

Abstract
The objective of this review was to analyze components of the vitamin D and their potential in preventing and treating colorectal cancer. The active form of vitamin D, 1α25 (OH2) D3, targets the wnt/β-catenin pathway by up regulating key tumor suppressor genes such as e-cadherin, which promotes an epithelial phenotype, but this effect is only useful when the vitamin D receptor (VDR) is present. Colorectal cell lines have shown that VDR expression levels decrease in the later stages of colon cancer. In colorectal cancers with low VDR expression, treatments could target the genomic and epigenomic level alterations to increase VDR expression through modulating transcription factors such as SNAIL1 or utilizing histone deacetyltransferases (HDAC) inhibitors, respectively. Finally, epidemiological studies suggest that the current RDA should be raised to 2000IU in order to raise serum 25(OH)D3 levels above 30ng/ml, this increase in vitamin D status can most efficiently be obtained from sun exposure or vitamin D supplementation. In summary, vitamin Dand its metabolites could be utilized for treatment and preventive strategies for colon cancer.
Keywords: Vitamin D, colon cancer, vitamin D receptor
Introduction
Colon Cancer
According to the American Cancer Society, colon cancer is the third most commonly diagnosed cancer and the third leading cause of cancer death. Colon carcinogenesis begins with the development of adenomatous polyps, which are usually benign but if left untreated or undetected can develop into metastatic cancer. As with most cancers, colon cancer exhibits disrupted signaling. One of the key pathways that is disrupted in colon cancer is the wnt/β-catenin signaling pathway, which is often regarded as part of the initial event leading to colon cancer 1, 2. In a normal cell, the wnt/β-catenin signaling pathway is tightly regulated. β-catenin is normally regulated by the phosphorylation of the NH2 terminal region by glycogen synthase kinase-3β (GSK3). The cytosolic proteins axin and adenomatous polyposis coli (APC) are required for GSK3 to properly phosphorylate β-catenin, which can then be targeted for degradation 3–5. In colon cancer, the wnt/β-catenin pathway is disrupted due to mutations in β-catenin or APC; for example, APC is mutated in 80–90% of colon cancers 6. These mutations prevent the phosphorylation of β-catenin and contribute to its accumulation in the cytosol of the cells, un-phosphorylated β-catenin then is able to migrate and accumulate in the nucleus7, 8. Once in the nucleus, β-catenin dimerizes with DNA-bound T cell factor (TCF1–4), which lead to the expression of genes (e.g., c-myc, cyclin D1) capable of inducing the transformation of normal cells into an oncogenic phenotype 9–12. Recent research has suggested that components of the vitamin D pathway can modulate the unregulated wnt/β-catenin signaling.
Vitamin D(1α25 (OH2) D3) and Vitamin D Receptor (VDR) and Colon Cancer
Vitamin D3 (cholecalciferol) is a fat soluble vitamin that can be obtained both endogenously and exogenously. Mammals have the ability to generate vitamin D3 by exposing the skin to ultraviolet light, which causes7-dehydrocholesterol to convert into vitamin D3. Alternatively, mammals can obtain vitamin D3 from dietary sources, specifically dairy products13. In order to form the active form of vitamin D3, 1α25-dihydroxycholecaliferol (1α25 (OH2) D3), vitamin D3 is hydroxylated by 25-hydroxylase and 1α hydroxylase in the liver and kidney, respectively14. 1α25 (OH2) D3 can then act as a steroid messenger to carry out multiple cellular functions by mediating its effects through the vitamin D receptor (VDR).
Unbound 1α25 (OH2) D3 can enter a cell and bind to a VDR present in the cytoplasm or the nucleus15. VDR is classified as a class II nuclear receptor, and can heterodimerize with retinoid X receptor (RXR). Once this heterodimer is formed, it can bind to the vitamin D response elements (VDREs), which are located in the promoter region of key genes 16. Many vitamin D target genes have been found to regulate cell cycle arrest and cell differentiation, p21, p27, and e-cadherin 17, 18. Therefore, it has been proposed that 1α25 (OH2) D3 can possibly be used as a therapeutic for cancer by mediating its effects through the VDR and up-regulating the above genes.
The above processes require the presence of VDRs. VDRs are expressed in normal colonic cells, but it has been shown that VDR expression levels decrease in the later stages of colon cancer; the mechanism behind this phenomenon is not fully understood 19. Therefore, treatment with 1α25 (OH2) D3 may not be as effective in the later stages of colon carcinogenesis. It has been suggested that both genomic and epigenetic modifications might be involved in the reduction of VDR expression. 20–25 For instance, Malinen et al. demonstrated that the down regulation of 25 (OH)D3 due to HDACs can be overcome with the use of HDAC inhibitors. 26 Furthermore, it is proposed that an individual could reduce their risk to colorectal cancer approximately 30–50% by either increasing vitamin D intake to 2000 IU/day or increasing their sun exposure to raise blood levels of 25 (OH)D3 to greater than 30 ng/ml 27. Recently, case control studies have shown an inverse relationship between serum 25 (OH)D3 and the incidence of polpys or adenomas in the colon.28, 29 Taken to gather, these studies suggest that a greater understanding of the protective molecular actions of vitamin D against colon cancer should be pursued. The purpose of this paper is to determine how better treatments and preventative strategies for colorectal cancer might be developed utilizing components of the vitamin D pathway.
Possible mechanisms of 1α25 (OH2) D3 in the differentiation phenotype of colon cancer cells
The wnt/β-catenin signaling pathway is highly unregulated in most colon cancers due to the over expression of cell division/metastatic genes by accumulated β-catenin, contributing to a highly proliferative undifferentiated cell phenotype 30. Recently, it has been shown that 1α25 (OH2) D3 promotes the differentiation of colon cancer cells by stimulating e-cadherin and inhibiting β-catenin signaling 18. E-cadherin is a key protein that is involved in the adhesion properties of epithelial cells 31, 32. The loss of e-cadherin is associated with the modulation of a normal epithelial cell phenotype to a more invasive metastatic phenotype; therefore, it is considered a tumor-suppressor 33, 34. However, β-catenin is considered a proto-oncogene because if unregulated it could contribute to the constitutive transcription of cellular proliferative genes 35. In Palmer et al., investigators used SW480 cells, a well characterized human colon cancer cell line, to determine the effects of 1α25 (OH2) D3 on e-cadherin and β-catenin 25. The SW480-ADH colon cancer cells express low levels of e-cadherin and accumulate elevated amounts of nuclear β-catenin. In addition, Palmer et al. showed that with the addition of 1α25 (OH2) D3 the VDR positive SW480 (SW480-ADH) cells developed a differentiated phenotype by increasing the mRNA expression of e-cadherin and accelerating the translocation of β-catenin from the nucleus to the cytoplasm. Moreover, 1α25 (OH2) D3 decreased β-catenin-TCF-4 transcription activity by increasing ligand-activated VDR 25. Mariadson et al. also demonstrated that the down-regulation of β-catenin-TCF interaction is related to the differentiation of colonic cells 36. The above experiments have been repeated in a sub-line of SW480 cells. The SW480-R cells express residual levels of VDR compared to the SW480-ADH cells, so the addition of 1α25 (OH2) D3 has no effect. Moreover, experiments conducted in the metastatic derivative of the SW480 cells, SW620, also have very low levels of VDR and did not respond to the addition of 1α25 (OH2) D3 25, 37.
It has been discussed that 1α25 (OH2) D3 can promote a differentiated phenotype in colon cancer cells by up-regulating e-cadherin 25. However, 1α25 (OH2) D3 could be aiding differentiation via other mechanisms. Recently, it has been found that 1α25 (OH2) D3 can induce the wnt antagonist DICKKOPF-1 (DKK-1) gene in colon cancer cells 38. The wnt/β-catenin pathway requires the presence of both the frizzled receptor and LDL receptor-related protein (LRP5/6); both receptors form a complex that accepts a wnt ligand and induces the wnt/β-catenin canonical pathway 39, 40. DKK-1 is a protein that can bind to the LRP 5/6 receptor and induce endocytosis; therefore, preventing the formation of the wnt-frizzled-LRP5/6 receptor complex, which decreases wnt/β-catenin canonical signaling 41, 42. It has been shown that DKK-1 transcription is increased by β-catenin/TCF-4, and should provide a negative feedback to wnt/β-catenin signaling; however, DKK-1 expression is low in colon cancer suggesting this feedback mechanism is disrupted 43. Moreover, it has been shown that key CpG island promoters of two wnt-inhibitors (DKK-1 and Wnt inhibitory factor-1 (WIF-1))are hypermethylated in colon cancer, which contribute to their decreased expression and lack of activity 44. Aguilera et al. has shown that 1α25 (OH2) D3 increases both DKK-1 mRNA and protein expression in SW480-ADH colon cancer cells. Moreover, researchers used immuno-deficient mice supplemented with EB1089 (vitamin D analog) and showed an increased expression of DKK-1 45. A less studied member of the DKK family is DKK-4. Interestingly, DKK-4 has contrasting effects compared to DKK-1. Franco et al. reported that DKK-4 was up-regulated in human colorectal tumors and had an inverse relationship with VDR expression. Moreover, it was demonstrated that DKK-4 was a down-stream target of TCF/β-catenin. However, in the presence of 1α25 (OH2) D3, DKK-4 expression was decreased 45. Therefore, it could be suggested that 1α25 (OH2) D3 can simultaneously hinder the wnt/β-catenin pathway and promote an epithelial phenotype by increasing the wnt antagonists (DKK-1) and decreasing the wnt agonist (DKK-4).
1α25 (OH2) D3 may also mediate its effects by interacting with transcription factors involved in epithelial cell function such as inhibitors of DNA-binding proteins (Id). Ids are important regulators of development, but if uncontrolled can promote tumorigenesis 46. Moreover, Ids have been found to contribute to colon carcinogenesis 47. Understanding the fact that 1α25 (OH2) D3 promotes colonic cellular differentiation, Fernandez-Garcia et al. proposed that 1α25 (OH2) D3 may alter Id expression in colon carcinoma cells 48. Using SW480 colon cancer cells, researchers demonstrated that treatment with 1α25 (OH2) D3 increased Id1 expression, which remained high in the differentiated phenotype. However, the presence of 1α25 (OH2) D3 decreased Id2 expression and promoted an anti-proliferative effect possibly by decreasing c-myc and the number of TCF/β-catenin complexes48. Interestingly, researchers also found that 1α25 (OH2) D3 blocked angiogenesis by decreasing such factors as vascular endothelial growth factor (VEGF)48. This area of research is very new and should be explored more in order to further characterize the roles of Id1 and Id2 in colon carcinogenesis. 1α25 (OH2) D3 may be promoting a normal epithelial phenotype in colon cancer cells through the above mechanisms; this is summarized in figure 1. However, many of these protective mechanisms are dependent on the presence of VDR, which is known to decrease as the progression of colon cancer advances. However, it is possible to explore reasons why VDR expression decreases in order to provide interventions so that treatment with 1α25 (OH2) D3 can be more effective.
Figure 1. Possible mechanisms of 1α25 (OH2) D3 in the differentiation phenotype of colon cancer cells.

1α25 (OH2) D3 maybe promoting a more differentiated phenotype in cancer cells by preventing the translocation of β-catenin or its ability to form complexes with TCF4, thus decreasing mRNA levels of genes involved in cell survival and proliferation. Additionally, 1α25 (OH2) D3 can interact with VDR and promote the mRNA expression of cell differentiation and arrest genes. Finally, 1α25 (OH2) D3 can alter Id expression in colon carcinoma cells, by increasing the expression of Id1 and decreasing the levels of Id2; therefore, promoting colon cancer cell differentiation and decreasing proliferation, respectively.
Low VDR expression in colon cancer: explanations at a genomic and epigenomic level
Low VDR expression in Colon Cancer: a Genomic Explanation
As mentioned previously, most colon carcinomas express very low levels of VDR in the later stages of development; therefore, this makes treatment with 1α25 (OH2) D3 challenging. Little is known on how or why the VDR expression levels decrease in colon carcinogenesis. However, if the mechanism is discovered then treatment strategies can be targeted to increasing VDR expression in the later stages of colon carcinogenesis. One potential mechanism for the low VDR expression in colon cancer involves a transcription factor called SNAIL1. SNAIL1 has been correlated with low VDR expression in human colon cancers 49. SNAIL1 is a zinc-finger transcription factor that binds to the VDR gene promoter and represses activity and therefore decreases VDR mRNA expression levels 23, 49; this is summarized in figure 2a. SNAIL1 has been well characterized in tumor invasion and epithelial mesenchymal transition (EMT), and has also been shown to suppress e-cadherin gene expression 50, 51. In Larriba et al., severe immune-deficient female scid mice were injected with either 5×10^6 SNAIL1 or mock-infected SW480 cells and treated with either a placebo or EB1089 (vitamin D analog) 21. Also, they used SW480- ADH cells and 1α25 (OH2) D3 for numerous in vitro experiments 21. Collectively, for the in vitro studies, the authors discovered that SNAIL1 diminished the translocation of β-catenin from the nucleus that was induced by 1α25 (OH2) D3 in the SW480 cells; therefore, the expression of genes activated by β-catenin were increased. Also, the presence of SNAIL1 removed 1α25 (OH2) D3 inhibition on cell proliferation and decreased VDR protein expression. For the animal studies, Larriba et al. found that in the animals injected with the SNAIL1 SW480 cells, the nuclear exportation of β-catenin induced by EB1089 was significantly decreased compared to the mock-infected animals 21. In short, the above study concluded that SNAIL1 positively regulated the wnt/β-catenin pathway, decreased VDR expression, and potentially abolished the abilities of 1α25 (OH2) D3 to differentiate colonic cells.
Figure 2. Low VDR expression in Colon Cancer: a Genomic Explanation.
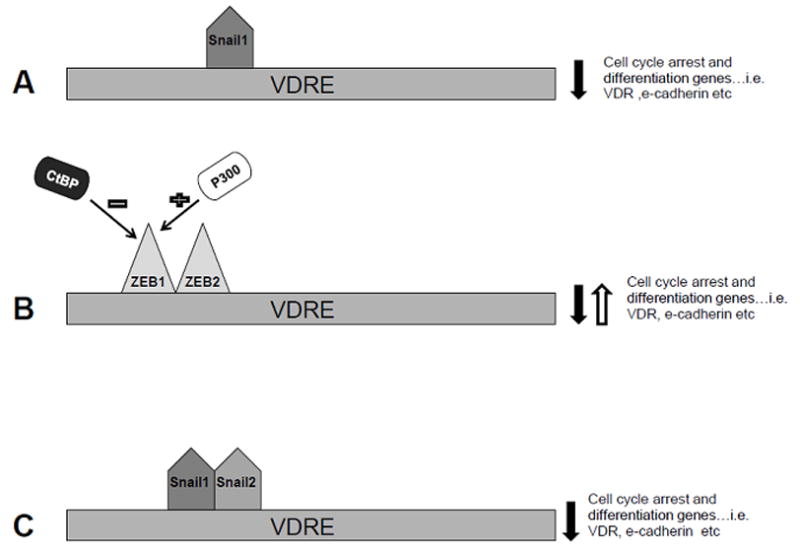
(A) SNAIL1 is a zinc-finger transcription factor that binds to the VDR gene promoter and represses activity and therefore decreases VDR mRNA expression levels. (B) ZEB1 and 2 are transcription factors that can dimerize and bind to the VDR promoter region and activate or repress the transcription of VDR mRNA depending on the presence of co-factors, such as p300 and CtBP.
It is well known that transcription factors work with other transcription factors in order to fine-tune the transcription of target genes. Therefore, it is of little surprise that researchers discovered that SNAIL1 was coordinating with other transcription factors in order to regulate VDR expression levels. One of these potential transcription factors is ZEB1. This unique transcription factor has the ability to down-regulate e-cadherin expression and stimulate VDR protein levels 52, 53. ZEB1 homodimerizes with ZEB2 and both are zinc-finger transcription factors that can bind to the VDR promoter region 54. ZEB1 has the ability to activate or repress transcription of key genes (e-cadherin, VDR), but this is dependent on the recruitment of co-activators or co-repressors, respectively. ZEB1 has an N-terminal region that can bind the co-activator, p300, an acetyltransferase which can alter chromatin structure so that it is in an open conformation 55, 56. On the other hand, in order for ZEB1/ZEB2 to repress transcription they need to recruit a co-repressor, CtBP 57, this is summarized in figure 2b. In the case of colon cancer, Pena et al. found an interesting relationship between SNAIL1, ZEB1, VDR, e-cadherin, P300, and CtBP 20. Using tumors collected from colon carcinoma patients, researchers measured mRNA levels of the above by using RT-PCR. Results showed that when SNAIL1 was over expressed, e-cadherin and VDR expression levels were decreased. However, both ZEB1 and e-cadherin expression levels correlated with elevated VDR levels, but if the co-repressor, CtBP, was expressed, then ZEB1 and e-cadherin had an inverse relationship. Moreover, high levels of p300 promoted a stronger correlation between ZEB1 and VDR expression; it could be proposed that designing p300 analogs in order to promote this relationship over the ZEB1-CtBP relationship might be beneficial 20. Overall, this study suggests that activities of SNAIL1 and ZEB1 are tightly regulated and dependent on the presence of co-activators or co-repressors.
Researchers later discovered a potential role for SNAIL2 in the suppression of VDR in colon cancer. SNAIL2, also known as SLUG, like SNAIL1, is a zinc-finger transcription factor that contributes to EMT 58. SNAIL2 decreases e-cadherin gene expression and other epithelial genes and is associated with a poor prognosis 59, this is summarized in figure 2c. Approximately, 60–70% of human colon tumors express SNAIL1 mRNA and have a low VDR expression 22, 49. However, VDR expression is low in approximately 80–90% of colon cancers; therefore, it could be suggested that another transcription factor could contribute to the down regulation of VDR in colon cancer. Therefore, Larriba et al. proposed that SNAIL2 represses VDR promoter activity and consequently decreases VDR mRNA and protein expression 22. Using SW480-ADH cells, it was found that SNAIL 2 decreased VDR activity and when present with SNAIL1 had an additive effect of the inhibition of VDR promoter activity. Like SNAIL1, SNAIL 2 blocked the effects of 1α25 (OH2) D3 on increased e-cadherin expression and exportation of β-catenin from the nucleus. Using colon tumors collected from humans, Larriba et al. found that SNAIL2 was up-regulated in 58% of the tumors 22. Furthermore, if both SNAIL1 and SNAIL2 were present, then VDR expression was significantly lower compared to a tumor expressing only one of the above transcription factors 22. Since SNAIL1 and SNAIL2 are present in the later stages of carcinogenesis, when malignant transformation is prevalent and VDR expression is low, they can contribute to the constitutive activation of the wnt/β-catenin pathway and can be potential targets for treatment.
As discussed thus far, 1α25 (OH2) D3 has the ability to promote differentiation in colon carcinoma cells; however, this is only possible if VDR expression is sufficient. Previously, we have discussed possible genomic explanations on why VDR expression might be down regulated in colon cancer, but it is possible that low VDR expression levels might be explained by epigenetics.
Low VDR expression in Colon Cancer: An Epigenetic Explanation
Recently, epigenetics and cancer have become a popular field of study. Epigenetics is the study of changes in gene expression caused by mechanisms unrelated to changes in the DNA sequence60. Unlike genetics, epigenetics is highly influenced by diet and lifestyle. Since carcinogenesis has become a topic of interest, many researchers are considering epigenetics as a potential contributor to cancer incidence and its progression. Colon cancer is one of the few cancers that has a well established relationship between specific genetic mutations and specific carcinogenic events 61. Slattery et al. has linked colon tumor mutations and epigenetic alterations that are associated with some of the key genetic polymorphisms that occur in colon carcinoma 24. Slattery’s results show that Fok1 VDR polymorphisms were associated with CIMP positive (CpG island methylator phenotype) and Ki-ras mutated colorectal tumors. However, the VDR polyA polymorphism was associated with a lower risk of developing Ki-ras mutations 24. In short, this study showed that the progression of colorectal cancer was dependent upon polymorphisms of VDR. However, other researchers believe that the VDR may not be mutated, but simply dys-functional.
Recently, it has been suggested that the relationship between 1α25 (OH2) D3 and VDR is possibly skewed in colon carcinogenesis due to the presence of histone deacetyltransferases (HDACs). HDACS are extremely active in carcinogenesis 62. Methylation often recruits other co-repressors such as HDACs in order to silence key regulatory genes such as tumor suppressors. HDAC’s alter nucleosome structure by removing acetyl groups from the n-terminal of the tails of histone octamers. By removing the acetyl groups, the nucleosome structure alters to a tight conformation and transcription is hindered 63. SNAIL1 often acts as a repressing transcription factor and is highly activated during EMT; at which time HDAC activity also increases 64. In colon cancer, it has been found that HDACs 1,2, and 3 have an increased expression, but the mechanism(s) behind this phenomenon are unknown. Godman et al. used SW480 and HCT116 cells (another human colon carcinoma cell line) and RNAi in order to determine the relationship between HDAC3 and vitamin D signaling. Results showed that knockdown of HDAC3, decreased β-catenin translocation to the nucleus, increased expression of key wnt inhibitors (TLE1 and TLE4), and increased expression of VDR in SW480 and HCT116 cells. Cells with HDAC3 shRNA were also more sensitive to the actions of 1α25 (OH2) D3 on cell cycle inhibition 25. A similar study was conducted using Caco2 cells (an immortalized line of human colorectal adenocarcinoma cells). The researchers treated cells with butyrate compounds (short-chain fatty acids produced by the colon during fermentation by intestinal bacteria that can acts as HDAC inhibitors) and found that VDR expression was increased 65. Therefore, HDAC inhibitors are another potential mechanism in which scientists can alter VDR expression in colon carcinogenesis in order to create a more effective treatment regime.
By understanding the mechanisms behind the decreased VDR expression observed in colon cancer, we can target these mechanisms and potentially restore VDR expression. Therefore, a likely future direction for this area is to create a treatment regime that includes both VDR ligands (1α25 (OH2) D3) and HDAC inhibitors.
Utilizing Vitamin D to prevent Colon Cancer
The best treatment for cancer is prevention. Many epidemiological studies have shown that high levels of serum 25-hydroxyvitamind D (25(OH)D3) are related to lower incidence rates in many cancers, particularly colon cancer 66–68. 25(OH)D3 is a metabolite in the vitamin D pathway that precedes the 1α hydroxylase in the kidneys in order to form 1α25 (OH2) D3. The vitamin D pathway includes both endogenous and exogenous sources of vitamin D. In other words, vitamin D3 from either exogenous or endogenous sources will both cause a significant increase in serum 25(OH)D3. According to a study using data from NHANES 2000–2004, up to 78% of Americans have a serum levels less than 30ng/ml of 25(OH)D3 69. Moreover, it has been shown that individuals who have 25(OH)D3 serum levels greater than 30ng/ml, which is considered an adequate amount of 25(OH)D3, have a 25% reduced risk from dying of colorectal cancer 27. Moreover, Gorham et al. demonstrated that there is a dose response relationship between serum 25(OH)D3 and the odds ratio of colon cancer. In short, his work illustrates that when 25(OH)D3 serum levels reach 38ng/ml there is a 55% reduction in colon cancer risk 70. Freedman et al. further proved the dose response relationship, by showing that individuals with 25(OH)D3 serum levels between 50–80ng/ml and greater than 80ng/ml had a relative risk of colon cancer mortality of 0.44 and 0.28, respectively 71. Currently, the recommended dietary allowance (RDA) for vitamin D3 is 600 IU/day and the upper limit tolerance is 4000 IU/day. It has been estimated that if Americans increase their intake of vitamin D3 to 2000IU/day, then this would lead to a 27% decrease in colorectal cancer incidence 72. An intake of 2000 IU/day would lead to approximately 40–60ng/ml 25(OH)D3 serum concentration levels, which can act has a protective factor against both the incidence and mortality of colorectal cancer 73. Furthermore, it has been proposed that raising the RDA to 2000 IU/day would prevent approximately 49,000 colon cancers per year for North America and Canada 71. Again, vitamin D3 intake can be obtained through dietary sources, sun exposure, or dietary supplements.
One of the major vitamin D dietary sources for Americans is milk. One cup of milk contains approximately 50 IU of vitamin D3, and the average American consumes about 1¼c of milk/day, with adults consuming less 74. Studies have shown conflicting results between milk consumption and colorectal cancer. However, Cho et al conducted a meta-analysis of ten cohort studies from five different countries and found that individuals who consumed more than one cup of milk per day had a 15% reduced risk of developing colorectal cancer. Moreover, he showed that for every 500g/day increase in milk consumption (2c of milk); colon cancer risk decreased by 12% 75. Since most Americans will only receive a small amount of vitamin D3 from dietary sources, the more logical source for vitamin D3 is through sun exposure. Spending approximately thirty minutes outside at noon will produce approximately 10,000 IU of vitamin D3 27. However, this can vary from person, place, and time of day. Dietary supplements are another alternative for those individuals who live in the very Northern hemisphere or are concerned about skin cancer risk, since sun block does block the synthesis of vitamin D3 in the skin.
Conclusion
In this review, it has been demonstrated that vitamin D and its metabolites have a potent effect on colorectal cancer, this is summarized in figure 3. The components of the vitamin D pathway can be used both as treatment and preventive strategies for colorectal cancer. The metabolite 1α25 (OH2) D3 targets the wnt/β-catenin by up regulating key tumor suppressor genes such as e-cadherin, which promotes an epithelial phenotype, but is only useful when the VDR is present. In colorectal cancers with low VDR expression, treatments could target the genomic and epigenomic level alterations to increase VDR expression by modulating transcription factors such as SNAIL1 or utilizing HDAC inhibitors, respectively. Finally, epidemiological studies suggest that the current RDA should be raised to 2000IU in order to raise serum 25(OH)D3 levels above 30ng/ml, this increase in vitamin D status can most efficiently be obtained from sun exposure or vitamin D supplements. In summary, vitamin D3 and its metabolites appear promising for developing treatment and preventive strategies for colon cancer.
Figure 3. Vitamin D and its metabolites have a potent effect on colorectal cancer.
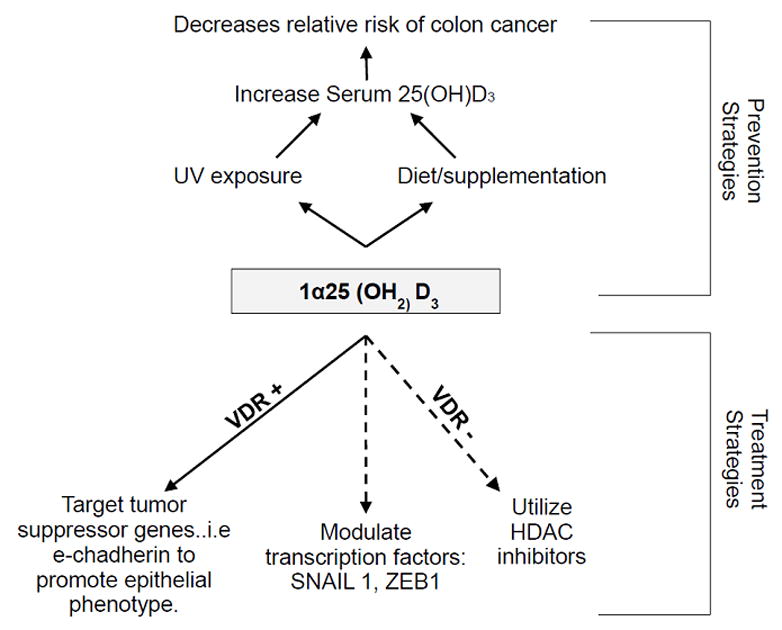
Here we summarize the findings discussed in this review. In short, colorectal cancer can be treated by targeting transcription factors that down-regulate the vitamin d receptor, which is essential in order for vitamin d to mediate its effects in promoting an epithelial phenotype. Furthermore, we show that their our potential preventative measures to protect individuals from colon cancer, such as increasing serum levels of 25(OH)D3 by either sun exposure, diet or supplements.
Acknowledgments
This work was supported by American Cancer Society grant ACS RSG CNE-113703 and by grants from the National Institutes of Health: National Cancer Society grant NCI 1K22CA127519-01A1 and National Institute of Environmental Health Sciences Center grants ES09145 and ES007784
Footnotes
Disclosure Statement: The authors have nothing to disclose
References
- 1.Balmain A, Gray J, Ponder B. The genetics and genomics of cancer. Nat Genet. 2003;33 (Suppl):238–44. doi: 10.1038/ng1107. [DOI] [PubMed] [Google Scholar]
- 2.ER F, GT B. Molecular Biology of Colorectal Cancer. Philadelphia: Lippincott Williams & Wilkins; 2008. [Google Scholar]
- 3.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–6. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 4.Behrens J, Jerchow B, Würtele M, Grimm J, Asbrand C, Wirtz R, et al. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–9. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- 5.Kishida S, Yamamoto H, Ikeda S, Kishida M, Sakamoto I, Koyama S, et al. Axin, a negative regulator of the wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of beta-catenin. J Biol Chem. 1998;273:10823–6. doi: 10.1074/jbc.273.18.10823. [DOI] [PubMed] [Google Scholar]
- 6.Powell S, Zilz N, Beazer-Barclay Y, Bryan T, Hamilton S, Thibodeau S, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–7. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 7.Inomata M, Ochiai A, Akimoto S, Kitano S, Hirohashi S. Alteration of beta-catenin expression in colonic epithelial cells of familial adenomatous polyposis patients. Cancer Res. 1996;56:2213–7. [PubMed] [Google Scholar]
- 8.Korinek V, Barker N, Morin P, van Wichen D, de Weger R, Kinzler K, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/-colon carcinoma. Science. 1997;275:1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 9.He T, Chan T, Vogelstein B, Kinzler K. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–45. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crawford H, Fingleton B, Rudolph-Owen L, Goss K, Rubinfeld B, Polakis P, et al. The metalloproteinase matrilysin is atarget of beta-catenin transactivation in intestinal tumors. Oncogene. 1999;18:2883–91. doi: 10.1038/sj.onc.1202627. [DOI] [PubMed] [Google Scholar]
- 11.Mann B, Gelos M, Siedow A, Hanski M, Gratchev A, Ilyas M, et al. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectalcarcinomas. Proc Natl Acad Sci U S A. 1999;96:1603–8. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He T, Sparks A, Rago C, Hermeking H, Zawel L, da Costa L, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 13.DeLuca H. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80:1689S–96S. doi: 10.1093/ajcn/80.6.1689S. [DOI] [PubMed] [Google Scholar]
- 14.Bouillon R. Vitamin D and human health. Presse Med. 2009;38:3–6. doi: 10.1016/j.lpm.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Norman A, Mizwicki M, Norman D. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov. 2004;3:27–41. doi: 10.1038/nrd1283. [DOI] [PubMed] [Google Scholar]
- 16.Carlberg C. Mechanisms of nuclear signalling by vitamin D3. Interplay with retinoid and thyroid hormone signalling. Eur J Biochem. 1995;231:517–27. [PubMed] [Google Scholar]
- 17.Freedman L. Transcriptional targets of the vitamin D3 receptor-mediating cell cycle arrest and differentiation. J Nutr. 1999;129:581S–86S. doi: 10.1093/jn/129.2.581S. [DOI] [PubMed] [Google Scholar]
- 18.Pálmer H, González-Sancho J, Espada J, Berciano M, Puig I, Baulida J, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154:369–87. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vandewalle B, Adenis A, Hornez L, Revillion F, Lefebvre J. 1,25-dihydroxyvitamin D3 receptors in normal and malignant human colorectal tissues. Cancer Lett. 1994;86:67–73. doi: 10.1016/0304-3835(94)90181-3. [DOI] [PubMed] [Google Scholar]
- 20.Peña C, García J, García V, Silva J, Domínguez G, Rodríguez R, et al. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int J Cancer. 2006;119:2098–104. doi: 10.1002/ijc.22083. [DOI] [PubMed] [Google Scholar]
- 21.Larriba M, Valle N, Pálmer H, Ordóñez-Morán P, Alvarez-Díaz S, Becker K, et al. The inhibition of Wnt/beta-catenin signalling by 1alpha,25-dihydroxyvitamin D3 is abrogated by Snail1 in human colon cancer cells. Endocr Relat Cancer. 2007;14:141–51. doi: 10.1677/ERC-06-0028. [DOI] [PubMed] [Google Scholar]
- 22.Larriba M, Martín-Villar E, García J, Pereira F, Peña C, de Herreros A, et al. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis. 2009;30:1459–68. doi: 10.1093/carcin/bgp140. [DOI] [PubMed] [Google Scholar]
- 23.Peña C, García J, Silva J, García V, Rodríguez R, Alonso I, et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum Mol Genet. 2005;14:3361–70. doi: 10.1093/hmg/ddi366. [DOI] [PubMed] [Google Scholar]
- 24.Slattery M, Wolff R, Curtin K, Fitzpatrick F, Herrick J, Potter J, et al. Colon tumor mutations and epigenetic changes associated with genetic polymorphism: insight into disease pathways. Mutat Res. 2009;660:12–21. doi: 10.1016/j.mrfmmm.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Godman C, Joshi R, Tierney B, Greenspan E, Rasmussen T, Wang H, et al. HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biol Ther. 2008;7:1570–80. doi: 10.4161/cbt.7.10.6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malinen M, Saramäki A, Ropponen A, Degenhardt T, Väisänen S, Carlberg C. Distinct HDACs regulate the transcriptional response of human cyclin-dependent kinase inhibitor genes to Trichostatin A and 1alpha,25-dihydroxyvitamin D3. Nucleic Acids Res. 2008;36:121–32. doi: 10.1093/nar/gkm913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holick M. Vitamin D and sunlight: strategies for cancer prevention and other health benefits. Clin J Am Soc Nephrol. 2008;3:1548–54. doi: 10.2215/CJN.01350308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong SN, Kim JH, Choe WH, Lee SY, Seol DC, Moon HW, et al. Circulating Vitamin D and Colorectal Adenoma in Asymptomatic Average-Risk Individuals Who Underwent First Screening Colonoscopy: A Case-Control Study. Dig Dis Sci. 2011 doi: 10.1007/s10620-011-1926-1. [DOI] [PubMed] [Google Scholar]
- 29.Ashktorab H, Nguza B, Fatemi M, Nouraie M, Smoot DT, Schäffer AA, et al. Case-control study of vitamin D, dickkopf homolog 1 (DKK1) gene methylation, VDR gene polymorphism and the risk of colon adenoma in African Americans. PLoS One. 2011;6:e25314. doi: 10.1371/journal.pone.0025314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goss K, Groden J. Biology of the adenomatous polyposis coli tumor suppressor. J Clin Oncol. 2000;18:1967–79. doi: 10.1200/JCO.2000.18.9.1967. [DOI] [PubMed] [Google Scholar]
- 31.Takeichi M. Morphogenetic roles of classic cadherins. Curr Opin Cell Biol. 1995;7:619–27. doi: 10.1016/0955-0674(95)80102-2. [DOI] [PubMed] [Google Scholar]
- 32.Gumbiner B. Cell adhesion: the molecular basisof tissue architecture and morphogenesis. Cell. 1996;84:345–57. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 33.Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. 1994;1198:11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 34.Perl A, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–3. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- 35.Morin P. beta-catenin signaling and cancer. Bioessays. 1999;21:1021–30. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 36.Mariadason J, Bordonaro M, Aslam F, Shi L, Kuraguchi M, Velcich A, et al. Down-regulation of beta-catenin TCF signaling is linked to colonic epithelial cell differentiation. Cancer Res. 2001;61:3465–71. [PubMed] [Google Scholar]
- 37.Thomas M, Tebbutt S, Williamson R. Vitamin D and its metabolites inhibit cell proliferation in human rectal mucosa and a colon cancer cell line. Gut. 1992;33:1660–3. doi: 10.1136/gut.33.12.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguilera O, Peña C, Garcia J, Larríba M, Ordóñez-Morán P, Navarro D, et al. The Wnt antagonist DICKKOPF-1 gene is induced by 1alpha,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis. 2007;28:1877–84. doi: 10.1093/carcin/bgm094. [DOI] [PubMed] [Google Scholar]
- 39.Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor development. J Cancer Res Clin Oncol. 2003;129:199–221. doi: 10.1007/s00432-003-0431-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cadigan K, Liu Y. Wnt signaling: complexity at the surface. J Cell Sci. 2006;119:395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- 41.Bafico A, Liu G, Yaniv A, Gazit A, Aaronson S. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nat Cell Biol. 2001;3:683–6. doi: 10.1038/35083081. [DOI] [PubMed] [Google Scholar]
- 42.Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, et al. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001;411:321–5. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- 43.González-Sancho J, Aguilera O, García J, Pendás-Franco N, Peña C, Cal S, et al. The Wnt antagonist DICKKOPF-1 gene is adownstream target of beta-catenin/TCF and is downregulated in human colon cancer. Oncogene. 2005;24:1098–103. doi: 10.1038/sj.onc.1208303. [DOI] [PubMed] [Google Scholar]
- 44.Aguilera O, Fraga M, Ballestar E, Paz M, Herranz M, Espada J, et al. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene. 2006;25:4116–21. doi: 10.1038/sj.onc.1209439. [DOI] [PubMed] [Google Scholar]
- 45.Pendás-Franco N, García J, Peña C, Valle N, Pálmer H, Heinäniemi M, et al. DICKKOPF-4 is induced by TCF/beta-catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25-dihydroxyvitamin D3. Oncogene. 2008;27:4467–77. doi: 10.1038/onc.2008.88. [DOI] [PubMed] [Google Scholar]
- 46.Zebedee Z, Hara E. Id proteins in cell cycle control and cellular senescence. Oncogene. 2001;20:8317–25. doi: 10.1038/sj.onc.1205092. [DOI] [PubMed] [Google Scholar]
- 47.Wilson J, Deed R, Inoue T, Balzi M, Becciolini A, Faraoni P, et al. Expression of Id helix-loop-helix proteins in colorectal adenocarcinoma correlates with p53 expression and mitotic index. Cancer Res. 2001;61:8803–10. [PubMed] [Google Scholar]
- 48.Fernandez-Garcia N, Palmer H, Garcia M, Gonzalez-Martin A, del Rio M, Barettino D, et al. 1alpha,25-Dihydroxyvitamin D3 regulates the expression of Id1 and Id2 genes and the angiogenic phenotype of human colon carcinoma cells. Oncogene. 2005;24:6533–44. doi: 10.1038/sj.onc.1208801. [DOI] [PubMed] [Google Scholar]
- 49.Pálmer H, Larriba M, García J, Ordóñez-Morán P, Peña C, Peiró S, et al. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat Med. 2004;10:917–9. doi: 10.1038/nm1095. [DOI] [PubMed] [Google Scholar]
- 50.Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–9. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 51.Cano A, Pérez-Moreno M, Rodrigo I, Locascio A, Blanco M, del Barrio M, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 52.Grooteclaes M, Frisch S. Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene. 2000;19:3823–8. doi: 10.1038/sj.onc.1203721. [DOI] [PubMed] [Google Scholar]
- 53.Lazarova D, Bordonaro M, Sartorelli A. Transcriptional regulation of the vitamin D(3) receptor gene by ZEB. Cell Growth Differ. 2001;12:319–26. [PubMed] [Google Scholar]
- 54.Postigo A, Dean D. Differential expression and function of members of the zfh-1 family of zinc finger/homeodomain repressors. Proc Natl Acad Sci U S A. 2000;97:6391–6. doi: 10.1073/pnas.97.12.6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bannister A, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–3. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 56.Ogryzko V, Schiltz R, Russanova V, Howard B, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–9. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 57.Postigo A, Dean D. ZEB represses transcription through interaction with the corepressor CtBP. Proc Natl Acad Sci U S A. 1999;96:6683–8. doi: 10.1073/pnas.96.12.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 59.Elloul S, Elstrand M, Nesland J, Tropé C, Kvalheim G, Goldberg I, et al. Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer. 2005;103:1631–43. doi: 10.1002/cncr.20946. [DOI] [PubMed] [Google Scholar]
- 60.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 61.Markowitz S, Bertagnolli M. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–60. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marks P, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;14:1497–511. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]
- 63.Thiagalingam S, Cheng K, Lee H, Mineva N, Thiagalingam A, Ponte J. Histone deacetylases: unique players in shaping the epigenetichistone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- 64.Patra S, Patra A, Dahiya R. Histone deacetylase and DNA methyltransferase in human prostate cancer. Biochem Biophys Res Commun. 2001;287:705–13. doi: 10.1006/bbrc.2001.5639. [DOI] [PubMed] [Google Scholar]
- 65.Gaschott T, Werz O, Steinmeyer A, Steinhilber D, Stein J. Butyrate-induced differentiation of Caco-2 cells is mediated by vitamin D receptor. Biochem Biophys Res Commun. 2001;288:690–6. doi: 10.1006/bbrc.2001.5832. [DOI] [PubMed] [Google Scholar]
- 66.Garland C, Comstock G, Garland F, Helsing K, Shaw E, Gorham E. Serum 25-hydroxyvitamin D and colon cancer: eight-year prospective study. Lancet. 1989;2:1176–8. doi: 10.1016/s0140-6736(89)91789-3. [DOI] [PubMed] [Google Scholar]
- 67.Tangrea J, Helzlsouer K, Pietinen P, Taylor P, Hollis B, Virtamo J, et al. Serum levels of vitamin D metabolites and the subsequent risk of colon and rectal cancer in Finnish men. Cancer Causes Control. 1997;8:615–25. doi: 10.1023/a:1018450531136. [DOI] [PubMed] [Google Scholar]
- 68.Feskanich D, Ma J, Fuchs C, Kirkner G, Hankinson S, Hollis B, et al. Plasma vitamin D metabolites and risk of colorectal cancer in women. Cancer Epidemiol Biomarkers Prev. 2004;13:1502–8. [PubMed] [Google Scholar]
- 69.Yetley E. Assessing the vitamin D status of theUS population. Am J Clin Nutr. 2008;88:558S–64S. doi: 10.1093/ajcn/88.2.558S. [DOI] [PubMed] [Google Scholar]
- 70.Gorham E, Garland C, Garland F, Grant W, Mohr S, Lipkin M, et al. Optimal vitamin D status for colorectal cancer prevention: a quantitative meta analysis. Am J Prev Med. 2007;32:210–6. doi: 10.1016/j.amepre.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 71.Freedman D, Looker A, Chang S, Graubard B. Prospective study of serum vitamin D and cancer mortality in the United States. J Natl Cancer Inst. 2007;99:1594–602. doi: 10.1093/jnci/djm204. [DOI] [PubMed] [Google Scholar]
- 72.Garland C, Gorham E, Mohr S, Garland F. Vitamin D for cancer prevention: global perspective. Ann Epidemiol. 2009;19:468–83. doi: 10.1016/j.annepidem.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 73.Garland C, Gorham E, Mohr S, Grant W, Giovannucci E, Lipkin M, et al. Vitamin D and prevention of breast cancer: pooled analysis. J Steroid Biochem Mol Biol. 2007;103:708–11. doi: 10.1016/j.jsbmb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 74.Storey M, Forshee R, Anderson P. Beverage consumption in the US population. J Am Diet Assoc. 2006;106:1992–2000. doi: 10.1016/j.jada.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 75.Cho E, Smith-Warner S, Spiegelman D, Beeson W, van den Brandt P, Colditz G, et al. Dairy foods, calcium, and colorectal cancer: a pooled analysis of 10 cohort studies. J Natl Cancer Inst. 2004;96:1015. doi: 10.1093/jnci/djh185. [DOI] [PubMed] [Google Scholar]