

Abstract
BACKGROUND AND PURPOSE
In osteosarcoma (OS) patients, only a limited number of drugs are active and the regimens currently in use include a combination of at least two of these drugs: doxorubicin, cisplatin, methotrexate and ifosfamide. Today, 30–40% of patients still die of OS highlighting the urgent need for new treatments. Invariant NKT (iNKT) cells are a lymphocyte lineage with features of both T and NK cells, playing important roles in tumour suppression. Our aim was to test whether the cytoxicity induced by cisplatin, doxorubicin and methotrexate against OS cells can be enhanced by iNKT cell treatment.
EXPERIMENTAL APPROACH
iNKT cells were purified from human peripheral blood mononuclear cells by cell sorting (Vα24Vβ11+ cells) and used as effector cells against OS cells (U2-OS, HOS, MG-63). Cell death (calcein-AM method), perforin/granzyme B and Fas/FasL expressions were determined by flow cytometry. CD1d expression was analysed at both the gene and protein level.
KEY RESULTS
iNKT cells were cytotoxic against OS cells through a CD1d-dependent mechanism. This activity was specific for tumour cells, because human CD1d+ mesenchymal stem cells and CD1d- osteoblasts were not affected. iNKT cell treatment enhanced drug-induced OS cell death in a concentration-dependent manner and this effect was reduced in CD1d-silenced OS cells.
CONCLUSION AND IMPLICATIONS
iNKT cells kill malignant, but not non-malignant, cells. iNKT cell treatment enhances the cytotoxicity of anti-neoplastic drugs against OS cells in a CD1d-dependent manner. The present data encourage further studies on the use of iNKT cells in OS therapy.
Keywords: iNKT cells, osteosarcoma, cisplatin, doxorubicin, methotrexate, combination therapy
Introduction
Osteosarcoma (OS) is an aggressive, highly malignant bone tumour and the most common primary solid tumour of bone in childhood and adolescence, with 70–75% of cases occurring between the ages of 10 and 25. Although OS can occur in any bone, it especially affects the long bones, where it starts intramedullary and grows towards the cortex (Ta et al., 2009). Local therapy alone is insufficient to control malignancy, as 80–90% of patients with seemingly localized disease will develop metastases, mostly in the lungs, and will die if chemotherapy is not implemented. The use of multi-agent chemotherapy, in the 1970s, led to a significant increase in event-free and overall survival (Ferrari et al., 2005); today, 5 year event-free and overall survival for localized OS are 60–65% and 75–80% respectively (Janeway and Grier, 2010). OS treatment typically encompasses pre-operative (neo-adjuvant) chemotherapy, surgical resection and post-operative (adjuvant) chemotherapy (Ritter and Bielack, 2010). A variety of combination chemotherapy regimens have been tested and it appears that only a limited number of cytotoxic drugs are active in OS; the most active are doxorubicin, cisplatin, methotrexate and ifosfamide. The regimens currently in use include at least two of these drugs (Whelan et al., 2012), but there is no consensus on their optimum combination (Hogendoorn et al., 2010). Given the intensity of OS therapy, most patients experience acute (e.g. alopecia, myelosuppression, mucositis, nausea and vomiting) and/or rare (e.g. cardiac, renal, neuronal, reproductive) toxicities, as well as late effects, including secondary malignancies (Janeway and Grier, 2010). In addition, despite the aggressive treatments, 30–40% of patients still die of OS (Clark et al., 2008), highlighting the urgent need for novel modalities to either prevent or treat chemotherapy refractory and recurrent diseases.
In the last few years, many investigators have focused on immunotherapy and its applicability to OS (Marina and Gorlick, 2009). IFN has been employed either alone or in combination with chemotherapy, and ILs (e.g. IL-2 or IL-12), liposomal muramyl tripeptide phosphatidyl ethanolamine, as well as inhibitors of the mammalian target of rapamycin have been proposed to treat OS (Ta et al., 2009). Furthermore, the identification in OS of several human tumour antigens [e.g. melanoma-associated antigen, squamous cell carcinoma antigen recognized by T-cells (SART) 1 and 3, and papillomavirus binding factor] has provided the rationale to develop cellular therapies (Mori et al., 2006). SART3-derived peptides induce the production of SART3-specific cytotoxic T lymphocytes (CTLs) (Tsuda et al., 2001), while tumour-specific T lymphocytes are toxic against allergenic tumour cells in patients with bone-associated tumours (Théoleyre et al., 2005). OS patient-derived NK cells are functionally and phenotypically unimpaired (Luksch et al., 2003), and OS cells, including chemoresistant variants, are highly susceptible to lysis by IL-15-induced NK cells from both allogeneic and autologous origin (Buddingh et al., 2011).
To the best of our knowledge, the antitumor effects of type I NKT, or invariant NKT (iNKT) cells, a lymphocyte lineage with features of both T and NK cells, have not been investigated so far in human OS.
iNKT cells are a unique autoreactive CD1d-restricted T-cell subpopulation, characterized by the expression of an invariant T-cell receptor (TCR) alpha chain (Vα14-Jα18 in mice and Vα24-Jα18 in humans) (Kronenberg, 2005), paired with a limited array of TCR-β chains (Vβ8, Vβ7 or Vβ2 in mice; Vβ11 in humans). Other CD1d-restricted NKT cells exist that use different TCR α- and β-chains and can recognize distinct lipid-based antigens presented by CD1d; these are often referred to as ‘diverse’ or ‘type II’ NKT cells (Godfrey et al., 2004).
Phenotypically, iNKT cells express surface receptors typical for NK cells (e.g. NK1.1 and Ly49), in addition to T-cell markers (e.g. CD44, CD69, CD122) (Molano et al., 2008). iNKT cells recognize, through their semi-invariant TCR, glycolipid antigens, such as the agelasphins, a family of glycolipid extracts from the marine sponge Agelas mauritianus, or the agelasphin derivative α-galactosylceramide (α-GalCer) and its analogues, presented by antigen presenting cells in the context of CD1d presenting molecules. After TCR ligation, iNKT cells promptly produce a large amount of both Th1 (e.g. IFN-γ) and Th2 (e.g. IL-4) cytokines, and can bias adaptive immune responses towards Th1, Th2, Th17 or regulatory T-cell (Treg) differentiation (Taniguchi et al., 2010). iNKT cells are also capable of extensive talk with a variety of other cell types, including dendritic cells (DCs), NK cells, macrophages, neutrophils, conventional T- and B-cells, and different subsets of Treg cells (van der Vliet et al., 2004).
Based on the surface expression of CD4 and CD8 molecules, human iNKT cells can be divided in four subsets: CD4+; CD8+; CD4-CD8-[double negative (DN)]; and CD4+CD8+[double positive (DP)] (Montoya et al., 2007). Each cellular subtype shares different cytokine profiles when analysed directly ex vivo; in particular, human CD4+ iNKT cells produce both Th1- and Th2-types of cytokines, whereas CD4- iNKT cells primarily produce Th1 cytokines (Lee et al., 2002). In vitro, human NKT cells can be expanded and skewed towards IFN-γ production with the potent synthetic agonist α-GalCer, loaded onto either monocyte-derived DCs (Nieda et al., 2001), total peripheral blood mononuclear cell (PBMCs) (Rogers et al., 2004) or synthetic CD1d tetramers (Metelitsa et al., 2003), in the presence of single/combined growth factors, such as IL-2, IL-7, IL-12 or IL-15 (van der Vliet et al., 2001; Lin et al., 2004).
Although iNKT cells only constitute approximately 0.02–0.2% of the human peripheral blood T lymphocytes (Metelitsa, 2004), they play important roles in a variety of immune responses, such as infections, autoimmunity and cancer (van der Vliet et al., 2004).
In the context of cancer, iNKT cells have a primarily protective role, which has been demonstrated in transplantable, chemically-induced and genetic tumour models (Terabe and Berzofsky, 2008). α-GalCer-expanded iNKT cells can induce either direct cytotoxicity against tumour cells in a CD1d-dependent manner (Bourgeois et al., 2009) or tumour lysis by activation of other immune mediators, such as human NK cells (indirect cytotoxicity) (Ishihara et al., 2000). Direct cytotoxicity can be mediated by both the release of perforine/granzyme B lytic granules and the up-regulation of cell-death-inducing effector molecules, such as Fas ligand (FasL) and tumour-necrosis factor-related apoptosis-inducing ligand (TRAIL) (Nicol et al., 2000; Lisbonne et al., 2004).
In vivo, a defect in iNKT cell number and function has been observed in progressive malignant multiple myeloma patients (Dhodapkar et al., 2003), in prostate cancer patients (Tahir et al., 2001) and in patients with a number of solid tumours as well (Crough et al., 2004). The loss of iNKT cell function to produce IFN-γ appears to correlate with the clinical stage of tumours and may be, at least in part, reversible.
Several clinical trials have been performed demonstrating that iNKT cell-based immunotherapy, with α-GalCer itself or autologous α-GalCer-loaded DC, is well tolerated and effective in mediating iNKT cell expansion and activation in cancer patients (Giaccone et al., 2002; Motohashi et al., 2006). This therapy led to prolonged disease stabilization or an extended time to tumour progression in some patients (Fujii, 2008).
On this basis, the iNKT cell-based immunotherapy, alone or in combination with surgery or chemotherapy, has been proposed as a new valuable therapeutic approach in tumour patients (Molling et al., 2008).
The aim of our study was to investigate in vitro whether the cytoxicity induced by some anti-neoplastic drugs against OS cells can be enhanced by iNKT cells.
We provide evidence, for the first time, that: (i) human iNKT cells kill malignant OS cells (U2-OS, HOS, MG-63) in a CD1d-dependent manner, while they are not toxic against human non-malignant CD1d+ mesenchymal stem cells (MSC-BM) and CD1d- osteoblasts; (ii) the cytotoxicity induced by cisplatin, doxorubicin and methotrexate against OS cells is significantly potentiated by iNKT cell treatment; and (iii) the iNKT cell-induced enhancement of drug effects is dependent on CD1d expression in OS cells and mediated by both perforine/granzyme B and Fas/FasL pathways.
The overall data encourage further studies on iNKT cells in OS therapy.
Methods
Cell cultures
The human U2-OS cell line, obtained from American Type Culture Collection (Manassas, VA, USA), and the human HOS cell line, obtained from Interlab Cell Line Collection (IST, Genova, Italy), were maintained in DMEM, supplemented with 10% FBS, 2 mM L-glutamine, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin. The human MG-63 cell line and the primary osteoblast cultures, kindly provided by Dr. Michela Bosetti (Department Of Pharmaceutical Sciences, Univ. ‘Piemonte Orientale’, Novara, Italy), were cultured in supplemented MEM. The human primary MSC-BM cell cultures, a gift from Dr M. Serafini (Tettamanti Research Center, Monza, Italy), were maintained in supplemented low-glucose DMEM.
Twice a week, cells were detached with trypsin/EDTA, counted and re-seeded in a fresh culture medium at different densities.
Human PBMCs were isolated by gradient centrifugation onto Ficoll-Hystopaque of venous blood obtained from healthy volunteers after their informed consent, and human monocytes were isolated from PBMCs, as previously described (Fallarini et al., 2008). The purity of monocytes was assessed with the anti-CD14 mAb. PBMCs and monocytes were maintained in RPMI medium, supplemented with 10% FBS, 2 mM L-glutamine, 1% non-essential amino acids, 1 mM sodium pyruvate and 10 µg·mL−1 kanamycin, and used on the day of isolation under endotoxin-free conditions.
All cells were incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2.
iNKT cell expansion and analysis
Human iNKT cells were expanded and purified from human PBMCs, as previously described (Fallarini et al., 2010). Briefly, human PBMCs (5 × 106 cells per well) were plated in six-well plates and immediately treated with 10 ng·mL−1 of α-GalCer (KRN-7000). About 20 U·mL−1 of human recombinant IL-2 was added after 48 h, followed by 40 U·mL−1 of IL-2 every 2 days until the end of cell proliferation. At day 12, human iNKT cells were sorted by staining with anti-Vα24Vβ11 and anti-CD3 mAbs by FACSVantage-SE® flow cytometer (Becton Dickinson, Milan, Italy). Sorted human iNKT cells were suspended in RPMI-1640 complete medium (see above) before using in functional studies. To evaluate the frequency and the phenotype of iNKT cells, α-GalCer-treated PBMCs and resting PBMCs (day 12) were labelled with phycoerythrin (PE)-conjugated anti-Vα24Vβ11, peridinin-chlorophyll protein (PerCP)-conjugated anti-CD3, PE-conjugated anti-CD19, FITC-conjugated anti-CD56, Alexa Fluor® (Life Technologies EU, Monza, Italy) 647-conjugated anti-CD14, FITC-conjugated anti-CD4 and allophycocyanin (APC)-conjugated anti-CD8 mAbs, and analysed by FACSVantage-SE flow cytometer. Non-specific background fluorescence was evaluated with the appropriate isotype-matched control mAb. Appropriate combinations of PE-, PerCP-, FITC-, Alexa Fluor 647- and APC-conjugated mAbs were used. The lymphocytes were gated using side scatter/forward scatter analysis and the increase in iNKT cells was evaluated by cytofluorimetric identification with the specific Vα24Vβ11 mAb.
Reverse transcriptase-PCR (RT-PCR)
RNA extraction and reverse transcriptase-PCR (RT-PCR) analyses were performed as previously described (Paoletti et al., 2009). Briefly, cells were harvested and centrifuged at 1000×g for 5 min at 4°C. Total RNA was isolated using the GenElute™ (Sigma Aldrich, St Louis, MO, USA) mammalian total RNA miniprep kit and reverse-transcribed using the ThermoScript™ (Life Technologies EU) RT-PCR kit according to the manufacturer's instructions. For amplification, 3 µL of cDNA was added to GoTaq FlexiDNA Polymerase in 25 µL reaction buffer, containing 0.5 mM of forward and reverse primers (Table 1). RT-PCR amplicons were resolved in a 2% agarose gel by electrophoresis, and signals were quantified with densitometric analysis software (NIH Image 1.32; National Institutes of Health, Bethesda, MD, USA).
Table 1.
PCR primers and protocols used in this study
| Template | Primers | Exon | Size (bp) | Denaturation | Annealing | Extension | Cycles |
|---|---|---|---|---|---|---|---|
| HmCD1d | forward 5'- GCTCAACCAGGACAAGTGGACGAG-3' | 4 | 535 | 94°C for 60 s | 60°C for 2 min | 72°C for 2 min | 30 |
| NM_001766a | reverse 5'- GGAGGTAAAGCCCACAATGAGGAG-3' | 6 | |||||
| GAPDH | forward 5'-GGTCGGAGTCAACAACGGATTTGG-3' | 2 | 1000 | 94°C for 30 s | 60°C for 30 s | 72°C for 60 s | 28 |
| NM_002046a | reverse 5'-ACCACCCTGTTGCTGTAGCCA-3' | 9 |
Accession number NCBI sequence database (GenBank).
Data are expressed as the ratio of the signals obtained for each gene in one sample divided by that obtained for the reference gene [human glyceraldehyde-3-phosphate dehydrogenase (GAPDH)] in the same sample.
Assessment of iNKT cell cytotoxicity
Cytotoxicity of iNKT cells against target cells was determined by the calcein-AM (CAM) flow cytometric assay, as previously described (Metelitsa et al., 2003). Briefly, 1 µM CAM was added to the target cell, and the cells were incubated at 37°C for 15 min. After being washed, labelled target cells were seeded in a 24-well plate at a density of 0.6 × 105 cells in 500 µL per well, and allowed to adhere at 37°C in a humidified atmosphere with 5% CO2. iNKT cells at day 12 of culture were harvested, washed and added in 500 µL per well to reach the desiderate target : effector (T : E) ratios. Each plate included three wells of target cells alone, as controls, for spontaneous cell death measurements. Plates were incubated at 37°C in a humidified atmosphere with 5% CO2 for 24 h. After incubation, the cells of each well were harvested, washed and labelled with propidium iodide (PI), and the cytotoxicity was measured by FACS. Live target cells were identified as CAMhigh/PI- population, whereas killed target cells were CAMlow/PI+ and the effector iNKT cells were CAM- (at least 10-fold less fluorescent than in killed target cells). After gating on target cells, cytotoxicity was calculated as the % increase in CAMlow/PI+ population relative to target cells alone [cytotoxicity, % = (CAMlow/PI+ in experimental wells – CAMlow/PI+ in control wells)/CAMhigh/PI- in control well × 100]. The mean cytotoxicity % ± SEM for each condition was calculated from three replicate experimental wells.
To determine whether the blockade of Vα24Vβ11 TCR/CD1d interaction prevents the anti-tumour activity of iNKT cells, target cells were exposed to 20 µg·mL−1 anti-CD1-blocking mAb 42.1, 1 h before the co-culture with iNKT cells.
Assessment of combination treatment
For combination experiments (anti-neoplastic drugs plus iNKT cells), we used CAM-labelled OS cells as target cells; 0.6 × 105 target cells per well were treated with increasing concentrations (0.01–300 µmol·L−1) of cisplatin/doxorubicin (24 h), or methotrexate (72 h) in the presence/absence of iNKT cells at the T : E ratio 1:6. In each plate, target cells alone were considered as control for spontaneous cell death measurement, while target cells treated with increasing concentrations of each drug were used to determine drug-induced cell death. Following co-culture, cytotoxicity was determined by flow cytometry, as described above.
Blocking iNKT cell toxicity
Blocking agents were used to determine the mechanisms of iNKT cell-mediated toxicity. Effector cells were added to target cells untreated/treated with cisplatin/doxorubicin (3 µmol·L−1) or methotrexate (1 µmol·L−1) in the presence/absence of 100 ng·mL−1 concanamycin A (CMA) or 10 µg·mL−1 anti-FasL-blocking mAb. Afterwards, OS cytotoxicity was determined by flow cytometry, as described above.
Specific small interfering RNA (siRNA) and transfection
siRNA oligonucleotides targeting human CD1d, scrambled non-targeting siRNA (used for a negative control; Mock) and fluorescent siRNA were designed and synthesized by Invitrogen (Carlsbad, Ca, USA). The sequences were as follows: CD1d, 5'-ACA CCU CCA UGG GCU UGA UUG CCU U-3' (sense) and 5'-AAG GCA AUC AAG CCC AUG GAG GUG U-3' (antisense). OS cells were cultured in six-well plates and used at 50–70% density the day of transfection. The 25, 50 and 100 nM siRNAs were transfected into OS cells with Lipofectamine 2000 reagent, according to the manufacturer's protocol. Briefly, OS cells were transfected with Lipofectamine 2000: 0.6, 1.25 or 2.5 µL of siRNA stock (20 µM) and 2.5 µL of Lipofectamine 2000 were each diluted with 250 µL of culture medium without supplements. After 5 min at room temperature, they were combined and incubated for 20 min. The reaction mixtures were overlaid on the cell culture for 5 h. The medium was then changed to fresh complete growth medium. After transfection at 48 h, CD1d silenced (siCD1d)-OS cells were harvested, analysed for CD1d expression, and only OS cells with verified CD1d silencing were used for co-culturing experiments.
Protein extraction and Western blot analyses
Mok-silenced OS cells and siCD1d-OS were harvested, centrifuged at 1000×g for 5 min at 4°C, and the cell pellets washed once with ice-cold PBS. Cellular proteins were extracted with RIPA lysis buffer and the concentrations measured by the Bradford method using BCA Protein Assay Reagent (Fallarini et al., 2009). Protein samples (20 µg per well) were separated by 12% SDS-PAGE and electrophoretically transferred to nitrocellulose membrane; the membranes were blocked, then incubated with the mouse-anti-human CD1d (1:2000) or mouse-anti-human β-actin (1:5000) primary mAb overnight at 4°C, followed by the anti-mouse IgG HRP-conjugated (1:3000) secondary mAb for 2 h at room temperature. Proteins were visualized with an ECL detection kit, according to the manufacturer's instructions. Chemiluminescence signals were analysed under unsaturating conditions with an image densitometer (NIH Image 1.32). Protein expression was quantified by densitometry and normalized to β-actin expression.
FACS analysis of cytotoxicity mediators
iNKT cells were harvested, washed, stimulated with 10 ng·mL−1 PMA plus 3 µg·mL−1 ionomycin for 3 h, and labelled with PE-conjugated anti-Vα24Vβ11, PerCP-conjugated anti-CD3, FITC-conjugated anti-perforin, FITC-conjugated anti-granzyme B, and Alexa Fluor 647-conjugated anti-FasL mAbs, and analysed by FACS. Non-specific background fluorescence was evaluated with the appropriate isotype-matched control mAbs. Appropriate combinations of PE-, PerCP-, FITC-, and Alexa Fluor 647-conjugated mAbs were used. FasL is expressed as median fluorescence intensity by using FACSDiva software (BD Bioscience, Milan, Italy). OS cells, untreated/treated with increasing concentrations (0.1–100 µmol·L−1) of cisplatin, doxorubicin or methotrexate for 24 h, were harvested, washed, labelled with FITC-conjugated anti-Fas mAb and analysed by FACS. OS and siCD1d-OS cells were harvested, labelled with the mouse-anti-human CD1d primary mAb for 1 h at room temperature, followed by the anti-mouse IgG FITC-conjugated secondary mAb for 2 h at room temperature, and analysed by FACS.
Statistical analysis
Results are expressed as means ± SEM of at least three experiments. Statistical significance was evaluated by the one-way anova followed by the Student's t-test for paired populations using Graph Pad Prism 4 (Graph Pad Software, Inc., San Diego, CA, USA). Differences were considered statistically significant when P≤ 0.05. Data were fitted as sigmoidal concentration–response curves and analysed with a four-parameter logistic equation by using the software Origin version 6.0 (Microcal Software, Northampton, MA, USA).
Drugs, chemical reagents and other materials
DMEM, MEM, FBS, L-glutamine, penicillin, streptomycin, trypsin/EDTA, RPMI, non-essential amino acids and sodium pyruvate were purchased from Lonza, Milan, Italy. Ficoll-Hystopaque, kanamycin, GenElute mammalian total RNA miniprep kit, human CD1d primers, human GAPDH primers, HRP-conjugated anti-mouse IgG, CMA, PMA and ionomycin were purchased from Sigma-Aldrich, Milan, Italy. PE-conjugated anti-Vα24Vβ11 (clone 6B11) and human recombinant IL-2 were from Miltenyi Biotec, Bologna, Italy. PerCP-conjugated anti-CD3, PE-conjugated anti-CD19, FITC-conjugated anti-CD56, Alexa Fluor 647-conjugated anti-CD14, FITC-conjugated anti-CD4, APC-conjugated anti-CD8, FITC-conjugated anti-perforin, FITC-conjugated anti-granzyme B, Alexa Fluor 647-conjugated anti-FasL mAbs, mouse-anti-human CD1d, FITC-conjugated anti-mouse IgG and mouse-anti-human β-actin were purchased from Biolegend, DS Uithoorn, The Netherlands. FITC-conjugated anti-Fas and anti-FasL-blocking mAbs were from AbD Serotec, Oxford, UK. Anti-CD1d-blocking mAb (clone 42.1) was from Becton Dickinson. ThermoScript RT-PCR kit, CAM, siRNA targeting human CD1d, scrambled non-targeting siRNA, fluorescent siRNA and Lipofectamine 2000 were purchased from Invitrogen, Milan, Italy. GoTaq FlexiDNA Polymerase kit and BCA Protein Assay Reagent were from Promega, Madison, WI, USA. ECL detection kit was from Amersham Biosciences, Milan, Italy.
α-GalCer (KRN-7000) was kindly provided by Prof. Luigi Panza (Department of Pharmaceutical Sciences, University of ‘Piemonte Orientale’, Novara, Italy).
Cisplatin, doxorubicin and methotrexate were kindly provided by Dr Mario Botta (Unit of Medical Oncology, Casale Monferrato Hospital, Casale Monferrato, Alessandria, Italy).
Results
Selective expansion of human iNKT cells
In healthy donors iNKT cells are a small percentage (0.02–0.2%) of circulating T-cells (Metelitsa, 2004); therefore, to investigate their cytotoxic activity against OS cells we first had to expand them.
As shown in Figure 1B (middle panel), 12 days of PBMC treatment with 10 ng·mL−1α-GalCer + 40 U·mL−1 IL-2 resulted in a selective expansion of the starting cell population. FACS analysis revealed a significant (P≤ 0.01) increase in CD3 V−1α24Jα18-Vβ11-positive iNKT cells in stimulated PBMC (21 ± 2.6%) in comparison to controls (PBMCs treated with medium alone) (0.2 ± 0.1%).
Figure 1.
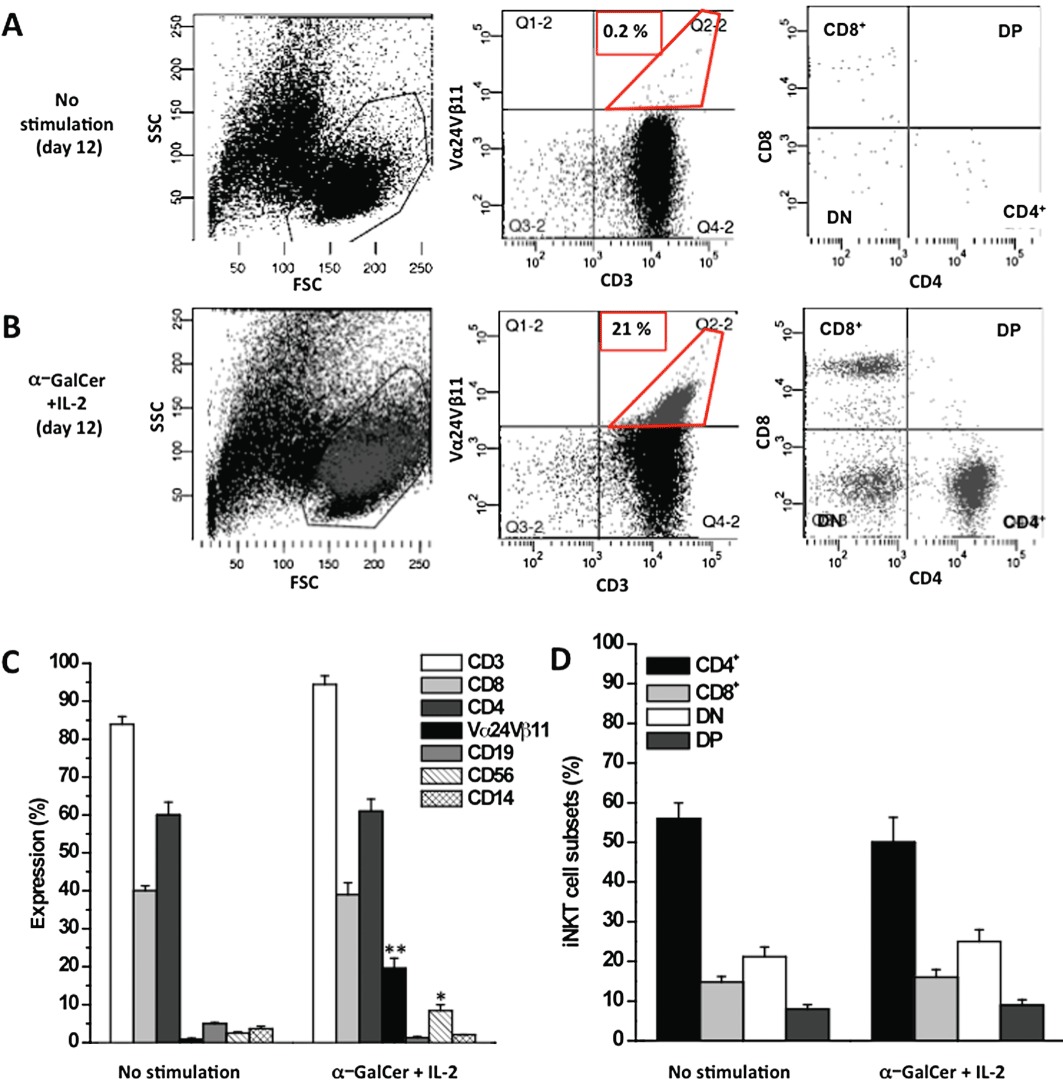
Ex vivo expansion of iNKT cells. PBMCs were stimulated with 10 ng·mL−1α-GalCer + 40 U·mL−1 IL-2 for 12 days. Unstimulated (day 12) and α-GalCer-stimulated (day 12) PBMCs were stained with specific mAbs and analysed by FACS. Representative FACS dot plots of unstimulated (A) and stimulated (B) PBMCs showing the gating strategy used to identify iNKT cells. Lymphocytes were gated based on a side scatter/forward scatter (SSC/FSC) dot plot (A and B, left panels). In the lymphocyte gate, iNKT cells were identified as CD3+Vα24Vβ11+ double-positive cells (A and B, middle panels); the percentage in the panels is representative of gated iNKT cells. The phenotypic composition of iNKT cells (CD4+, CD8+ DP and DN cells) was examined within the iNKT-positive cells (A and B, right panels). The dot plots are representative of at least five independent experiments. (C) Proliferation of T-cells (CD3+, CD4+, CD8+), iNKT cells, monocytes (CD14+), B-cells (CD19+) and NK cells (CD56+) population in unstimulated and α-GalCer + IL-2-stimulated PBMCs. (D) Distribution of CD4+, CD8+, DN and DP cells among unstimulated and α-GalCer-stimulated iNKT cells. The data represent mean ± SEM of at least five experiments. **P≤ 0.01; *P≤ 0.05 versus unstimulated cells.
A limited but statistically (P≤ 0.05) significant increase in CD56+ NK cells (from 2.5 ± 0.3% to 8.4 ± 1.5% of PBMC) was also determined, while no expansion of other PBMCs subpopulations (CD3+, CD4+, CD8+ T-cells, CD19+ B-cells, and CD14+ monocytes) was measured (Figure 1C).
These results were confirmed by 5(6)-carboxyfluorescein diacetate N-succinimidyl ester staining, demonstrating that the relative iNKT cell expansion is due to proliferation rather than to selective survival within the total CD3+ T-cell population (data not shown).
To analyse whether the experimental conditions altered or promoted the proliferation of any particular subset of iNKT cells, we then analysed the percentage of each cell subpopulation (CD4+, CD8+, DP and DN), before and after cell expansion, by FACS. No significant differences were measured as compared with controls (PBMCs treated with medium alone) (Figure 1A and B, right panel).
In the expanded iNKT cell population, CD4+ was the most frequent, while DP the least frequent, iNKT cell subset (50 ± 6.2% and 8 ± 1.3% over the total iNKT cells respectively) (Figure 1D).
The overall results demonstrate a significant increase in iNKT cells without variations in the relative cell subsets.
OS cells express CD1d
The CD1d molecule is widely expressed in both mammalian non-malignant (haematopoietic/non-haematopoietic) and malignant cells (Exley et al., 2011). Because the presence of functional CD1d on tumour cells is essential for direct iNKT cell-induced cytotoxicity (Nowak et al., 2010; Metelitsa, 2011), we investigated the expression of CD1d in malignant OS cell lines (U2-OS, HOS and MG-63) and in non-malignant cells (monocytes, osteoblasts and MSC-BM). As shown in Figure 2A, RT-PCR analysis demonstrated the presence of CD1d in all these cell cultures; densitometric measurement of PCR products showed that U2-OS and HOS cells express CD1d mRNA levels similar to that of human monocytes, taken as positive control (Fallarini et al., 2008). Conversely, the level of CD1d expression in MG-63 cells was comparable to that of MSC-BM cells, while no CD1d expression was measured in human primary osteoblasts, taken as negative control. This was further confirmed when by the consistent results obtained when CD1d protein expression was determined by FACS (Figure 2B).
Figure 2.
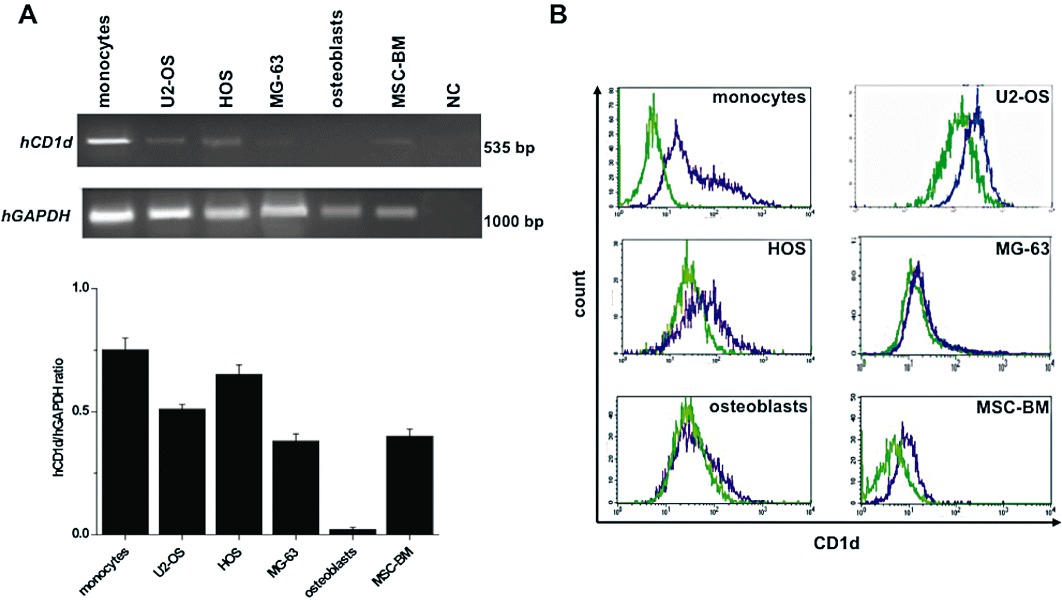
CD1d expression in human OS cell lines and primary non-malignant cells. (A) CD1d expression was quantified by RT-PCR in: human OS cells (U2-OS, HOS, MG-63), MSC-BM, osteoblasts and monocytes (CD1d+ control). The mRNA was extracted, reverse-transcribed into its related cDNA, and PCR was carried out to amplify CD1d cDNA by using specific primers (see Table 1). Expression of GAPDH was used as a loading control. PCR products were visualized with ethidium bromide on a 2% agarose gel. In the negative control (NC) reverse transcriptase was omitted. The signals are densitometrically analysed; data, calculated as mean ± SEM of at least three determinations, are expressed as the ratio (CD1d/GAPDH) of the signal obtained for each sample divided by that obtained for GAPDH in the same sample to permit between-sample comparison. (B) CD1d surface expression was examined in cells by indirect immunofluorescence using anti-CD1d mAb and FITC-anti-mouse IgG. Data are presented as overlapping histograms: isotype control, green dotted histograms; CD1d, violet histograms. Data represent one of at least three independent experiments.
Overall these results suggest that these OS cell lines are a good model for studying iNKT cell-induced direct cytotoxicity.
CD1d-dependent iNKT cell-induced cytotoxicity and its specificity for tumour cells
To determine whether CD1d+ OS cells are affected by direct iNKT cell cytoxicity, we measured cell death by FACS in OS cells after 24 h co-culture with iNKT cells at different T : E ratios (Figure 3A). As shown in Figure 3B, iNKT cells displayed a moderate but significant (P≤ 0.05) cytotoxicity against OS cells, which increased proportionally with the T : E ratio.
Figure 3.
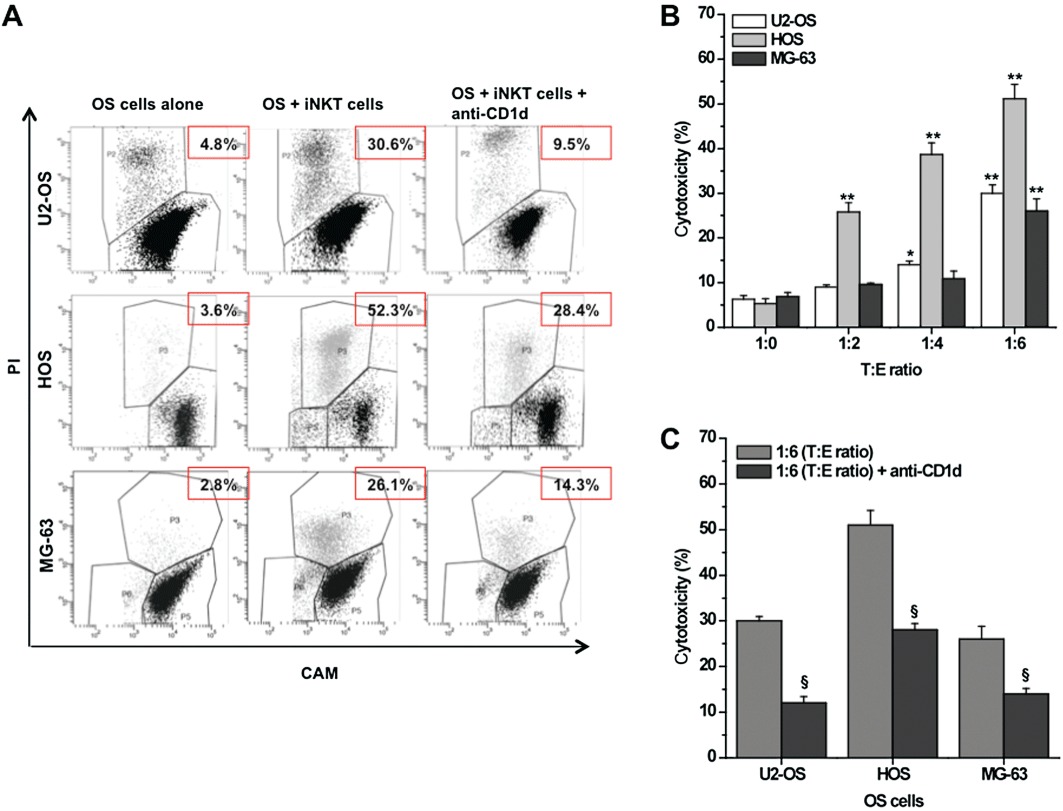
Cytotoxic activity of iNKT cells against CD1d+ OS cell lines. CAM-labelled (1 µM) U2-OS/HOS/MG-63 (target cells) was co-cultured (24 h) with unstained iNKT cells (effector cells) at indicated T : E ratio, in the presence/absence of specific anti-CD1d-blocking mAb (20 µg·mL−1). The calcein fluorescence retained was measured by FACS. (A) Representative dot plots showing the gating strategy used to identify target cells (CAM+ cells) and to discriminate between living target cells (CAMhigh/PI- cells; black cells) and dying target cells (CAMlow/PI+ cells; grey cells). The numbers in the panels indicate the percentage of dead cells. (B) OS cytotoxicity in the presence/absence of iNKT cells at indicated T : E ratio. (C) OS cytotoxicity in the presence/absence of anti-CD1d mAb (20 µg·mL−1) at 1:6 T : E ratio. The mean cytotoxicity % ± SEM for each condition was calculated as described in the Methods section from four replicate experimental wells. **P≤ 0.01; *P≤ 0.05 versus iNKT-untreated OS cells; §P≤ 0.05 versus iNKT-treated OS cells.
Then, because iNKT cells can exert their cytotoxicity either directly, via a CD1d-dependent recognition of malignant cells (Metelitsa et al., 2003; Morris et al., 2005), or indirectly, via activation of other immune cells (e.g. NK cells and T-cells), to determine the contribution of direct cytotoxicity we repeated the same co-culture experiments while blocking the interaction between CD1d and TCR. The presence of a specific anti-CD1d-blocking mAb significantly (P≤ 0.05) reduced the percentage of cell death in all three OS cell lines, confirming that, in our model, cytotoxicity is mainly mediated by direct iNKT cell activity (Figure 3C).
To evaluate the specificity of CD1d-dependent iNKT cell-induced toxicity against tumour cells, we repeated the above experiments using CD1d+ MSC-BM and CD1d- osteoblast cells as target cells (see Figure 2). As shown in Figure 4A and B, these cells were not killed by iNKT cells, demonstrating that the expression of CD1d is essential, but not sufficient, for iNKT cell toxicity, and confirming the specificity of this activity against tumour cells.
Figure 4.
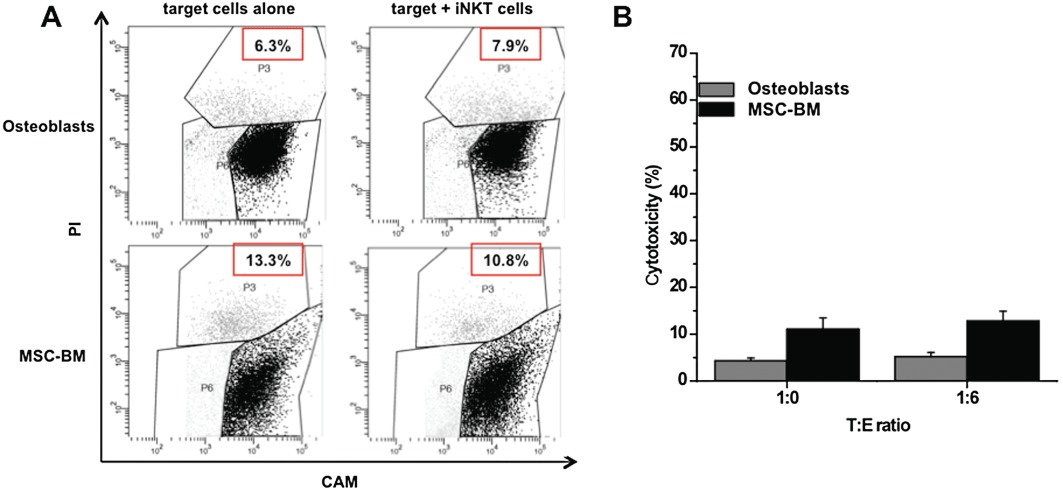
Cytotoxic activity of iNKT cells against human CD1d- osteoblasts and CD1d+ MSC-BM. (A) CAM-labelled (1 µM) CD1d- /CD1d+ target cells were co-cultured (24 h) with unstained iNKT cells (effector cells) at indicated T : E ratio. Retained calcein fluorescence was measured by FACS. Representative dot plots showing the gating strategy used to identify target cells (CAM+ cells) and to discriminate between living target cells (CAMhigh/PI- cells; black cells) and dying target cells (CAMlow/PI+ cells, grey cells). The number in the panels indicates the percentage of dead cells. (B) CD1d- osteoblast and CD1d+ MSC-BM cytotoxicity in the presence/absence of iNKT cells (T : E ratio = 1:6), calculated as described in the Methods section. The mean cytotoxicity % ± SEM for each condition was calculated from four replicate experimental wells.
iNKT cells enhance drug-induced OS cell death
There is accumulating evidence that the use of iNKT cell-based immunotherapy in combination with chemotherapy provides a useful option for improving the treatment of several neoplastic diseases (Mattarollo et al., 2006; Zitvogel et al., 2008; Dhodapkar and Richter, 2011). To explore this possibility in OS, we evaluated the ability of iNKT cells to enhance the cytotoxic effects of some anti-neoplastic drugs. For our experiments, we selected doxorubicin, cisplatin and methotrexate, the most clinically effective drugs in both pre- and post-surgery OS chemotherapy (Whelan et al., 2012).
Firstly, the sensitivity of OS cells to these agents was carefully evaluated at different exposure times: doxorubicin/cisplatin (24 h) and methotrexate (72 h) (Decker et al., 1999; Sun et al., 2007). All the drugs tested significantly (P≤ 0.05) induced OS cell death in a concentration-dependent manner (Figure 5B). Maximal cytotoxicity was obtained at a 100 µmol·L−1 drug concentration for all three cell lines. The calculated EC50 values were: 10.7, 5.2 and 1.4 µmol·L−1 in U2-OS cells; 4.9, 10.8 and 6.2 µmol·L−1 in HOS cells; and 11.2, 16.2 and 7.6 µmol·L−1 in MG-63 cells for cisplatin, doxorubicin and methotrexate respectively.
Figure 5.
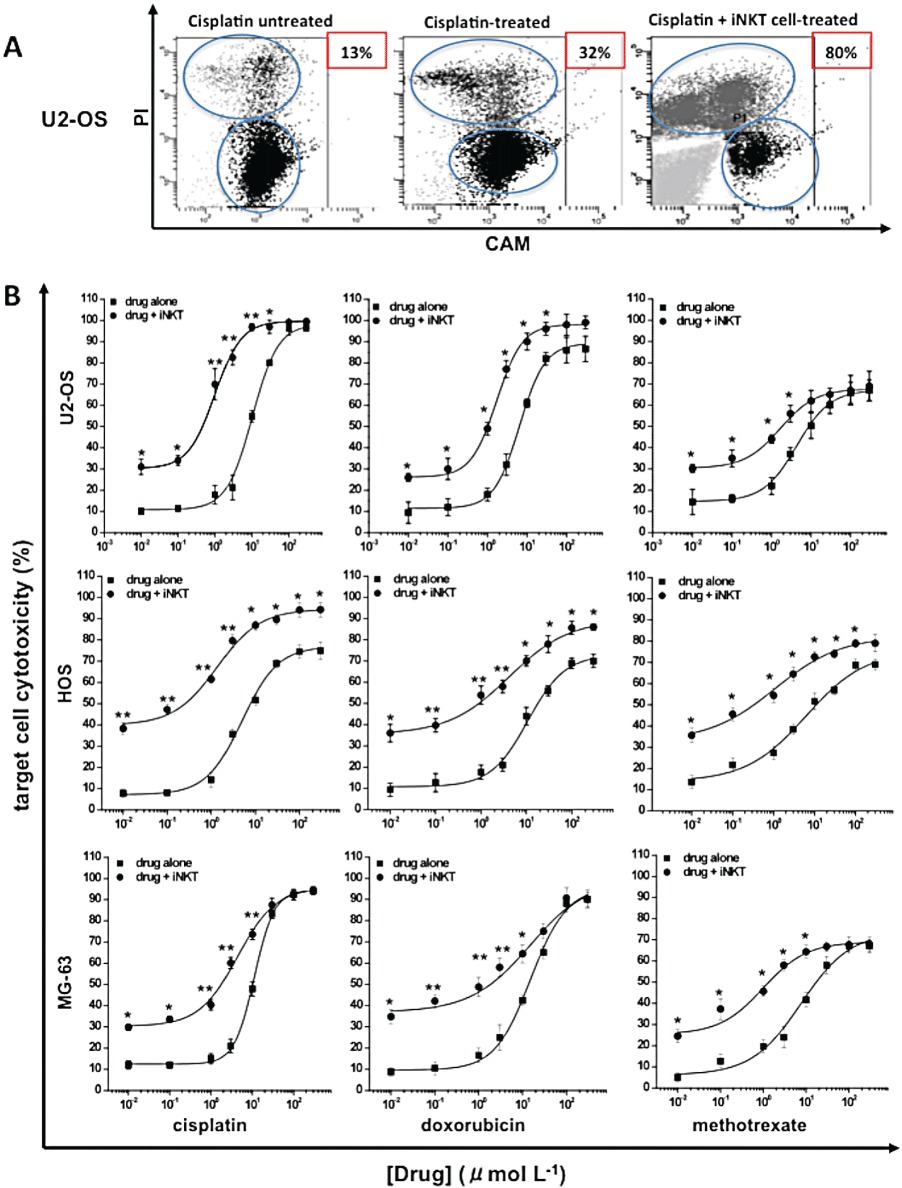
Effects of iNKT cells against drug-induced OS cytoxicity. CAM labelled (1 µM) U2-OS/ HOS/MG-63 cells were treated/untreated with increasing concentrations (0.01–300 µml·mL−1) of cisplatin, doxorubicin (24 h) or methotrexate (72 h) in the presence/absence of iNKT cells (T : E ratio = 1:6). (A) Representative dot plots showing the gating strategy used to identify target cells (CAM+ cells) and to discriminate between living target cells (CAMhigh/PI- cells) and dying target cells (CAMlow/PI+ cells) in U2-OS cells: untreated (left panel), treated with 3 µmol·L−1 of cisplatin (middle panel) or treated with 3 µmol·L−1 of cisplatin +iNKT cells (right panel). The numbers in the panels indicate the percentage of dead cells. (B) Concentration–response curves for cytotoxicity of drug and drug + iNKT cells in each OS cell line. The mean cytotoxicity % ± SEM for each condition was calculated from four replicate experimental wells. **P≤ 0.01; *P≤ 0.05 versus drug-treated OS cells.
Then, to explore whether the combination of chemotherapy and immunotherapy can enhance the level of cell death, OS cells were simultaneously treated with increasing concentrations (0.01–300 µmol·L−1) of each drug and iNKT cells (T : E ratio = 1:6). This co-treatment significantly increased the cytotoxic activity of each drug tested. The greatest increase was observed in U2-OS cells treated with 3 µmol·L−1 cisplatin and was +60 ± 5.1% over cells treated with drug alone (controls) (Figure 5A). The calculated EC50 values were: 0.9, 1.5 and 0.2 µmol·L−1 in U2-OS cells; 1.3, 4.1 and 1.1 µmol·L−1 in HOS cells; and 4.3, 4.5 and 0.9 µmol·L−1 in MG-63 cells for cisplatin, doxorubicin and methotrexate respectively.
Overall, these data clearly indicate that the cytotoxicity induced by anti-neoplastic drugs on OS cells can be enhanced by iNKT cell treatment.
The iNKT cell-induced enhancement of drug cytotoxicity is mediated by CD1d
Having demonstrated that a CD1d-dependent mechanism underlies the iNKT cell-induced OS cell death (see Figure 3), to verify if this mechanism is also involved in the iNKT cell-induced enhancement of anti-neoplastic drug effects, we repeated the above described co-culture experiments using siCD1d-OS cells (see Methods).
First of all, the transfection efficiency of siRNA in OS cells was evaluated by measuring the uptake of fluorescently-labelled scrambled siRNAs (25–100 nM) at 48 h post-transfection. Results show that more than 85% of cells displayed green fluorescence with 100 nM fluorescent siRNA, suggesting a high-efficient transfection (data not shown).
Subsequently, OS cells were treated with either a CD1d-targeting siRNA (100 nM) or a scramble siRNA (Mock; 100 nM) for 48 h, and the inhibition of CD1d expression was evaluated at both gene and protein levels. As shown in Figure 6A and B, Mock did not affect the CD1d expression, whereas CD1d-targeting siRNA abolished it.
Figure 6.
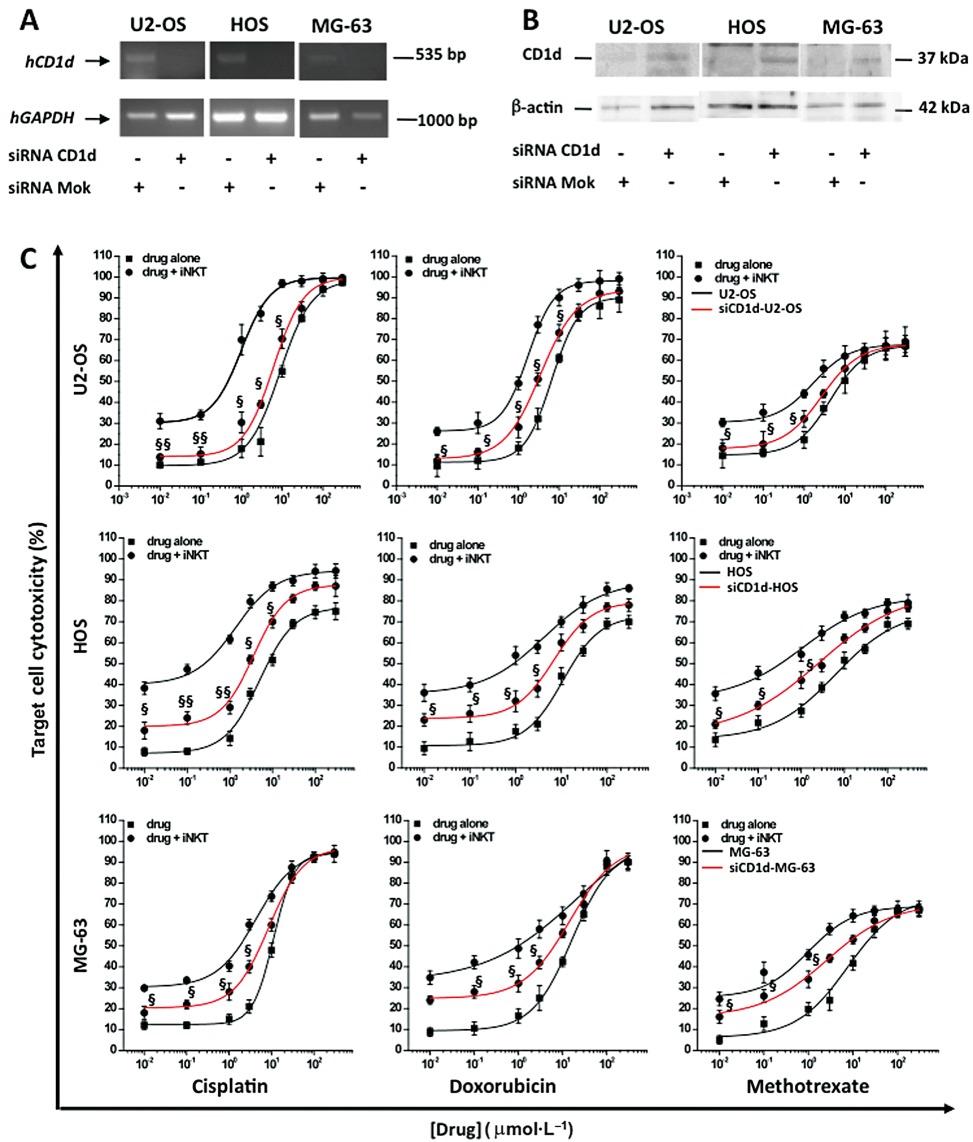
Effects of iNKT cells against drug-induced cytotoxicity in siCD1d-OS cells. (A) U2-OS/HOS MG63 cells were transfected (5 h) with siCD1d or Mok (100 nM) and Lipofectamine 2000 and analysed for CD1d gene and protein expression 48 h post-transfection. CD1d gene expression was quantified by semi-quantitative RT-PCR in CD1d and Mok-transfected OS cells. The mRNA was extracted, reverse-transcribed into its related cDNA, and PCR was carried out to amplify CD1d cDNA by using specific primers (see Table 1). Expression of GAPDH was used as a loading control. PCR products were visualized with ethidium bromide on a 2% agarose gel. (B) CD1d protein expression was evaluated by Western blot in CD1d and Mok-transfected U2-OS/HOS/MG-63 cells. Total cellular proteins (20 µg) were resolved by 12% polyacrylamide gel and immunoblotted with the specific mAb anti-CD1d (37 kDa). β-actin (42 kDa) was used as internal control. (C) Concentration–response curves for cytotoxicity of drug and drug + iNKT cells in naive (black line)/siCD1d (red line) OS cells. The mean cytotoxicity % ± SEM for each condition was calculated from three replicate experimental wells. §§P≤ 0.01; §P≤ 0.05 versus iNKT cells + drug-treated OS cells.
To validate the silencing procedure, we repeated the co-culture experiments using siCD1d-OS cells as target cells. As shown in Figure 6C, the iNKT cell-induced enhancement of drug cytotoxicity was significantly (P≤ 0.05) reduced using siCD1d-OS cells, confirming that the increase in OS cell death is mainly due to the direct, CD1d-dependent iNKT cell cytotoxicity.
Mechanism of iNKT cell toxicity
iNKT cells can directly kill tumour cells by at least two different mechanisms: the release of perforine/granzyme B lytic granules and the Fas/FasL interaction (Nicol et al., 2000; Lisbonne et al., 2004). The perforine/granzyme B pathway requires the synthesis and the accumulation of these lytic mediators into the cells, while the Fas/FasL pathway requires high expression levels of both FasL in iNKT cells and Fas in tumour cells (Chávez-Galán et al., 2009).
To evaluate the mechanisms involved in the iNKT cell-induced enhancement of OS cell death, we first determined by FACS the expression of both perforine/granzyme B and FasL in iNKT cells. Figure 7A shows that iNKT cells express detectable amounts of these proteins after 3 h of PMA-ionomycin treatment, and that the inhibition of these proteins (CMA or anti-FasL mAb cell treatment) leads to a significant reduction in iNKT cell-induced OS cytotoxicity (Figure 7B).
Figure 7.
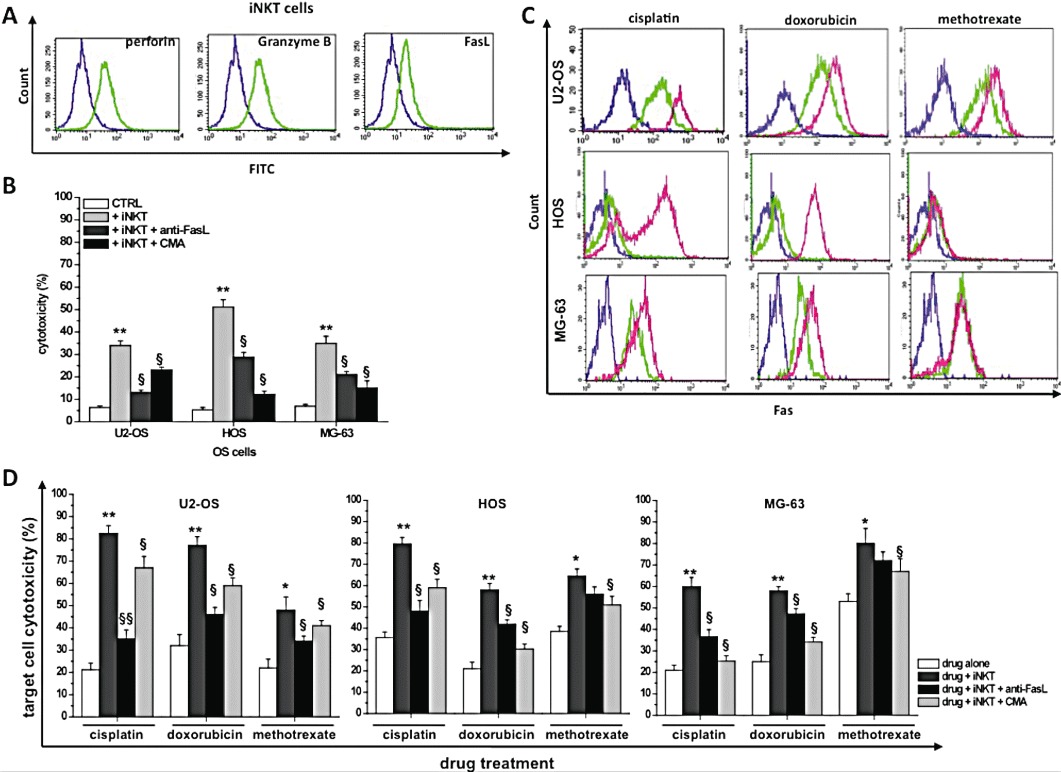
Mechanisms of iNKT cell toxicity. (A) α-GalCer-stimulated (day 12) iNKT cells were treated with 10 ng·mL−1 PMA plus 3 µg·mL−1 ionomycin for 3 h, stained with anti-perforin, anti-granzyme B, anti-FasL mAbs, and analysed by FACS. Representative overlay plots showing expression of perforine (left panel), granzyme B (middle panel) and FasL (right panel) in unstimulated (violet line) and stimulated (green line) iNKT cells. (B) OS cytotoxicity induced by iNKT cells (1:6 T : E ratio) in the presence/absence of anti-FasL (10 µg·mL−1) or CMA (100 ng·mL−1) compared to cells not treated with iNKT cells (CTRL). *P≤ 0.05; **P≤ 0.01 versus iNKT-treated OS cells; §P≤ 0.05; §§P≤ 0.01 versus iNKT-treated OS cells. (C) OS cells untreated/treated (24 h) with cisplatin/doxorubicin (3 µmol·L−1), or methotrexate (1 µmol·L−1), were stained with anti-Fas or isotype mAb and analysed by FACS. Representative overlay plots showing expression of Fas and appropriate isotype control (violet line) in each untreated (green line), drug-treated (pink line) OS cell line. (D) OS cells exposed to cisplatin/doxorubicin (3 µmol·L−1) or methotrexate (1 µmol·L−1) were untreated/treated with iNKT cells at 1:6 T : E ratio, in the presence/absence of anti-FasL-blocking (10 µg·mL−1) mAb or CMA (100 ng·mL−1). Cytotoxicity was calculated as described in the Methods section and represents the mean cytotoxicity % ± SEM for each condition calculated from three replicate experimental wells. *P≤ 0.05; **P≤ 0.01 versus drug-treated OS cells; §P≤ 0.05; §§P≤ 0.01 versus iNKT cells + drug-treated OS cells.
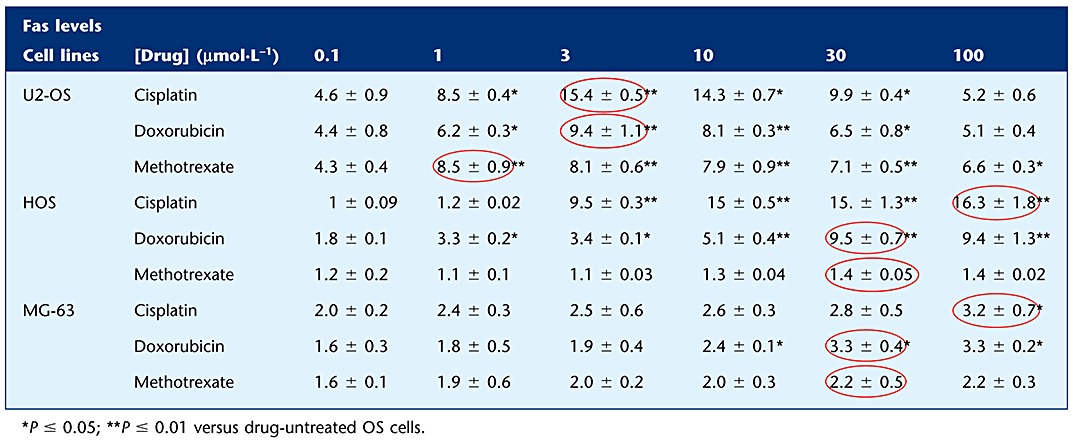
The expression of Fas was then studied in OS cells. The results demonstrated that U2-OS, HOS and MG-63 cells constitutively express Fas at their cell surface (Figure 7C), and that this expression can be significantly increased by anti-neoplastic drugs. When each OS cell line was treated with increasing concentrations (0.1–100 µmol·L−1) of each tested drug for 24 h, a significant increase in Fas expression was determined (Table 2).
Table 2.
Fas levels in drug-treated OS cell lines
 |
These results suggest that anti-neoplastic drugs, increasing the surface expression of Fas on OS cells, could synergistically act with iNKT cells, which express FasL, to kill tumour cells through this death pathway.
To validate this hypothesis, we repeated the same co-culture experiments in the presence of a specific anti-FasL-blocking mAb. In these conditions, the enhancement of OS cell death was significantly (P≤ 0.05) reduced (Figure 7D), confirming the involvement of the Fas/FasL pathway.
Finally, the contribution of the perforin-mediated pathway was evaluated by measuring cytotoxicity in cells treated with CMA. Figure 7D shows that this pathway significantly contributes to OS killing and suggests that perforine plays a major role in OS cells expressing low Fas levels (see Table 2).
Discussion
iNKT cell-based immunotherapy plays a primarily protective role in several tumours (Terabe and Berzofsky, 2008), but no evidence has been published yet in OS. This in vitro study demonstrated that iNKT cells are able to kill OS cells and to potentiate the effects of some anti-neoplastic drugs. These effects were shown to be dependent on CD1d expression on tumour cells and to be mediated by both perforine/granzyme B and Fas/FasL pathways.
The extremely variable frequency of circulating iNKT cells in human PBMCs (Metelitsa, 2004) led us to expand them before experiments. Following a preliminary set of experiments (data not shown), the highest iNKT cell proliferation rate (+100-fold increase over basal conditions) was obtained after 12 days of PBMCs stimulation with 10 ng·mL−1α-GalCer + 40 U·mL−1 IL-2. The levels of cell expansion were comparable to those from other laboratories, but at a lower concentration of α-GalCer (10 vs. 100 ng·mL−1) (Rogers et al., 2004; Croudace et al., 2008). This difference might be due to the high starting number of PBMCs (5 × 106 PBMC per well) that we have used in comparison to that from other studies (1–2 × 106 PBMCs per well) (Rogers et al., 2004; Motohashi et al., 2006). This result confirms that the generation of greater numbers of iNKT cells is possible with a higher starting number of PBMCs (Croudace et al., 2008).
At the end of proliferation, we observed a slight, but significant, expansion of NK cells. This is not surprising, because activated iNKT cells can produce and release a broad spectrum of both Th1 and Th2 cytokines that, in turn, stimulate the proliferation/activation of other leucocytes, including NK cells (Smyth et al., 2001). Of note, no expansion of CD8+ cytotoxic T-cells (CTL), which contribute to cell death in several tumour models (Terabe et al., 2006), was measured after α-GalCer treatment. However, we cannot exclude the possibility that a subset of CD1d-restricted NKT cells, which have negative immunoregulatory function on CD8+ T-cells, is present in our cell preparation.
Our iNKT cell population (FACS analysis) was composed of a greater number of CD4+ than CD4- cells, and this initial proportion was maintained at the end of the expansion. Because CD4- iNKT cells can mediate high tumour protection, whereas CD4+ iNKT cells are less protective (Crowe et al., 2005), our in vitro model could be subject to some criticisms. However, independent of their phenotype, expanded iNKT cells can induce equivalent toxicity against tumour cells (Takahashi et al., 2002), displaying an overall Th0 cytokine pattern in vitro, in which Th1 type is produced markedly more than Th2-type cytokines (van der Vliet et al., 2001). Experiments aimed to skew iNKT cells towards a preferential expansion of the CD4- subtype, for instance, by culturing the starting cell population in the presence of IL-15 (Buddingh et al., 2011), might be useful to elucidate this point.
Among the commercially available OS cell lines, we selected those considered a good tool to study drug responses in tumours (Chou and Gorlick, 2006): U2-OS cells, relatively resistant to cisplatin and methotrexate (Serra et al., 1993; Cole et al., 2002), and HOS/MG63, which do not exhibit this property (Robson et al., 2002).
We have reported here the first evidence that CD1d is expressed on OS cells: U2-OS, HOS and MG-63 cell lines expressed CD1d and this expression is essential for iNKT cell-induced direct tumour cytolysis in vitro and tumour immunity in vivo (Bagnara et al., 2009). With the exception of tumours arising from lymphoid and myeloid neoplasms (Metelitsa et al., 2003; Fais et al., 2004), certain neurological tumours (Exley et al., 2011) and prostate cancers (Nowak et al., 2010), the expression pattern of CD1d on malignant cells is largely unknown, and our data add new insights into this field.
Our results indicate that iNKT cells recognize and kill CD1d+ OS cells, but not CD1d- osteoblasts, confirming the CD1d restriction of NKT cell cytotoxicity, as mentioned above.
In addition, OS cell death was obtained without pulsing tumour cells with α-GalCer (see Figure 3B), and this suggests that the iNKT cell TCR can interact with lipid ligand(s), physiologically expressed on tumour cells (Song et al., 2009) and loaded on CD1d molecules (Brossay et al., 1998).
The existence of tumour-specific glycolipid antigen(s) suggests also that, to escape NKT cell-mediated immune surveillance, tumours may repress the synthesis of these antigens, lose their CD1d expression or activate inhibitory pathways/mediators. The isolation of a glycolipid (lysophosphatidylcholine), from plasma of myeloma patients, which activates type II NKT cells, that negatively regulate anti-tumour responses and suppress the protective function of type I NKT cells, confirms this possibility (Dhodapkar and Richter, 2011).
The identification of the specific antigen(s), expressed on different tumour cells, might be also useful to increase the effectiveness of immunotherapy. The ex vivo expansion of iNKT cells could, in fact, be performed using tumour-specific antigens, instead of the non-specific α-GalCer, so that a highly tumour-specific iNKT cell population can be generated.
Cell exposure to an anti-CD1d-blocking mAb, which prevents the interaction between tumour CD1d and iNKT cell TCR, drastically reduced, but did not abolish, the iNKT cell-induced OS cytotoxicity (see Figure 3C). This result is in complete agreement with observations from other laboratories (Carnaud et al., 1999; Eberl and MacDonald, 2000) and confirms that the overall cytotoxicity is due to both direct and indirect cytotoxicity.
It is noteworthy that the iNKT cells did not kill human normal CD1d+ MSC-BM (see Figure 4): iNKT cells might be able to specifically discriminate normal from malignant CD1d+cells, perhaps through the expression of specific endogenous lipid antigen(s), naturally loaded on tumour cells, but not on normal CD1d+cells. This property might confer a high degree of tumour specificity to iNKT cell-mediated immunotherapy.
We have explored the possibility that a combination of iNKT cell treatment plus chemotherapy might provide a valid option in OS, as it does in other neoplastic diseases (Zitvogel et al., 2008). We considered doxorubicin, cisplatin, methotrexate, but not ifosfamide, because ifosfamide, as a prodrug, requires hepatic metabolic activation before it can exert its effect (Johnstone et al., 2000).
Critical points of a combination therapy are the dosage and the timing of drug administration; frequently, in vitro studies have been performed using drug pretreatment followed by immunotherapy (Mattarollo et al., 2006). Conversely, in agreement with data from the literature (Lake and Robinson, 2005), we exposed OS cells to both each drug and iNKT cells simultaneously, and this is because we decided to measure all toxic effects, including drug-induced effects on effector cells.
Drug-induced OS cell toxicity was significantly enhanced by iNKT cell exposure; the greatest effect was obtained with cisplatin. Yet, the mechanisms underlying this effect have not been studied, but it may be related to a greater ability of cisplatin than other drugs to modulate the surface expression of proteins, involved in tumour immune cell recognition (Zitvogel et al., 2008), and/or co-inhibitory/stimulatory molecules, involved in T-cell activation. In particular, on tumour cells, cisplatin down-regulates the expression of programmed death receptor-ligand 2, a T-cell inhibitory molecule (Lesterhuis et al., 2011). The anti-neoplastic drugs, used in this study, may differently affect the expression of these molecules on OS cells and, through this mechanism, differently modulate the ability of iNKT cells, expressing several co-stimulatory (e.g. CD28 and ICOS) and co-inhibitory (e.g. CTLA-4 and PD-1) receptors (van den Heuvel et al., 2011), to kill tumour cells.
When we silenced the expression of CD1d on OS cells the potentiating effect was abolished, confirming that it was mainly due to direct cytotoxicity.
Anti-neoplastic drugs (e.g. cisplatin and cyclophosphamide) can promote immune cell-mediated tumour apoptosis via death receptor pathways, including Fas, TRAIL and NKG2d ligand (Zitvogel et al., 2008).
Cisplatin, doxorubicin and methotrexate differently induce Fas expression in different types of tumour cells: in the context of OS cells, all these drugs are able to induce an increase in Fas expression in HOS and MG-63 OS cells (Fellenberg et al., 2000), but not in SAOS-LM6 cells (Duan et al., 2004). We completed these data by showing that cisplatin, doxorubicin and methotrexate differently enhance Fas expression in U2-OS cells, with cisplatin being the most potent drug. The block of the Fas pathway significantly reduced the iNKT cell-induced enhancement of drug cytotoxicity (see Figure 7D), demonstrating that it was, at least in part, mediated by activation of the Fas/FasL pathway. Immediately after being activated, iNKT cells express very high levels of mRNA for perforine and granzyme B (primary pathway) (Metelitsa et al., 2003). From our data, this pathway not only significantly contributes to OS killing, but also plays a major role in OS cells expressing low Fas levels. These results should be investigated further. Also they are not consistent with the results of Wingender et al. (2010) who demonstrated that iNKT cells exert a CD1d-dependent tumour cytotoxicity, which requires Fas/FasL activation and is completely independent of the perforine/granzyme B pathway.
In conclusion, this study provides the first in vitro evidence that iNKT cells have the ability to recognize and kill the OS cells and to enhance drug-induced cytotoxicity.
Acknowledgments
We thank Dr Michela Bosetti, Department of Pharmaceutical Sciences, Univ. ‘Piemonte Orientale’, Novara, Italy, for supplying the human MG-63 cell line and the primary osteoblast cultures; Dr Marta Serafini, Tettamanti Research Center, Monza, Italy, for human primary MSC-BM cell cultures; Prof. Luigi Panza, Department of Pharmaceutical Sciences, University of ‘Piemonte Orientale’, Novara, Italy, for providing α-GalCer (KRN-7000); Dr Mario Botta, Unit of Medical Oncology, Casale Monferrato Hospital, Casale Monferrato, AL, Italy, for providing cisplatin, doxorubicin and methotrexate; and Dr Antonella Vallario for flow cytometry technical assistance.
This study was supported by grants from University of ‘Piemonte Orientale Amedeo Avogadro’ and ‘Regione Piemonte’, Italy.
Glossary
- α-GalCer
α-galactosylceramide
- APC
allophycocyanin
- CAM
calcein-AM
- CMA
concanamycin A
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- DN
double negative
- DP
double positive
- FasL
Fas ligand
- iNKT
invariant NKT
- MSC-BM
mesenchymal stem cell; OS, osteosarcoma
- PBMC
peripheral blood mononuclear cell
- PE
phycoerythrin
- PerCP
peridinin-chlorophyll protein
- PI
propidium iodide
- SART
squamous cell carcinoma antigen recognized by T-cell
- siCD1d
CD1d silenced
- siRNA
small interfering RNA
- TCR
T-cell receptor
- TRAIL
tumour-necrosis factor-related apoptosis-inducing ligand
- Treg
regulatory T-cell
Conflict of interest
None.
References
- Bagnara D, Ibatici A, Corselli M, Sessarego N, Tenca C, De Santanna A. Adoptive immunotherapy mediated by ex vivo expanded natural killer T cells against CD1d-expressing lymphoid neoplasms. Haematologica. 2009;94:967–974. doi: 10.3324/haematol.2008.001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois E, Van LP, Samson M, Diem S, Barra A, Roga S, et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur J Immunol. 2009;39:1046–1055. doi: 10.1002/eji.200838575. [DOI] [PubMed] [Google Scholar]
- Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med. 1998;188:1521–1528. doi: 10.1084/jem.188.8.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddingh EP, Schilham MW, Ruslan SE, Berghuis D, Szuhai K, Suurmond J, et al. Chemotherapy-resistant osteosarcoma is highly susceptible to IL-15-activated allogeneic and autologous NK cells. Cancer Immunol Immunother. 2011;60:575–586. doi: 10.1007/s00262-010-0965-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, et al. Cutting edge: cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol. 1999;163:4647–4650. [PubMed] [Google Scholar]
- Chávez-Galán L, Arenas-Del Angel MC, Zenteno E, Chávez R, Lascurain R. Cell death mechanisms induced by cytotoxic lymphocytes. Cell Mol Immunol. 2009;6:15–25. doi: 10.1038/cmi.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou AJ, Gorlick R. Chemotherapy resistance in osteosarcoma: current challenges and future directions. Expert Rev Anticancer Ther. 2006;6:1075–1085. doi: 10.1586/14737140.6.7.1075. [DOI] [PubMed] [Google Scholar]
- Clark JC, Dass CR, Choong PF. A review of clinical and molecular prognostic factors in osteosarcoma. J Cancer Res Clin Oncol. 2008;134:281–297. doi: 10.1007/s00432-007-0330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole PD, Smith AK, Kamen BA. Osteosarcoma cells, resistant to methotrexate due to nucleoside and nucleobase salvage, are sensitive to nucleoside analogs. Cancer Chemother Pharmacol. 2002;50:111–116. doi: 10.1007/s00280-002-0478-7. [DOI] [PubMed] [Google Scholar]
- Croudace JE, Curbishley SM, Mura M, Willcox CR, Illarionov PA, Besra GS, et al. Identification of distinct human invariant natural killer T-cell response phenotypes to alpha-galactosylceramide. BMC Immunol. 2008;9:71. doi: 10.1186/1471-2172-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crough T, Purdie DM, Okai M, Maksoud A, Nieda M, Nicol AJ. Modulation of human Valpha24(+) Vbeta11(+) NKT cells by age, malignancy and conventional anticancer therapies. Br J Cancer. 2004;91:1880–1886. doi: 10.1038/sj.bjc.6602218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe NY, Coquet JM, Berzins SP, Kyparissoudis K, Keating R, Pellicci DG, et al. Differential antitumor immunity mediated by NKT cell subsets in vivo. J Exp Med. 2005;202:1279–1288. doi: 10.1084/jem.20050953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker S, Winkelmann W, Nies B, van Valen F. Cytotoxic effect of methotrexate and its solvent on osteosarcoma cells in vitro. J Bone Joint Surg Br. 1999;81:545–551. doi: 10.1302/0301-620x.81b3.9167. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Richter J. Harnessing natural killer T (NKT) cells in human myeloma: progress and challenges. Clin Immunol. 2011;140:160–166. doi: 10.1016/j.clim.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med. 2003;197:1667–1676. doi: 10.1084/jem.20021650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Zhou Z, Jia SF, Colvin M, Lafleur EA, Kleinerman ES. Interleukin-12 enhances the sensitivity of human osteosarcoma cells to 4-hydroperoxycyclophosphamide by a mechanism involving the Fas/Fas-ligand pathway. Clin Cancer Res. 2004;10:777–783. doi: 10.1158/1078-0432.ccr-1245-02. [DOI] [PubMed] [Google Scholar]
- Eberl G, MacDonald HR. Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur J Immunol. 2000;30:985–992. doi: 10.1002/(SICI)1521-4141(200004)30:4<985::AID-IMMU985>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Exley MA, Lynch L, Varghese B, Nowak M, Alatrakchi N, Balk SP. Developing understanding of the roles of CD1d-restricted T cell subsets in cancer: reversing tumour-induced defects. Clin Immunol. 2011;140:184–195. doi: 10.1016/j.clim.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fais F, Morabito F, Stelitano C, Callea V, Zanardi S, Scudeletti M, et al. CD1d is expressed on B-chronic lymphocytic leukemia cells and mediates alpha-galactosylceramide presentation to natural killer T lymphocytes. Int J Cancer. 2004;109:402–411. doi: 10.1002/ijc.11723. [DOI] [PubMed] [Google Scholar]
- Fallarini S, Paoletti T, Panza L, Lombardi G. Alpha-galactosylceramide modulates the induction of indoleamine 2,3-dioxygenase in antigen presenting cells. Biochem Pharmacol. 2008;76:738–750. doi: 10.1016/j.bcp.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Fallarini S, Miglio G, Paoletti T, Minassi A, Amoruso A, Bardelli C, et al. Clovamide and rosmarinic acid induce neuroprotective effects in in vitro models of neuronal death. Br J Pharmacol. 2009;157:1072–1084. doi: 10.1111/j.1476-5381.2009.00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallarini S, Magliulo L, Paoletti T, de Lalla C, Lombardi G. Expression of functional GPR35 in human iNKT cells. Biochem Biophys Res Commun. 2010;398:420–425. doi: 10.1016/j.bbrc.2010.06.091. [DOI] [PubMed] [Google Scholar]
- Fellenberg J, Mau H, Nedel S, Ewerbeck V, Debatin KM. Drug-induced apoptosis in osteosarcoma cell lines is mediated by caspase activation independent of CD95-receptor/ligand interaction. J Orthop Res. 2000;18:10–17. doi: 10.1002/jor.1100180103. [DOI] [PubMed] [Google Scholar]
- Ferrari S, Smeland S, Mercuri M, Bertoni F, Longhi A, Ruggieri P, et al. Italian and Scandinavian sarcoma groups. neoadjuvant chemotherapy with high-dose ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian sarcoma groups. J Clin Oncol. 2005;23:8845–8852. doi: 10.1200/JCO.2004.00.5785. [DOI] [PubMed] [Google Scholar]
- Fujii S. Exploiting dendritic cells and natural killer T cells in immunotherapy against malignancies. Trends Immunol. 2008;29:242–249. doi: 10.1016/j.it.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res. 2002;8:3702–3709. [PubMed] [Google Scholar]
- Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what's in a name? Nat Rev Immunol. 2004;4:231–237. doi: 10.1038/nri1309. [DOI] [PubMed] [Google Scholar]
- van den Heuvel MJ, Garg N, Van Kaer L, Haeryfar SM. NKT cell costimulation: experimental progress and therapeutic promise. Trends Mol Med. 2011;17:65–77. doi: 10.1016/j.molmed.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogendoorn PC, Athanasou N, Bielack S, De Alava E, Dei Tos AP, Ferrari S, et al. ESMO/EUROBONET working group, bone sarcomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21:v204–v213. doi: 10.1093/annonc/mdq223. [DOI] [PubMed] [Google Scholar]
- Ishihara S, Nieda M, Kitayama J, Osada T, Yabe T, Kikuchi A, et al. Alpha-glycosylceramides enhance the antitumor cytotoxicity of hepatic lymphocytes obtained from cancer patients by activating CD3-CD56+ NK cells in vitro. J Immunol. 2000;165:1659–1664. doi: 10.4049/jimmunol.165.3.1659. [DOI] [PubMed] [Google Scholar]
- Janeway KA, Grier HE. Sequelae of osteosarcoma medical therapy: a review of rare acute toxicities and late effects. Lancet Oncol. 2010;11:670–678. doi: 10.1016/S1470-2045(10)70062-0. [DOI] [PubMed] [Google Scholar]
- Johnstone EC, Lind MJ, Griffin MJ, Boddy AV. Ifosfamide metabolism and DNA damage in tumour and peripheral blood lymphocytes of breast cancer patients. Cancer Chemother Pharmacol. 2000;46:433–441. doi: 10.1007/s002800000185. [DOI] [PubMed] [Google Scholar]
- Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol. 2005;23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742. [DOI] [PubMed] [Google Scholar]
- Lake RA, Robinson BW. Immunotherapy and chemotherapy–a practical partnership. Nat Rev Cancer. 2005;5:397–405. doi: 10.1038/nrc1613. [DOI] [PubMed] [Google Scholar]
- Lee PT, Benlagha K, Teyton L, Bendelac A. Distinct functional lineages of human V(alpha)24 natural killer T cells. J Exp Med. 2002;195:637–641. doi: 10.1084/jem.20011908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesterhuis WJ, Steer H, Lake RA. PD-L2 is predominantly expressed by Th2 cells. Mol Immunol. 2011;49:1–3. doi: 10.1016/j.molimm.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Lin H, Nieda M, Nicol AJ. Differential proliferative response of NKT cell subpopulations to in vitro stimulation in presence of different cytokines. Eur J Immunol. 2004;34:2664–2671. doi: 10.1002/eji.200324834. [DOI] [PubMed] [Google Scholar]
- Lisbonne M, Hachem P, Tonanny MB, Fourneau JM, Sidobre S, Kronenberg M, et al. In vivo activation of invariant V alpha 14 natural killer T cells by alpha-galactosylceramide sequentially induces Fas-dependent and -independent cytotoxicity. Eur J Immunol. 2004;34:1381–1388. doi: 10.1002/eji.200324828. [DOI] [PubMed] [Google Scholar]
- Luksch R, Perotti D, Cefalo G, Gambacorti Passerini C, Massimino M, Spreafico F, et al. Immunomodulation in a treatment program including pre- and post-operative interleukin-2 and chemotherapy for childhood osteosarcoma. Tumori. 2003;89:263–268. doi: 10.1177/030089160308900306. [DOI] [PubMed] [Google Scholar]
- Marina N, Gorlick R. Immune approaches to treating osteosarcoma. Cancer Biol Ther. 2009;8:981–983. doi: 10.4161/cbt.8.10.8602. [DOI] [PubMed] [Google Scholar]
- Mattarollo SR, Kenna T, Nieda M, Nicol AJ. Chemotherapy pretreatment sensitizes solid tumor-derived cell lines to V alpha 24+ NKT cell-mediated cytotoxicity. Int J Cancer. 2006;119:1630–1637. doi: 10.1002/ijc.22019. [DOI] [PubMed] [Google Scholar]
- Metelitsa LS. Flow cytometry for natural killer T cells: multi-parameter methods for multifunctional cells. Clin Immunol. 2004;110:267–276. doi: 10.1016/j.clim.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Metelitsa LS. Anti-tumor potential of type-I NKT cells against CD1d-positive and CD1d-negative tumors in humans. Clin Immunol. 2011;140:119–129. doi: 10.1016/j.clim.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metelitsa LS, Weinberg KI, Emanuel PD, Seeger RC. Expression of CD1d by myelomonocytic leukemias provides a target for cytotoxic NKT cells. Leukemia. 2003;17:1068–1077. doi: 10.1038/sj.leu.2402943. [DOI] [PubMed] [Google Scholar]
- Molano A, Illarionov PA, Besra GS, Putterman C, Porcelli SA. Modulation of invariant natural killer T cell cytokine responses by indoleamine 2,3-dioxygenase. Immunol Lett. 2008;117:81–90. doi: 10.1016/j.imlet.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molling JW, Moreno M, van der Vliet HJ, van den Eertwegh AJ, Scheper RJ, von Blomberg BM, et al. Invariant natural killer T cells and immunotherapy of cancer. Clin Immunol. 2008;129:182–194. doi: 10.1016/j.clim.2008.07.025. [DOI] [PubMed] [Google Scholar]
- Montoya CJ, Pollard D, Martinson J, Kumari K, Wasserfall C, Mulder CB, et al. Characterization of human invariant natural killer T subsets in health and disease using a novel invariant natural killer T cell-clonotypic monoclonal antibody, 6B11. Immunology. 2007;122:1–14. doi: 10.1111/j.1365-2567.2007.02647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Rédini F, Gouin F, Cherrier B, Heymann D. Osteosarcoma: current status of immunotherapy and future trends. Oncol Rep. 2006;15:693–700. [PubMed] [Google Scholar]
- Morris ES, MacDonald KP, Rowe V, Banovic T, Kuns RD, Don AL, et al. NKT cell-dependent leukemia eradication following stem cell mobilization with potent G-CSF analogs. J Clin Invest. 2005;115:3093–3103. doi: 10.1172/JCI25249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. 2006;12:6079–6086. doi: 10.1158/1078-0432.CCR-06-0114. [DOI] [PubMed] [Google Scholar]
- Nicol A, Nieda M, Koezuka Y, Porcelli S, Suzuki K, Tadokoro K, et al. Human invariant valpha24+ natural killer T cells activated by alpha-galactosylceramide (KRN7000) have cytotoxic anti-tumour activity through mechanisms distinct from T cells and natural killer cells. Immunology. 2000;99:229–234. doi: 10.1046/j.1365-2567.2000.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieda M, Nicol A, Koezuka Y, Kikuchi A, Lapteva N, Tanaka Y, et al. TRAIL expression by activated human CD4(+)V alpha 24NKT cells induces in vitro and in vivo apoptosis of human acute myeloid leukemia cells. Blood. 2001;97:2067–2074. doi: 10.1182/blood.v97.7.2067. [DOI] [PubMed] [Google Scholar]
- Nowak M, Arredouani MS, Tun-Kyi A, Schmidt-Wolf I, Sanda MG, Balk SP, et al. Defective NKT cell activation by CD1d+ TRAMP prostate tumor cells is corrected by interleukin-12 with alpha-galactosylceramide. Plos ONE. 2010;5:e11311. doi: 10.1371/journal.pone.0011311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti T, Fallarini S, Gugliesi F, Minassi A, Appendino G, Lombardi G. Anti-inflammatory and vascular protective properties of 8-prenylapigenin. Eur J Pharmacol. 2009;620:120–130. doi: 10.1016/j.ejphar.2009.08.015. [DOI] [PubMed] [Google Scholar]
- Ritter J, Bielack SS. Osteosarcoma. Ann Oncol. 2010;21(Suppl. 7):vii320–vii325. doi: 10.1093/annonc/mdq276. [DOI] [PubMed] [Google Scholar]
- Robson H, Meyer S, Shalet SM, Anderson E, Roberts S, Eden OB. Platinum agents in the treatment of osteosarcoma: efficacy of cisplatin vs. carboplatin in human osteosarcoma cell lines. Med Pediatr Oncol. 2002;39:573–580. doi: 10.1002/mpo.10076. [DOI] [PubMed] [Google Scholar]
- Rogers PR, Matsumoto A, Naidenko O, Kronenberg M, Mikayama T, Kato S. Expansion of human Valpha24+ NKT cells by repeated stimulation with KRN7000. J Immunol Methods. 2004;285:197–214. doi: 10.1016/j.jim.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Serra M, Scotlandi K, Manara MC, Maurici D, Lollini PL, De Giovanni C, et al. Establishment and characterization of multidrug-resistant human osteosarcoma cell lines. Anticancer Res. 1993;13:323–329. [PubMed] [Google Scholar]
- Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. 2001;13:459–463. doi: 10.1093/intimm/13.4.459. [DOI] [PubMed] [Google Scholar]
- Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. 2009;119:1524–1536. doi: 10.1172/JCI37869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Fu ZM, Fang CQ, Li JH. Induction of apoptosis in osteogenic sarcoma cells by combination of tumor necrosis factor-related apoptosis inducing ligand and chemotherapeutic agents. Chin Med J. 2007;120:400–404. [PubMed] [Google Scholar]
- Ta HT, Dass CR, Choong PF, Dunstan DE. Osteosarcoma treatment: state of the art. Cancer Metastasis Rev. 2009;28:247–263. doi: 10.1007/s10555-009-9186-7. [DOI] [PubMed] [Google Scholar]
- Tahir SM, Cheng O, Shaulov A, Koezuka Y, Bubley GJ, Wilson SB, et al. Loss of IFNgamma production by invariant NK T cells in advanced cancer. J Immunol. 2001;167:4046–4050. doi: 10.4049/jimmunol.167.7.4046. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Chiba S, Nieda M, Azuma T, Ishihara S, Shibata Y, et al. Cutting edge: analysis of human V alpha 24+CD8+ NK T cells activated by alpha-galactosylceramide-pulsed monocyte-derived dendritic cells. J Immunol. 2002;168:3140–3144. doi: 10.4049/jimmunol.168.7.3140. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Tashiro T, Dashtsoodol N, Hongo N, Watarai H. The specialized iNKT cell system recognizes glycolipid antigens and bridges the innate and acquired immune systems with potential applications for cancer therapy. Int Immunol. 2010;22:1–6. doi: 10.1093/intimm/dxp104. [DOI] [PubMed] [Google Scholar]
- Terabe M, Berzofsky JA. The role of NKT cells in tumor immunity. Adv Cancer Res. 2008;101:277–348. doi: 10.1016/S0065-230X(08)00408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terabe M, Khanna C, Bose S, Melchionda F, Mendoza A, Mackall CL, et al. CD1d-restricted natural killer T cells can down-regulate tumor immunosurveillance independent of interleukin-4 receptor-signal transducer and activator of transcription 6 or transforming growth factor-beta. Cancer Res. 2006;66:3869–3875. doi: 10.1158/0008-5472.CAN-05-3421. [DOI] [PubMed] [Google Scholar]
- Théoleyre S, Mori K, Cherrier B, Passuti N, Gouin F, Rédini F, et al. Phenotypic and functional analysis of lymphocytes infiltrating osteolytic tumors: use as a possible therapeutic approach of osteosarcoma. BMC Cancer. 2005;5:123–133. doi: 10.1186/1471-2407-5-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda N, Murayama K, Ishida H, Matsunaga K, Komiya S, Itoh K, et al. Expression of a newly defined tumor-rejection antigen SART3 in musculoskeletal tumors and induction of HLA class I-restricted cytotoxic T lymphocytes by SART3-derived peptides. J Orthop Res. 2001;19:346–351. doi: 10.1016/S0736-0266(00)90031-7. [DOI] [PubMed] [Google Scholar]
- van der Vliet HJ, von Blomberg BM, Nishi N, Reijm M, Voskuyl AE, van Bodegraven AA, et al. Circulating V(alpha24+) Vbeta11+ NKT cell numbers are decreased in a wide variety of diseases that are characterized by autoreactive tissue damage. Clin Immunol. 2001;100:144–148. doi: 10.1006/clim.2001.5060. [DOI] [PubMed] [Google Scholar]
- van der Vliet HJ, Molling JW, von Blomberg BM, Nishi N, Kölgen W, van den Eertwegh AJ, et al. The immunoregulatory role of CD1d-restricted natural killer T cells in disease. Clin Immunol. 2004;112:8–23. doi: 10.1016/j.clim.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Whelan JS, Jinks RC, McTiernan A, Sydes MR, Hook JM, Trani L, et al. Survival from high-grade localised extremity osteosarcoma: combined results and prognostic factors from three European osteosarcoma intergroup randomised controlled trials. Ann Oncol. 2012;23:1607–1616. doi: 10.1093/annonc/mdr491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender G, Krebs P, Beutler B, Kronenberg M. Antigen-specific cytotoxicity by invariant NKT cells in vivo is CD95/CD178-dependent and is correlated with antigenic potency. J Immunol. 2010;185:2721–2729. doi: 10.4049/jimmunol.1001018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Tesniere A, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of anticancer chemotherapy. Bull Acad Natl Med. 2008;192:1469–1478. [PubMed] [Google Scholar]