mimetic conditions.
Figure 5.
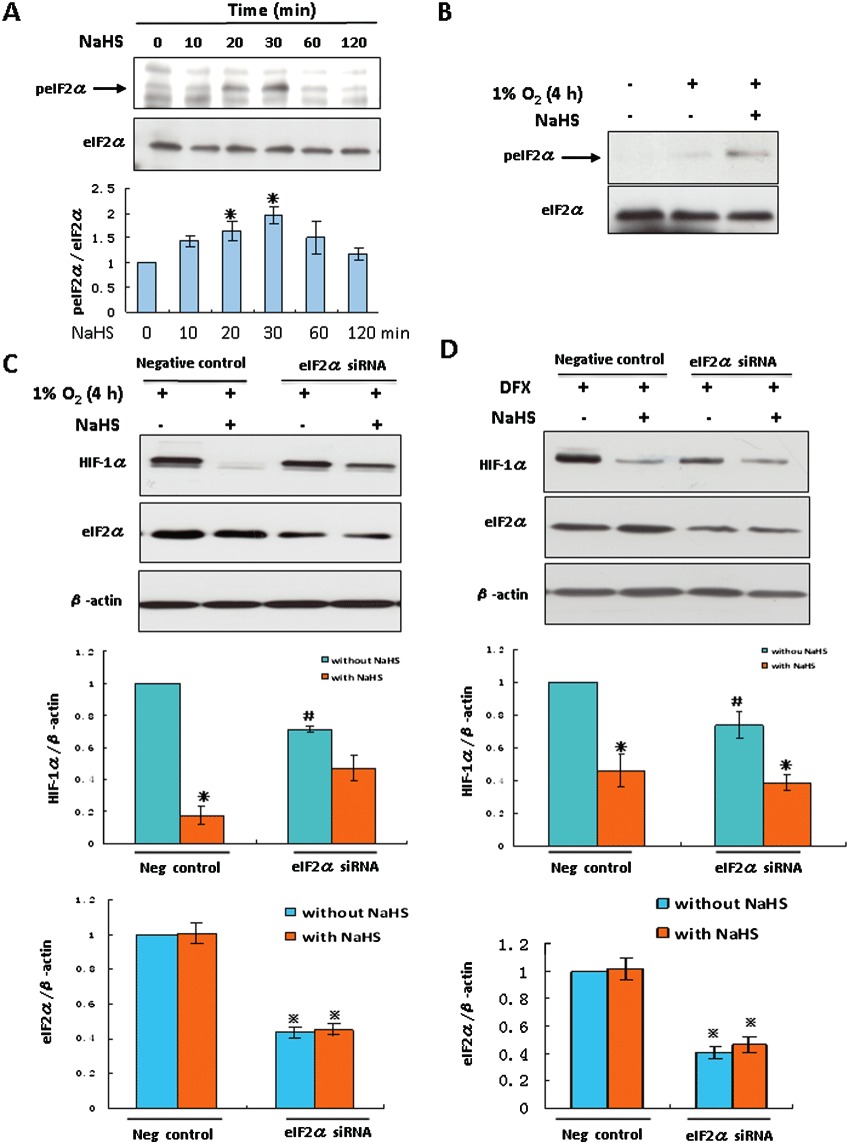
Phosphorylation of eIF2α was associated with H2S-induced HIF-1α translational repression in HEK293T cells. HEK293T cells were treated with 100 µM NaHS under normoxia (A) or hypoxia (B) for the indicated time. p-eIF2α were determined by Western blot analyses. The panel shown under (A) is the densitometric quantification of p-eIF2α normalized to total eIF2α, and presented as % of value at 0 min. *P < 0.05 versus 0 min, n= 5. Total eIF2α served as a loading control. HEK293T cells were transfected with a siRNA specific for eIF2α or a negative control siRNA. Transfected cells were exposed to hypoxia (C) or 200 µM DFX (D) for 4 h in the presence or absence of 100 µM NaHS. Levels of HIF-1α and eIF2α were examined by immunoblotting and β-actin served as a loading control. The middle and lower panels shown are the densitometric quantification of HIF-1α normalized to β-actin and eIF2α normalized to β-actin, respectively, and presented as % of negative control without NaHS from five (C) or seven (D) independent experiments. *P < 0.05 versus the same siRNA transfection group without NaHS. #P < 0.05 versus negative control group without NaHS.  P < 0.05 versus negative control group without NaHS.
P < 0.05 versus negative control group without NaHS.
H2S inhibited in vitro capillary tube formation under hypoxia
Among all the known genes regulated by HIF-1, VEGF plays a central role in stimulating angiogenesis and oxygen delivery (Forsythe et al., 1996). NaHS, at concentrations higher than 10 µM, evoked a marked decrease in hypoxia-induced HIF-1α accumulation in EA.hy926 cells (Figure 6A). Consistent with the result obtained after exogenous NaHS treatment, in recombinant defective adenovirus containing CSE gene (Ad-CSE) transfected endothelial cells, HIF-1α accumulation was inhibited under hypoxia compared with that of recombinant adenovirus encoding bacterial β-galactosidase (Ad-lacZ) transfected endothelial cells (Supporting Information Fig. S8). NaHS did not significantly decrease HIF-1α under hypoxia after eIF2α knockdown (Figure 6B). In the presence of MG132 to block the degradation of HIF-1α, NaHS still down-regulated HIF-1α under hypoxia (Supporting Information Fig. S9A). Additionally, H2S-induced eIF2α phosphorylation was also detected (Supporting Information Fig. S9B). The transcriptional expression of VEGF was significantly increased in hypoxia, and this was down-regulated by NaHS (Figure 6C). Under normoxic conditions, VEGF or NaHS alone stimulated tube formation of EA.hy926 cells. Hypoxia also increased tube formation compared with that under normoxic conditions. However, the application of NaHS reduced capillary tube formation (Figure 6D), and reduced cell viability and inhibited cell proliferation of EA.hy926 cells (Figure 6E,F). Neither eIF2α phosphorylation (Supporting Information Fig. S9) nor tube formation (Supporting Information Fig. S10) was changed by a lower concentration of NaHS (5 µM).
Discussion and conclusions
Both NO and CO have been shown to regulate HIF-1α expression and HIF-1α transcriptional activity (Hagen et al., 2003; Metzen et al., 2003; Choi et al., 2010). As the third gasotransmitter, H2S also regulates HIF-1α activity and expression. An earlier study it was reported that sulforaphane down-regulates the expression of HIF-1α under hypoxia (Yao et al., 2008). In this regard, sulforaphane has recently been reported to have H2S-donating properties (Pei et al., 2011). NaHS at 1 mM decreased HIF-1α protein level under hypoxia in several cell lines, including Hep3B, SH-SY5Y and Hela cells (Kai et al., 2012). Under normoxic conditions, H2S gas (50 p.p.m.) enhanced HIF-1 activity and HIF-1 protein concentration in C. elegans (Budde and Roth, 2009). NaHS (300 µM) also increases HIF-1α protein in CoCl2 (300 µM) treated vascular smooth muscle cells (Liu et al., 2010). Thus, the regulatory effects of H2S on HIF-1α protein levels might be different with different experimental setups, such as animal species and cell types, whether endogenous H2S levels are manipulated or exogenous H2S is given in different formulas, and the degree of hypoxia (Jaakkola et al., 2001; Berra et al., 2003).
NaHS at 10–100 µM has been used in different studies to mimic physiologically relevant concentrations of H2S in vivo (Yang et al., 2012). However, within hours of its administration, the NaHS concentration is decreased (Suzuki et al., 2011; Yang et al., 2012); we previously demonstrated a 55–76% loss of H2S from NaHS-containing medium during the incubation periods 30 min to 12 h (Zhao et al., 2001; Yang et al., 2012). In addition, the accuracy of conventional methods used in the measurement of H2S level has been challenged in recent years and the actual level of endogenous H2S in circulation or tissues in vivo has not been unambiguously established. With different H2S measurement techniques, endogenous levels of H2S in circulation have been reported from nanomolar to low micromolar range. Therefore, one should be cautious in correlating the concentrations of exogenously applied H2S to real physiological levels of H2S in vivo. In the present study, we found that NaHS (10–100 µM) treatment of HEK293T, Hep3B and EA.hy926 cells decreased HIF-1α protein levels under both hypoxia and hypoxia-mimetic conditions (DFX, DMOG and CoCl2) in a dose- and time-dependent manner. Down-regulation of HIF-1α protein was associated with a reduction in HIF-1α transcriptional activity. In addition, H2S-induced eIF2α phosphorylation was shown to contribute partially to the translational suppression of HIF-1α protein. Finally, H2S inhibited angiogenesis under hypoxic conditions and this may be involved in the down-regulation of HIF-1α. NaHS at 5 µM had no effect on HIF-1α protein levels, or on eIF2α phosphorylation or tube formation.
HIF-1α degradation is mainly mediated by prolyl hydroxylases and von Hippel Lindau protein in different types of cells in an oxygen-dependent manner (Rocha, 2007). The oxygen-independent HIF-1α degradation involves the receptor of activated PKC and heat-shock protein 90 (Hsp90) (Liu et al., 2007). These mechanisms regulating HIF-1α stability converge at the ubiquitin-proteasomal system, which ultimately determines HIF-1α proteolysis. NO and CO have been reported to regulate HIF-1α accumulation by different mechanisms that affect its degradation. NO-stimulated HIF-1α down-regulation under hypoxia results from an increase in its degradation mediated by prolyl hydroxylases (Hagen et al., 2003), whereas CO up-regulates HIF-1α partially by suppressing its oxygen-independent degradation (Choi et al., 2010). Kai et al. showed that H2S down-regulated HIF-1α under hypoxic, but not hypoxia-mimetic, conditions in Hep3B cells (Kai et al., 2012). These authors believed that this was due to an H2S-induced decrease in oxygen consumption under hypoxia. They found that H2S promoted HIF-1α degradation under hypoxia after CHX-induced HIF-1α translational inhibition. Moreover, H2S did not affect HIF-1α expression induced by MG132. Their work suggested that H2S-inhibited mitochondrial oxygen consumption resulted in increased oxygen level in hypoxic cells, which therefore promoted HIF-1α degradation. On the other hand, H2S had no effect on DFX- or CoCl2-induced HIF-1α accumulation because H2S could not significantly increase cellular oxygen partial pressure in the presence of hypoxia-mimetic agents (Kai et al., 2012). Unfortunately, most of the key observations that elucidated potential mechanisms for H2S-inhibited HIF-1α accumulation were obtained with NaHS at 1 mM, which is clearly not within the physiological range of endogenous H2S. Kai et al. performed a cytotoxicity assay and showed that 4 h treatment with 1 mM NaHS had no effect on cell viability (Kai et al., 2012). However, treatment for 4 h, a relatively short time, may not reflect the potential injury induced by 1 mM NaHS. We found that a 24 h treatment of HEK293T cells with NaHS at 1 mM decreased cell viability (Supporting Information Fig. S11). When we used NaHS at a concentration of 100 µM, which had no inhibitory effect on cell viability, we reached a different conclusion that H2S-induced HIF-1α down-regulation is independent of the ubiquitin-proteasomal degradation pathway. Firstly, in the presence of the proteasome inhibitor MG132 to inhibit HIF-1α degradation, H2S still induced HIF-1α down-regulation. Secondly, co-immunoprecipitation assays showed that H2S did not induce HIF-1α ubiquitination which is a key step for HIF-1α degradation. Thirdly, H2S had no effect on re-oxygenation-induced reduction in HIF-1α levels. Fourthly, after HIF-1α translation had been blocked with CHX, NaHS ceased to affect HIF-1α protein levels at all of the time points during the observation. Finally, NaHS lowered HIF-1α protein levels even with hypoxia-mimetic agents, which did not lower oxygen partial pressure in the culture medium. It is worthy of mention that Kai et al. performed their experiments by adding hypoxia-mimetic agents and NaHS at the same time (Kai et al., 2012), whereas we pretreated cells with hypoxia-mimetic agents, 2–4 h before the addition of NaHS.
Several lines of evidence have demonstrated that the PI3K-Akt-mTOR pathway, which regulates the formation of translational complex eIF4F, is involved in regulating HIF-1α protein translation (Rocha, 2007). YC-1 (Sun et al., 2007). Also KC7F2 (Narita et al., 2009) and the promyelocytic leukaemia tumour suppressor (Bernardi et al., 2006), negatively regulate HIF-1α protein synthesis through inhibition of the PI3K-Akt-mTOR pathway. In our present study, pretreatment with rapamycin did not reverse H2S-induced HIF-1α protein reduction under either hypoxic or hypoxia-mimetic conditions, suggesting that the mTOR pathway is not involved in this event. On the other hand, H2S did not alter the affinity of 4E-BP1 with eIF4E, ruling out the possibility that the eIF4F complex is involved in H2S-induced HIF-1α translational inhibition under hypoxia.
Another critical regulatory molecule for eukaryotic initiation of translation is eIF2α, of which the phosphorylation at Ser51 arrests protein synthesis by preventing the re-formation of eIF-2 ternary complex (Rocha, 2007). Previous studies have shown that translational suppression of HIF-1α by certain anti-tumour compounds is closely correlated with increased eIF2α phosphorylation (Jung et al., 2009; 2011; Zhang et al., 2010). Our data showed that H2S led to the phosphorylation of eIF2α under both normoxic and hypoxic conditions. Restoration of H2S-induced decrease in HIF-1α proteins under hypoxia was observed after knockdown of the eIF2α gene, demonstrating that HIF-1α translational suppression is associated with H2S-induced eIF2α phosphorylation. What is more interesting is that under hypoxia-mimetic conditions, where only the utilization of oxygen is blocked but the actual oxygen partial pressure is not reduced, eIF2α knockdown did not significantly restore H2S-induced reduction of HIF-1α. This phenomenon indicates that eIF2α phosphorylation may not be a major contributor to H2S-induced decrease in HIF-1α expression under hypoxia-mimetic conditions. Although our data showed that the ubiquitin-proteasomal degradation pathway was not responsible for H2S-induced HIF-1α down-regulation, we cannot rule out the possibility that the calcium/calpain-mediated HIF-1α degradation pathway might be involved in this event (Zhou et al., 2006). H2S has been found to be associated with the regulation of intracellular calcium (Wang, 2011). Thus, under hypoxic-mimetic conditions, whether H2S inhibits HIF-1α accumulation by promoting the calcium/calpain pathway needs to be investigated.
EA.hy926 cell line is a useful model for in vitro study of angiogenic processes, which has maintained the phenotype of endothelial cells (Bauer et al., 1992; Aranda and Owen, 2009; Schwalm et al., 2010). This cell line has also been reported to produce more consistent responses to specific variables and greater reproducibility of data (Aranda and Owen, 2009). H2S exhibited an inhibitory effect on the angiogenic activity of EA.hy926 cells under hypoxia, which was associated with H2S-induced HIF-1α down-regulation and VEGF inhibition. H2S stimulated capillary tube formation under normoxia but without an effect on HIF-1α and VEGF expression, indicating that HIF-1α is not essential for H2S-stimulated angiogenic activity under normoxia. In line with this, H2S has been demonstrated to increase angiogenesis under normoxia through KATP channels and the MAPK pathway (Papapetropoulos et al., 2009; Szabo and Papapetropoulos, 2011). Thus, our results suggest that H2S may play a dual role in the regulation of angiogenesis, which is associated with oxygen levels.
In summary, H2S-induced repression of HIF-1α protein translation, rather than an effect on the ubiquitin-proteasomal degradation pathway, is the key mechanism for H2S-mediated HIF-1α down-regulation under both acute moderate hypoxia and hypoxia-mimetic conditions. This inhibitory effect of H2S on HIF-1α translation is partially associated with an increase in eIF2α phosphorylation. Additionally, H2S-inhibited HIF-1α expression is involved in H2S-induced inhibition of angiogenic activity under hypoxia. These findings elucidate a novel eIF2α phosphorylation-related mechanism for the interaction of H2S and HIF-1 α under hypoxic conditions and its functional consequence in vascular endothelial cells. They also pave the way for further elucidation of the oxygen-sensing role of H2S in different physiological and pathophysiological situations.
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada.
Glossary
- CHX
cycloheximide
- CSE
cystathionine γ-lyase
- DFX
desferrioxamine
- DMOG
dimethyloxalyl glycine
- eIF2α
eukaryotic translation initiation factor 2α
- H2S
hydrogen sulfide
- mTOR
mammalian target of rapamycin
- NaHS
sodium hydrosulfide
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Endogenous H2S decreased HIF-1α protein levels in HEK293 cells under hypoxia. HEK293 cell line stably expressing CSE (HEK293 + CSE) and control cell line (HEK293 + vector) were exposed to hypoxia for 4h. Western blot analyses of HIF-1α, CSE and β-actin were shown.
Figure S2 H2S decreased HIF-1α protein levels in HEK293T cells treated with CoCl2. HEK293T cells were challenged with 200 μM CoCl2 for 4h followed by 100 μM NaHS treatment for the indicated time. Western blot analyses of HIF-1α and β-actin were shown.
Figure S3 Oxygen partial pressure in cell-culture medium in different conditions. HEK293T cells were treated as indicated in the presence or absence of 100 μM NaHS. Oxygen partial pressure in cell-culture medium was measured by using GEM Premier 3000 blood gas analyser. n = 5 for each independent experiment. *P < 0.05 versus control group.
Figure S4 H2S down-regulated HIF-1α protein levels in other cell lines. Hep3B cells (A) or EA.hy926 cells (B) were treated with hypoxia or 200 μM DFX for 4h in the presence or absence of 100 μM NaHS. Western blot analyses of HIF-1α and β-actin were shown.
Figure S5 H2S did not induce any HIF-1α accumulation under normoxia in HEK293T cells. HEK293T cells were treated with increasing concentrations of NaHS for 1 h (A) or 4 h (B) under normoxia. Western blot analyses of HIF-1α and β-actin were shown.
Figure S6 H2S had no effect on HIF-1α mRNA levels under hypoxia in HEK293T cells. Total RNA was extracted from HEK293T cells subjected to normoxia or hypoxia for 4 h in the presence or absence of 100 μM NaHS. Real-time quantitative PCR was performed to analyse HIF-1α mRNA levels. Results are representative of an average of three independent experiments.
Figure S7 H2S did not affect the ubiquitination of HIF-1α in HEK293T cells. HEK293T cells co-transfected with plasmid HA-HIF1α-pcDNA3 and plasmid HA-Ubiquitin were exposed to 100 μM NaHS under normoxia for 4 h. Cell extracts were subjected to immunoprecipitation with antibody to ubiquitin (IP) followed by immunoblotting with anti-HIF-1α antibody (IB). An aliquot of cell lysates that was reserved prior to IP was also analysed (input) using anti-HIF-1α or anti-β-actin antibody. Ub, ubiquitin; Ub-HIF-1α, ubiquitinated HIF-1α.
Figure S8 Endogenous H2S decreased HIF-1α protein levels in EA.hy926 cells under hypoxia. EA.hy926 cells infected with recombinant CSE adenovirus (Ad-CSE) or Ad-lacZ were treated under hypoxia for 4 h. Western blot analyses of HIF-1α, CSE and β-actin were shown.
Figure S9 H2S-induced HIF-1αprotein translation inhibition was indicated in EA.hy926 cells under hypoxia. (A) EA.hy926 cells were treated with or without 100μM NaHS in the presence or absence of 20 μM MG132 for 4 hunder hypoxia. HIF-1α levels were determined by Western blot analyses. β-actin serves as a loading control. (B) EA.hy926 cells were treated with 5 or 100 μM NaHS under normoxia for the indicated time. p-eIF2α was determined by Western blot analyses. Total eIF2α serves as a loading control.
Figure S10 NaHS at a concentration of 5 μM did not affect in vitro tube formation in EA.hy926 cells. EA.hy926 cells were seeded onto 96-well plates coated with Matrigel, followed by incubation with 5 μM NaHS for 8 h undernormoxia or hypoxia. 50 ng mL−1 VEFG was used as apositive control. Images of tube formation were taken under aninverted light microscope and tube branch points from each of four randomly chosen fields were quantified. Data represent the mean± SEM, n = 3.
Figure S11 The effects of NaHS at different concentrations on cell viability in HEK293T cells. Cell viability in HEK293T cells were detected in the presence of 100 μM or 1 mM NaHS for 24 h. *P < 0.05 versus control group, n = 4.
References
- Aranda E, Owen GI. A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the EA.hy926 endothelial cell line. Biol Res. 2009;42:377–389. [PubMed] [Google Scholar]
- Bauer J, Margolis M, Schreiner C, Edgell CJ, Azizkhan J, Lazarowski E, et al. In vitro model of angiogenesis using a human endothelium-derived permanent cell line: contributions of induced gene expression, G-proteins, and integrins. J Cell Physiol. 1992;153:437–449. doi: 10.1002/jcp.1041530302. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, et al. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 2006;442:779–785. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde MW, Roth MB. Hydrogen sulfide increases hypoxia-inducible factor-1 activity independently of von Hippel-Lindau tumor suppressor-1 in C. elegans. Mol Biol Cell. 2009;21:212–217. doi: 10.1091/mbc.E09-03-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A, et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res. 2010;16:5424–5435. doi: 10.1158/1078-0432.CCR-10-1102. [DOI] [PubMed] [Google Scholar]
- Choi YK, Kim CK, Lee H, Jeoung D, Ha KS, Kwon YG, et al. Carbon monoxide promotes VEGF expression by increasing HIF-1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J Biol Chem. 2010;285:32116–32125. doi: 10.1074/jbc.M110.131284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekundi-Valentim E, Santos KT, Camargo EA, Denadai-Souza A, Teixeira SA, Zanoni CI, et al. Differing effects of exogenous and endogenous hydrogen sulphide in carrageenan-induced knee joint synovitis in the rat. Br J Pharmacol. 2010;159:1463–1474. doi: 10.1111/j.1476-5381.2010.00640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Weinberg F, Liu JL, Mak TW, Chandel NS. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a) Proc Natl Acad Sci U S A. 2008;105:2622–2627. doi: 10.1073/pnas.0706790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esechie A, Enkhbaatar P, Traber DL, Jonkam C, Lange M, Hamahata A, et al. Beneficial effect of a hydrogen sulphide donor (sodium sulphide) in an ovine model of burn- and smoke-induced acute lung injury. Br J Pharmacol. 2009;158:1442–1453. doi: 10.1111/j.1476-5381.2009.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil V, Gallego D, Jimenez M. Effects of inhibitors of hydrogen sulphide synthesis on rat colonic motility. Br J Pharmacol. 2011;164:485–498. doi: 10.1111/j.1476-5381.2011.01431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jung HJ, Park JW, Lee JS, Lee SR, Jang BC, Suh SI, et al. Silibinin inhibits expression of HIF-1alpha through suppression of protein translation in prostate cancer cells. Biochem Biophys Res Commun. 2009;390:71–76. doi: 10.1016/j.bbrc.2009.09.068. [DOI] [PubMed] [Google Scholar]
- Jung HJ, Suh SI, Suh MH, Baek WK, Park JW. Pentamidine reduces expression of hypoxia-inducible factor-1alpha in DU145 and MDA-MB-231 cancer cells. Cancer Lett. 2011;303:39–46. doi: 10.1016/j.canlet.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Kai S, Tanaka T, Daijo H, Harada H, Kishimoto S, Suzuki K, et al. Hydrogen sulfide inhibits hypoxia- but not anoxia-induced hypoxia-inducible factor 1 activation in a von hippel-lindau- and mitochondria-dependent manner. Antioxid Redox Signal. 2012;16:203–216. doi: 10.1089/ars.2011.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani T, Kito K, Nguyen HP, Yeh ET. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem. 1997;272:28557–28562. doi: 10.1074/jbc.272.45.28557. [DOI] [PubMed] [Google Scholar]
- Keely S, Glover LE, MacManus CF, Campbell EL, Scully MM, Furuta GT, et al. Selective induction of integrin beta1 by hypoxia-inducible factor: implications for wound healing. FASEB J. 2009;23:1338–1346. doi: 10.1096/fj.08-125344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–246. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25:7029–7040. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- Liu X, Pan L, Zhuo Y, Gong Q, Rose P, Zhu Y. Hypoxia-inducible factor-1alpha is involved in the pro-angiogenic effect of hydrogen sulfide under hypoxic stress. Biol Pharm Bull. 2010;33:1550–1554. doi: 10.1248/bpb.33.1550. [DOI] [PubMed] [Google Scholar]
- Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell. 2007;25:207–217. doi: 10.1016/j.molcel.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14:3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita T, Yin S, Gelin CF, Moreno CS, Yepes M, Nicolaou KC, et al. Identification of a novel small molecule HIF-1alpha translation inhibitor. Clin Cancer Res. 2009;15:6128–6136. doi: 10.1158/1078-0432.CCR-08-3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y, Wu B, Cao Q, Wu L, Yang G. Hydrogen sulfide mediates the anti-survival effect of sulforaphane on human prostate cancer cells. Toxicol Appl Pharmacol. 2011;257:420–428. doi: 10.1016/j.taap.2011.09.026. [DOI] [PubMed] [Google Scholar]
- Pouokam E, Diener M. Mechanisms of actions of hydrogen sulphide on rat distal colonic epithelium. Br J Pharmacol. 2011;162:392–404. doi: 10.1111/j.1476-5381.2010.01026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha S. Gene regulation under low oxygen: holding your breath for transcription. Trends Biochem Sci. 2007;32:389–397. doi: 10.1016/j.tibs.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Schwalm S, Pfeilschifter J, Huwiler A. Sphingosine kinase 1 is critically involved in nitric oxide-mediated human endothelial cell migration and tube formation. Br J Pharmacol. 2010;160:1641–1651. doi: 10.1111/j.1476-5381.2010.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Regulation of physiological responses to continuous and intermittent hypoxia by hypoxia-inducible factor 1. Exp Physiol. 2006;91:803–806. doi: 10.1113/expphysiol.2006.033498. [DOI] [PubMed] [Google Scholar]
- Spagnuolo RD, Recalcati S, Tacchini L, Cairo G. Role of hypoxia-inducible factors in the dexrazoxane-mediated protection of cardiomyocytes from doxorubicin-induced toxicity. Br J Pharmacol. 2011;163:299–312. doi: 10.1111/j.1476-5381.2011.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell. 2010;40:228–237. doi: 10.1016/j.molcel.2010.09.028. [DOI] [PubMed] [Google Scholar]
- Sun HL, Liu YN, Huang YT, Pan SL, Huang DY, Guh JH, et al. YC-1 inhibits HIF-1 expression in prostate cancer cells: contribution of Akt/NF-kappaB signaling to HIF-1alpha accumulation during hypoxia. Oncogene. 2007;26:3941–3951. doi: 10.1038/sj.onc.1210169. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gero D, et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:13829–13834. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol. 2011;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng H, Ballim RD, Mowla S, Prince S. Phosphorylation of histone H3 by protein kinase C signaling plays a critical role in the regulation of the developmentally important TBX2 gene. J Biol Chem. 2009;284:26368–26376. doi: 10.1074/jbc.M109.021360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood. 1993;82:3610–3615. [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal. 2003;5:493–501. doi: 10.1089/152308603768295249. [DOI] [PubMed] [Google Scholar]
- Wang R. Hydrogen sulfide: the third gasotransmitter in biology and medicine. Antioxid Redox Signal. 2010;12:1061–1064. doi: 10.1089/ars.2009.2938. [DOI] [PubMed] [Google Scholar]
- Wang R. Signaling pathways for the vascular effects of hydrogen sulfide. Curr Opin Nephrol Hypertens. 2011;20:107–112. doi: 10.1097/MNH.0b013e3283430651. [DOI] [PubMed] [Google Scholar]
- Wiley M, Sweeney KR, Chan DA, Brown KM, McMurtrey C, Howard EW, et al. Toxoplasma gondii activates hypoxia-inducible factor (HIF) by stabilizing the HIF-1alpha subunit via type I activin-like receptor kinase receptor signaling. J Biol Chem. 2010;285:26852–26860. doi: 10.1074/jbc.M110.147041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DI, Chen SD, Yang YT, Ju TC, Xu JM, Hsu CY. Carbamoylating chemoresistance induced by cobalt pretreatment in C6 glioma cells: putative roles of hypoxia-inducible factor-1. Br J Pharmacol. 2004;141:988–996. doi: 10.1038/sj.bjp.0705687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Yang W, Wu L, Wang R. H2S, endoplasmic reticulum stress, and apoptosis of insulin-secreting beta cells. J Biol Chem. 2007;282:16567–16576. doi: 10.1074/jbc.M700605200. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Li H, Tang G, Wu L, Zhao K, Cao Q, et al. Increased neointimal formation in cystathionine gamma-lyase deficient mice: role of hydrogen sulfide in alpha5beta1-integrin and matrix metalloproteinase-2 expression in smooth muscle cells. J Mol Cell Cardiol. 2012;52:677–688. doi: 10.1016/j.yjmcc.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Yao H, Wang H, Zhang Z, Jiang BH, Luo J, Shi X. Sulforaphane inhibited expression of hypoxia-inducible factor-1alpha in human tongue squamous cancer cells and prostate cancer cells. Int J Cancer. 2008;123:1255–1261. doi: 10.1002/ijc.23647. [DOI] [PubMed] [Google Scholar]
- Yee Koh M, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci. 2008;33:526–534. doi: 10.1016/j.tibs.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Zhang J, Cao J, Weng Q, Wu R, Yan Y, Jing H, et al. Suppression of hypoxia-inducible factor 1alpha (HIF-1alpha) by tirapazamine is dependent on eIF2alpha phosphorylation rather than the mTORC1/4E-BP1 pathway. PLoS ONE. 2010;5:e13910. doi: 10.1371/journal.pone.0013910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Kohl R, Herr B, Frank R, Brune B. Calpain mediates a von Hippel-Lindau protein-independent destruction of hypoxia-inducible factor-1alpha. Mol Biol Cell. 2006;17:1549–1558. doi: 10.1091/mbc.E05-08-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.