

Abstract
There has been significant progress in understanding the process of protein translation in recent years. One of the best examples is the discovery of usage bias in successive synonymous codons and its role in eukaryotic translation efficiency. We observed here a similar type of bias in the other two life domains, bacteria and archaea, although the bias strength was much smaller than in eukaryotes. Among 136 prokaryotic genomes, 98 were found to have significant bias from random use of successive synonymous codons with Z scores larger than three. Furthermore, significantly different bias strengths were found between prokaryotes grouped by various genomic or biochemical characteristics. Interestingly, the bias strength measured by a general Z score could be fitted well (R = 0.83, P < 10−15) by three genomic variables: genome size, G + C content, and tRNA gene number based on multiple linear regression. A different distribution of synonymous codon pairs between protein-coding genes and intergenic sequences suggests that bias is caused by translation selection. The present results indicate that protein translation is tuned by codon (pair) usage, and the intensity of the regulation is associated with genome size, tRNA gene number, and G + C content.
Keywords: successive synonymous codon bias, comparative analysis, prokaryotic genomes, Z scores, translation efficiency
1. Introduction
Recent studies show that protein-coding genes use codon patterns to fine-tune translation and increase protein synthesis efficiency.1–7 Three types of codon usages have been proposed to influence translation. First, use of single codons may influence the speed and accuracy of translation.3,5–11 Frequent use of ‘preferred codons’ is believed to maximize translation efficiency.12 This hypothesis takes previous observations in Escherichia coli and several other unicellular microbes as supporting evidence.13 In these small organisms, highly expressed genes are found to have more extreme codon bias, where codon bias denotes non-equilibrium usage of up to six synonymous codons encoding for the same amino acids, with ‘preferred codons’ in highly expressed genes corresponding to the most abundant isoaccepting tRNAs.12
Second, codon pair usage is associated with translation efficiency, wherein a codon pair indicates two successive codons.14–17 The biased use of codon pairs is a common phenomenon in a wide range of species.18 The observed codon pair frequency often deviates from expected values predicted from two single codons. Some codon pairs are overrepresented and others underrepresented. A variety of selective or non-selective factors are responsible for such bias.19 One such factor is that codon pair usage affects translation.15,20 In fact, peptide bond formation requires simultaneous accommodation of two codons and of two tRNAs in the ribosomal A and P sites.21 For spatial reasons, it is thought that not all codon and tRNA combinations are equally compatible on the ribosome surface.22 Some combinations of codon pairs and tRNAs would be advantageous for translation efficiency.20 Structural features that regulate tRNA geometry within the ribosome govern codon pair patterns, driving enhanced translational fidelity and/or rate.14 Experimental results support such a mechanism.17,22
Third, an interesting bias of successive synonymous codon pairs was found in eight eukaryotic genomes.1,2,4 Synonymous codon pair denotes a codon that recurs after its synonymy within a gene, regardless of how many codons encode other amino acids, and requiring only that there are not other synonymies between the two. In this study, bias of synonymous codon pairs denotes a difference between actual and expected frequencies when they are independent. Cannarozzi et al.1 found a strong tendency to use the same codon a second time as for the first synonymy. There is a bias towards the most closely related synonymous wobble codons, if the same codon is not reused. This predisposition towards selecting particular codons, rather than arbitrarily choosing one from the successive synonymous set, is termed ‘autocorrelation’ or ‘codon reuse’, and has important implications for protein translation. Based on comprehensive analyses of highly expressed genes, it was suggested that codon reuse may provide an effective mechanism to speed up translation.1 Through wet-bench experiments, it was successfully demonstrated that translation on autocorrelated mRNA was substantially (30%) faster than on anti-correlated mRNA.1 Therefore, this result reinforces the speculation that codon reuse could benefit translation efficiency.1
Due to its intimate relationship with translation efficiency, biased use of single codons and codon pairs has been studied extensively. However, biased use of successive codon pairs is relatively new.1,2 Cannarozzi and colleagues observed this phenomenon in eukaryotes.1 Here, we performed cross-species analysis of ‘codon reuse’ in 136 prokaryotes. We not only show the existence of ‘codon reuse’ in various prokaryotic genomes and hence illustrate it as a universal mechanism among the three domains of life, but also compare the level of biases among various prokaryotes. Most importantly, we observed that the overall bias intensity for successive synonymous codon pairs is positively correlated with several genomic factors. Using genomic G + C content, genome size, and tRNA gene numbers as limiting factors, the bias value could be predicted with high accuracy. Thus, these data reinforce the notion that the genome contains all the information necessary for regulating protein translation.
2. Materials and methods
We randomly picked one strain from each of prokaryotic species sequenced. Genome sequences and annotations were downloaded from the NCBI RefSeq project (ftp://ftp.ncbi.nih.gov/genomes/Bacteria) for 136 prokaryotic strains before June 2010. These 136 strains consisted of 109 bacteria and 27 archaea. Information on genome size, G + C content, and tRNA gene number was extracted from the RefSeq annotation.23 For all genomes, tRNA was assigned to 64 codons according to the extended wobble rule.24 According to the wobble rule, we adopt the consistent pattern for assigning codons to isoaccepting tRNAs for all 136 prokaryotic genomes, and it is similar with Cannarozzi et al.1 In Supplementary Table S1, the correspondence of codons to tRNAs is illustrated with E. coli as an example. In fact, there may exist some modifications of the wobble rule. For example, large bacterial genomes with high G + C% usually have tRNAs with a C-starting anticodon solely responsible for a G-ending codon, and the number of this tRNA gene is often multiple and larger than that for the respective isoaccepting tRNA responsible for both G- and A-ending codons. Another example is the presence of I in Arg tRNAs found in a wide range of bacteria. While A-ending codons are thought to be recognized by I-containing tRNA on the basis of the extended wobble rule, the efficiency of I-A recognition is low and there are often (but not always) other tRNAs responsible for A-ending (and G-ending) codons. However, we do not know the modification will appear in which specific genome. Therefore, we do not consider any of the modifications when calculating the bias of successive synonymous codon pairs.
For comparison, correlation of synonymous codon pairs in eight eukaryotes was also investigated (Arabidopsis thaliana, Ashbya gossypii, Caenorhabditis elegans, Candida glabrata, Drosophila melanogaster, Homo sapiens, Saccharomyces cerevisiae, and Schizosaccharomyces pombe).
We focused only on pairs of consecutive synonymous codons, which may be separated by any number of codons from other amino acids, in each prokaryotic genome. We used the Z score25 defined in Equation (1) to evaluate the difference in the actual number from the expected number of consecutive synonymous codon pairs and isoaccepting tRNA pairs. Similar to Cannarozzi et al.,1 the number of synonymous codon pairs of the nine amino acids with at least two tRNAs was counted. And, the expected number of synonymous codon pairs was calculated as the products of the frequencies of the individual codons of each pair in each prokaryotes. A negative Z score means that the actual frequency is below the expected frequency, whereas a positive Z score means that the former is above the latter.25 The more positive a score is, the stronger is the translation selection in the considered synonymous codon pair.
| (1) |
The standard deviation in Equation (1) is calculated based on actual numbers and expected numbers of the collection of all synonymous codon pairs or isoaccepting tRNA pairs. The distribution of codon pairs in Fig. 1 and genomes in Fig. 2 is fitted by the Gauss function.25 A linear relationship between various genomic factors and the general Z score is fitted with single variable or multiple linear regression.25 Differences in Z scores between two groups of prokaryotic genomes were statistically validated by t-tests.25 All statistical analyses were implemented with the freely available R package (http://www.r-project.org/).
Figure 1.
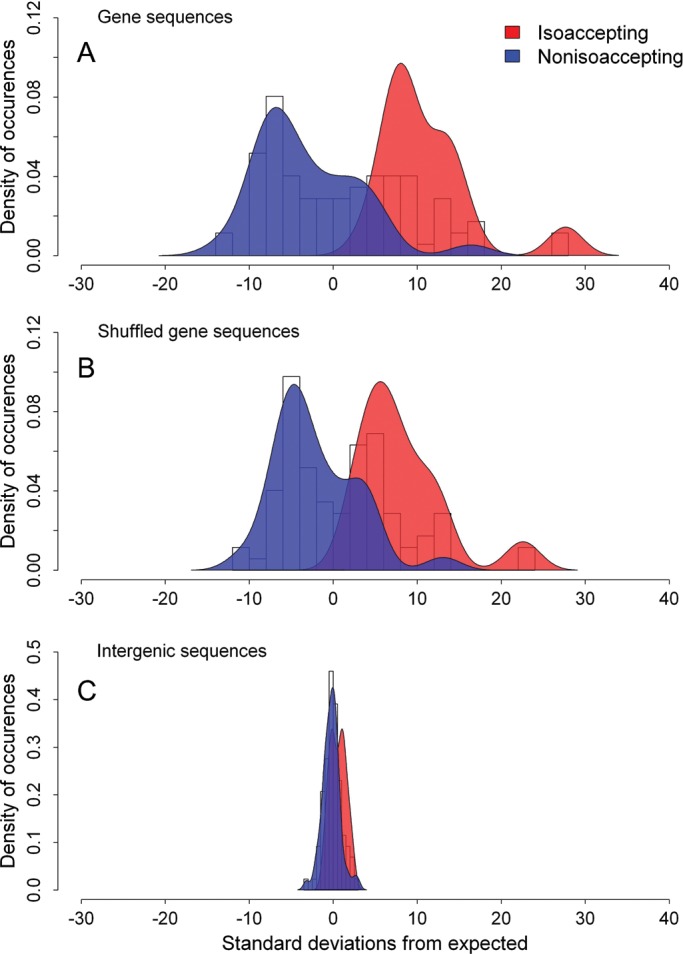
Z score histograms for two groups (isoaccepting and non-isoaccepting) of codon pairs in three types of sequences in E. coli K12. (A) Z score histograms for two groups (isoaccepting and non-isoaccepting) of codon pairs in gene sequences. The means of the two distributions are different with a P-value = 2.2e-14. (B) Z score histograms for two groups (isoaccepting and non-isoaccepting) of codon pairs in sequences generated by randomly shuffling. The means of the two distributions are different with a P-value = 3.3e-11. (C) Z score histograms for two groups (isoaccepting and non-isoaccepting) of codon pairs in intergenic sequences. The means of the two distributions are different with a P-value = 1.0e-3. The difference between the two types of codon pairs for intergenic sequences is not only much smaller than that for the gene sequences but also quite smaller than that for the shuffled gene sequences. Therefore, the pattern of codon reuse is present in protein-coding sequences, and the conserved pattern appears to be rooted in translation selection.
Figure 2.
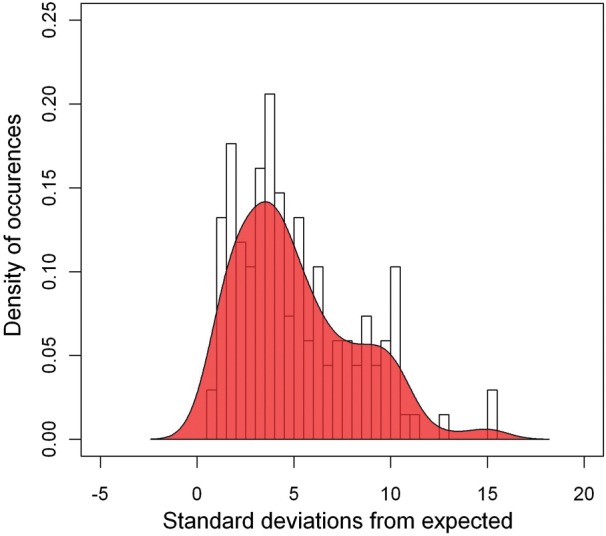
Histogram of the general Z scores among 136 prokaryotic genomes.
3. Results
3.1. Synonymous codon correlation in E. coli K12 genome
We evaluated 107 bacterial and 29 archaeal genomes. In the following two sections, E. coli K12 is taken as an example. We evaluated all pairs of consecutive synonymous codons in the E. coli K12. Pairs coding for nine amino acids (alanine, arginine, glycine, isoleucine, proline, leucine, serine, threonine, and valine), which have at least two tRNAs, were considered for further analysis. The frequency of all combinations (e.g. TCCTCT as one combination) was then calculated. Assuming a random distribution, the expected number of all combinations could be estimated from the actual single codon frequencies. According to Equation (1), the Z score quantifies the difference between the actual frequency of a combination from the expected frequency, in terms of the number of standard deviations.25 We classified each synonymous codon pair as favoured, if the difference is larger than 3 s.d., as neutral if between −3 and +3 s.d., or disfavoured if less than −3 s.d. The numbers for three groups of synonymous codon pairs for each amino acid are summarized in Table 1. Among codon pairs with isoacceptors (sharing a tRNA), favoured numbers are all larger than neutral or disfavoured ones. However, the opposite result is obtained for synonymous codon pairs without isoacceptors. These results indicate that the reuse of codons sharing the same tRNA is a universal phenomenon for the nine E. coli amino acids examined. This observation is similar to that seen in eukaryotes.1 It is worth noting that the strength of synonymous codon correlation in E. coli is much smaller than that in yeast. For example, the mean Z score for 10 groups of synonymous codon pairs encoding the amino acid serine is 8.6469 in yeast1 and only 5.8762 in E. coli. As mentioned below, Z scores of synonymous codon pairs in prokaryotes are generally lower than those seen in eukaryotes.
Table 1.
The numbers of three kinds of synonymous codon pairs for nine amino acids in E. coli
| Isoaccepting |
Non-isoaccepting |
|||||
|---|---|---|---|---|---|---|
| Grouped by | Favoured | Neutral | Disfavoured | Favoured | Neutral | Disfavoured |
| Ala | 4 | 2 | 2 | 1 | 1 | 6 |
| Arg | 6 | 2 | 4 | 11 | 3 | 10 |
| Gly | 4 | 2 | 0 | 2 | 1 | 7 |
| Ile | 3 | 0 | 2 | 0 | 2 | 2 |
| Leu | 5 | 3 | 2 | 6 | 13 | 7 |
| Pro | 6 | 0 | 2 | 2 | 2 | 4 |
| Ser | 8 | 2 | 0 | 1 | 11 | 14 |
| Thr | 4 | 2 | 0 | 1 | 3 | 6 |
| Val | 4 | 2 | 0 | 2 | 4 | 4 |
| Total | 44 | 15 | 12 | 26 | 40 | 60 |
Codon pairs are grouped into those with and without isoacceptors (sharing a tRNA), by parsimony. Within each group, pairs were classified as favoured (≥3 s.d.), neutral (between −3 and +3 s.d.), or disfavoured (≤−3 s.d.).
3.2. Confirming the hypothesis of translation selection
One reason may account for the codon correlation illustrated above. There is selection pressure for codon ordering in protein-coding genes, and, hence, synonymous codons sharing the same tRNA are successively used.1 However, there also exists a second explanation.1 Different genes may be enriched in different codons, and the correlation observed at the genomic level may be due to the accumulation of given codons in specific genes. If the second case is real, the synonymous codon correlation should remain, if codon distribution is shuffled within each gene individually.1 In the first case, such codon shuffling would reduce the difference of codon correlation between isoacceptor pairs and non-isoacceptor pairs. We performed shuffle experiments in E. coli K12 to test which hypothesis is correct. Results are shown in Fig. 1, where the vertical axis denotes synonymous codon pair frequency in the corresponding range of Z scores. Similar to Cannarozzi et al.,1 codon correlations were found to decrease for isoaccepting pairs for the shuffled genes and increase for non-isoaccepting pairs. The difference between the isoaccepting pairs and non-isoaccepting pairs without shuffle (Fig. 1A, t-test, P = 2.2 × 10−14) is significantly larger than the difference after shuffle (Fig. 1B, t-test, P = 3.3 × 10−11). Thus, autocorrelation was not simply due to codon bias at the gene level, but due to codon ordering within genes. To reinforce the hypothesis of translation selection, we also analysed synonymous codon correlation in non-coding regions (i.e. intergenic sequences). The Z scores histograms for isoaccepting and non-isoaccepting pairs are shown in Fig. 1C (t-test, P = 1.0 × 10−3). The difference between the two types of codon pairs for intergenic sequences is not only much smaller than that for the gene sequences but also smaller than that for the shuffled gene sequences. Based on this analysis and that by Cannarozzi et al.,1 the pattern of codon reuse exists in protein-coding sequences and reinforces the hypothesis that the conserved pattern is rooted in translation selection.
3.3. Varied strengths of codon reuse in prokaryotic genomes
We investigated synonymous codon correlation in 107 bacteria and 29 archaea. Taxonomic distribution of these genomes is summarized in Table 2. As can be seen, 107 bacteria are widely distributed across 14 phyla, and 29 archaea are distributed across 5 phyla. Therefore, most known prokaryotic phyla have representatives in our dataset. To compare the strength of synonymous codon correlation among different genomes, we calculated the Z general score for each genome. This value is equal to the Z score averaged for nine amino acids, and for each amino acid, the Z score is averaged for all isoaccepting tRNA pairs. First, we compared the general Z sores in the three domains of life. Among the eight eukaryotes, the mean value of the general Z scores is 15.0. However, the value is as small as 5.6 in bacteria and 3.2 in archaea. This indicates that the strength of codon reuse is much smaller in prokaryotes than in eukaryotes, although the strength in the former is also significant. The 136 prokaryotic genomes, collectively, have a mean Z score of 5.1 and standard deviation of 3.1. Streptomyces coelicolor A3 has the largest Z score of 15.1. Note that this genome has the highest G + C content and is one of largest of the 136 prokaryote genomes. In contrast, Nanoarchaeum equitans Kin4-M has the smallest Z score of 0.72 and is also the smallest genome. Ninety-eight prokaryotic genomes have Z scores larger than 3.0, indicating that 72% of the genomes have significant usage bias of successive synonymous codons. Histogram of the general Z scores is shown in Fig. 2, and, as can be seen, the genera distribution could not be well fitted by a simple Gaussian function because it has two peaks.
Table 2.
Taxonomic distribution of 136 prokaryotic genomes analysed in this study
| Phylum no. | Class no. | Order no. | Family no. | Genus no. | Species no. | |
|---|---|---|---|---|---|---|
| Bacteria | 14 | 19 | 35 | 55 | 76 | 107 |
| Archaea | 5 | 14 | 18 | 27 | 29 | 29 |
The phylogenetic relationship among the 136 prokaryotic genomes is shown in Supplementary Fig. S1. This tree is constructed using the neighbor-joining method26,27 based on 16S rRNA sequences. To comprehend the phylogenetic tree, some factors (e.g. taxonomy ID, organism name, general Z score, genome size, and G + C content) of 136 prokaryotic genomes are listed in Supplementary Table S2. For each genome, the general Z score is marked on the right side of the name. When visualized in this manner, it is clear that the strength of synonymous codon correlation changes with phylogeny. In fact, there is no consistent pattern for Z score variation among different phylogenetic groups. Some groups have very similar scores, whereas others do not. To illustrate the trend from the phylogenetic order, Tables 3, 4 and 5 list the mean Z scores and standard deviation at three levels: phylum, family, and genus. As can be seen, the mean standard deviation changes from 1.53 to 1.42 to 0.90, when the classifying level changes from phylum to family to genus. Correspondingly, the mean variance coefficient changes from 0.34 to 0.24 to 0.21. Therefore, it appears to naturally follow that the Z scores will be more similar at a lower phylogenetic level.
Table 3.
The mean Z scores and SD at the level of phylum
| Phylum | Mean | SD | Genome no. | VC (SD/mean) |
|---|---|---|---|---|
| Actinobacteria | 7.49 | 3.74 | 8 | 0.50 |
| Chlamydiae | 1.61 | 0.41 | 5 | 0.26 |
| Firmicutes | 4.60 | 1.37 | 21 | 0.30 |
| Proteobacteria | 6.70 | 3.31 | 54 | 0.49 |
| Spirochaetes | 3.03 | 0.31 | 4 | 0.10 |
| Tenericutes | 1.99 | 0.81 | 5 | 0.41 |
| Crenarchaeota | 3.16 | 0.71 | 6 | 0.23 |
| Euryarchaeota | 3.39 | 1.57 | 20 | 0.46 |
| 4.00 | 1.53 | 15.4 | 0.34 |
VC denotes variance coefficient in Table 3, 4 and 5, respectively.
Table 4.
The mean Z scores and SD at the level of family
| Family | Mean | SD | Genome no. | VC (SD/Mean) |
|---|---|---|---|---|
| Mycobacteriaceae | 5.01 | 1.08 | 3 | 0.22 |
| Chlamydiaceae | 1.44 | 0.19 | 4 | 0.13 |
| Bacillaceae | 4.84 | 2.29 | 4 | 0.47 |
| Lactobacillaceae | 4.84 | 1.14 | 3 | 0.24 |
| Streptococcaceae | 4.92 | 1.08 | 5 | 0.22 |
| Brucellaceae | 5.33 | 0.14 | 3 | 0.03 |
| Burkholderiaceae | 8.78 | 1.76 | 3 | 0.20 |
| Neisseriaceae | 10.72 | 3.85 | 3 | 0.36 |
| Enterobacteriaceae | 7.43 | 4.29 | 7 | 0.58 |
| Pasteurellaceae | 4.32 | 0.68 | 3 | 0.16 |
| Vibrionaceae | 8.02 | 0.66 | 5 | 0.08 |
| Xanthomonadaceae | 9.75 | 1.56 | 4 | 0.16 |
| Spirochaetaceae | 2.98 | 0.37 | 3 | 0.12 |
| Mycoplasmataceae | 1.99 | 0.81 | 5 | 0.41 |
| 5.74 | 1.42 | 3.9 | 0.24 |
Table 5.
The mean Z scores and SD at the level of genus
| Genus | Mean | SD | Genome no. | VC (SD/Mean) |
|---|---|---|---|---|
| Mycobacterium | 5.01 | 1.08 | 3 | 0.22 |
| Chlamydophila | 1.36 | 0.11 | 3 | 0.08 |
| Bacillus | 4.84 | 2.29 | 4 | 0.47 |
| Lactobacillus | 4.84 | 1.14 | 3 | 0.24 |
| Streptococcus | 5.03 | 1.22 | 4 | 0.24 |
| Brucella | 5.33 | 0.14 | 3 | 0.03 |
| Vibrio | 8.08 | 0.74 | 4 | 0.09 |
| Xanthomonas | 10.51 | 0.43 | 3 | 0.04 |
| Mycoplasma | 2.07 | 0.91 | 4 | 0.44 |
| 5.23 | 0.90 | 3.4 | 0.21 |
3.4. Comparative analyses of codon reuse strengths among different groups
The 136 prokaryotic genomes could be classified into two groups based on the Gram type, oxygen metabolism, growth rate, G + C content, genome size, and tRNA gene number, respectively. We performed comparative analyses of the general Z scores between any two groups based on the six classifying criteria and results are listed in Table 6. For all criteria except Gram type and oxygen metabolism, the two groups are divided equally based on median criterion values. Z scores of the two groups are generally significantly different based on all classifying criteria, except Gram type. Among the five indices with significant differences, genome size and tRNA gene number are the most sensitive as the P-value is the smallest. Based on the descending order of statistical difference, the other three indices will be G + C content, oxygen metabolism, and growth rate. It is interesting that the two most frequently used classifying criteria, oxygen metabolism and Gram type, are not associated with variance of synonymous codon pair strength as are genomic features such as G + C content, genome size, and tRNA gene number.
Table 6.
Comparison of the general Z scores between any two groups based on six classification criteriaa
| Classifying criteria | Mean | SD | Genome number | P-value |
|---|---|---|---|---|
| Gram type | ||||
| Gram negative | 6.01 | 3.43 | 70 | 0.064 |
| Gram positive | 4.87 | 2.64 | 35 | |
| Growth rateb | ||||
| Fast | 6.28 | 3.05 | 54 | 0.028 |
| Slow | 4.92 | 3.27 | 53 | |
| Oxygen metabolismc | ||||
| Aerobic | 6.39 | 3.05 | 35 | 0.017 |
| Anaerobic | 4.58 | 2.04 | 16 | |
| G + C content | ||||
| Low GC (<46.2%) | 3.57 | 2.19 | 68 | 1.21e-09 |
| High GC (>46.2%) | 6.62 | 3.11 | 68 | |
| tRNA gene number | ||||
| Less tRNA (<32) | 3.30 | 2.15 | 68 | 1.37e-13 |
| More tRNA (>32) | 6.89 | 2.84 | 68 | |
| Genome size | ||||
| Small size (<2.55 Mb) | 3.25 | 1.97 | 68 | 3.08e-14 |
| Large size (>2.55 Mb) | 6.93 | 2.90 | 68 | |
aBecause information of the upper three factors is not available for some of the genomes, the total genome number is less than 136 for these factors. Detailed information of each prokaryotic genomes is shown in Supplementary Table S2.
bOriginal growth rate data were obtained from Vieira-Silva and Rocha40. Genomes with generation time longer than 2 h are taken as slow growing, otherwise as fast growing.
cOriginal data on oxygen metabolism were obtained from NCBI at ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/lproks_0.txt.
3.5. Linear correlation between the general Z scores and genomic characteristics
The strength of synonymous codon correlation varied largely across prokaryotic genomes, as shown above. Furthermore, we have identified potential determinant factors of this observation. It would be interesting to identify a quantitative relationship between Z scores and genomic factors. Three factors that could be directly extracted from chromosomal DNA sequences are chromosome size, G + C content, and tRNA gene number. After obtaining values of each factor for all 136 prokaryotic genomes, linear fitting was performed between them and the general Z scores. The least squares method25 was used for linear fitting between them and the general Z scores. Their correlation coefficient (R) and significance (P-value) were computed, respectively, with R package. The scatter plot of general Z scores against the three factors and linear fitting are shown in Fig. 3. All three factors are significantly correlated (0.55 < = R < = 0.78, P < 10−11) with the general Z scores. Genome size has the strongest correlation with general Z score, according to both R coefficient and P-value. This fact is consistent with the above comparative analysis, where genome size is shown to be the most effective distinguishing feature. In general, a phenomenon is often associated with multiple factors. The multiple linear regression method is more actual and effective than linear fitting method. To obtain a stronger correlation, multiple linear regression was also performed. Chromosome size, G + C content, and tRNA gene number constitute the three explanatory variables, with general Z score as the dependent variable. Using R package, the regression equation is defined as Equation (2).
| (2) |
where Z denotes the general Z score, S denotes chromosome size, G denotes G + C content, T denotes tRNA gene number, and chromosome size is measured in millions of base pairs (Mb). The Pearson's coefficient (r value) of the multiple regression is 0.8334 and the P = 2.2 × 10−16. Using Equation (2), we seek to predict the strength of successive synonymous codon pairs in any sequenced prokaryotic genome with some reliability. The prediction error is only 1.72, and roughly speaking, the prediction accuracy is higher than 80%, given that the Pearson's coefficient is 0.8334.
Figure 3.

Scatter plot of general Z scores against three factors (genome size, G + C content, and tRNA gene number) for 136 prokaryotic genomes. In the figure, each point corresponds to a prokaryotic genomes. (A) Scatter plot of general Z scores against genome size: linear fitting by least squares method. (B) Scatter plot of general Z scores against G + C content: linear fitting by least squares method. (C) Scatter plot of general Z scores against tRNA gene number: linear fitting by least squares method.
4. Discussion
Cannarozzi et al. first observed the phenomenon of successive synonymous codon pairs in eukaryotic genomes in 2010.1 Fredrick and Ibba comment that this work is one of the best examples illustrating how codon usage patterns control ribosome speed and fine-tune translation to increase protein synthesis efficiency.2 They call on immediate work on bacterial genomes in which translation rates are substantially higher than eukaryotes.2 Interestingly, we found that most prokaryotic genomes have significant biases in successive synonymous codon pairs, suggesting that this pattern is universal to the three domains of life. Significantly, different distributions of synonymous codon pairs between protein-coding genes and intergenic sequences suggest that this bias is a result from translation selection. Combined with the previous work,1 we conclude that the bias of successive synonymous codon pairs, as universal pattern in all living organisms, would be a translation-associated effect and could be used to fine-tune protein synthesis.
Furthermore, the strength of the bias varies strongly across different genomes. Eukaryotic cells have the strongest bias, whereas archaeal cells have the least. Among bacteria, there is also a range of differences. Usually, large bacterial genomes and those with high G + C content tend to have a stronger bias. This type of variation reflects the diversity of living species. A better understanding of the precise reason for varied strength may be clarified by comparing these results with single codon bias. Generally, highly expressed genes tend to have more of a bias with single codons8,11,28–35 in unicellular organisms. Codon bias is thought to maximize translation efficiency, including speed and/or fidelity.9,11,12,28,30–35 The strength of the codon bias is determined by the strength of translation selection exerted on the genome.13 For example, species exposed to selection for rapid growth tend to have more strongly selected codon usage bias.36,37 At the same time, fast-growing bacteria with low generation time generally have more tRNA genes to increase translation speed.13,38 And, tRNA gene number is positively correlated with genome size and G + C content.39 Because the general Z scores of bacteria are also associated with these factors, the correlation between the bias strength of successive codon pairs and various genomic or biochemical characters in prokaryotes may be caused by the translation selection as the single codon bias. One lingering question is why single codon bias is almost absent, or at least smaller, in higher eukaryotic genomes.37 However, the strength of successive codon pair bias is much stronger in higher eukaryotes than in prokaryotes. Our current thought is that this may be due to different translation mechanisms among the three domains of life.
Another noticeable point is how translation influences synonymous codon pair usage or why codon reuse favours translation efficiency. Cannarozzi et al. have proposed that tRNA diffusion is slower than both reloading and translation.1 When the next amino acid that is incorporated is the same, a recently used tRNA would be more likely than any of its isoacceptors to still be in the vicinity of the ribosome.1 Therefore, reuse of isoaccepting codons would accelerate the translation process.1 Direct observation of slower tRNA diffusion than reloading and translation would be the most potent proof for this hypothesis.
Successive synonymous codon pair represents the order of protein-coding sequences rather than their composition, which is different from single codon use.2,4 Therefore, the observed bias of successive synonymous codon pairs, as a newly observed phenomenon, illustrates that regulatory information of protein translation is retained not only in nucleotide species but also in nucleotide order. Widely observed association of single codon,3,5,6,8,9,11–13 codon pair14–22 or successive synonymous codon pair1,2 use and translation efficiency suggests that the latter exerts influence on the various types of codons. On the other hand, genome size, G + C content and tRNA gene number are all significantly associated with the bias strength of synonymous codon pairs, illustrating that translation selection exerts influence on different genomic features. Taking all factors into consideration, we conclude that translation selection is exerted on genome sequences at multiple levels and by various mechanisms. In other words, protein translation is a complex process and is associated with various factors such as usage of single codons, codon pairs (in particular successive synonymous codon pair), genome size, genomic G + C content, and tRNA gene number.
Conflict of Interest statement
The authors have no conflicts of interest to declare.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by the National Natural Science Foundation of China (grant 31071109), the special fund of China Postdoctoral Science Foundation (grant 201104687), and the program for New Century Excellent Talents in University (grant number NCET-11-0059). Funding for open access charge: National Natural Science Foundation of China.
Supplementary Material
Acknowledgements
The authors are grateful to the reviewers for their valuable comments that have led to improvements to this paper. We thank Miss Meng-Ze Du for useful discussions.
Footnotes
Edited by Naotake Ogasawara
References
- 1.Cannarozzi G., Schraudolph N.N., Faty M., et al. A role for codon order in translation dynamics. Cell. 2010;141:355–67. doi: 10.1016/j.cell.2010.02.036. doi:10.1016/j.cell.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 2.Fredrick K., Ibba M. How the sequence of a gene can tune its translation. Cell. 2010;141:227–9. doi: 10.1016/j.cell.2010.03.033. doi:10.1016/j.cell.2010.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kudla G., Murray A.W., Tollervey D., Plotkin J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–8. doi: 10.1126/science.1170160. doi:10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plotkin J.B., Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 2011;12:32–42. doi: 10.1038/nrg2899. doi:10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuller T., Carmi A., Vestsigian K., et al. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141:344–54. doi: 10.1016/j.cell.2010.03.031. doi:10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 6.Tuller T., Waldman Y.Y., Kupiec M., Ruppin E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. USA. 2010;107:3645–50. doi: 10.1073/pnas.0909910107. doi:10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hershberg R., Petrov D.A. General rules for optimal codon choice. PLoS Genet. 2009;5:e1000556. doi: 10.1371/journal.pgen.1000556. doi:10.1371/journal.pgen.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grantham R., Gautier C., Gouy M., Jacobzone M., Mercier R. Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res. 1981;9:r43–74. doi: 10.1093/nar/9.1.213-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. doi:10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 10.Hershberg R., Petrov D.A. Selection on codon bias. Ann. Rev. Genet. 2008;42:287–99. doi: 10.1146/annurev.genet.42.110807.091442. doi:10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- 11.Sharp P.M., Li W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986;24:28–38. doi: 10.1007/BF02099948. doi:10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 12.Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 13.Sharp P.M., Bailes E., Grocock R.J., Peden J.F., Sockett R.E. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 2005;33:1141–53. doi: 10.1093/nar/gki242. doi:10.1093/nar/gki242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buchan J.R., Aucott L.S., Stansfield I. tRNA properties help shape codon pair preferences in open reading frames. Nucleic Acids Res. 2006;34:1015–27. doi: 10.1093/nar/gkj488. doi:10.1093/nar/gkj488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fedorov A., Saxonov S., Gilbert W. Regularities of context-dependent codon bias in eukaryotic genes. Nucleic Acids Res. 2002;30:1192–7. doi: 10.1093/nar/30.5.1192. doi:10.1093/nar/30.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutman G.A., Hatfield G.W. Nonrandom utilization of codon pairs in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1989;86:3699–703. doi: 10.1073/pnas.86.10.3699. doi:10.1073/pnas.86.10.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irwin B., Heck J.D., Hatfield G.W. Codon pair utilization biases influence translational elongation step times. J. Biol. Chem. 1995;270:22801–6. doi: 10.1074/jbc.270.39.22801. doi:10.1074/jbc.270.39.22801. [DOI] [PubMed] [Google Scholar]
- 18.Moura G., Pinheiro M., Silva R., et al. Comparative context analysis of codon pairs on an ORFeome scale. Genome Biol. 2005;6:R28. doi: 10.1186/gb-2005-6-3-r28. doi:10.1186/gb-2005-6-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu T., Tan S., Gou X., Araki H., Tian D. Avoidance of long mononucleotide repeats in codon pair usage. Genetics. 2010;186:1077–84. doi: 10.1534/genetics.110.121137. doi:10.1534/genetics.110.121137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boycheva S., Chkodrov G., Ivanov I. Codon pairs in the genome of Escherichia coli. Bioinformatics. 2003;19:987–98. doi: 10.1093/bioinformatics/btg082. doi:10.1093/bioinformatics/btg082. [DOI] [PubMed] [Google Scholar]
- 21.Nierhaus K.H., Wadzack J., Burkhardt N., et al. Structure of the elongating ribosome: arrangement of the two tRNAs before and after translocation. Proc. Natl. Acad. Sci. USA. 1998;95:945–50. doi: 10.1073/pnas.95.3.945. doi:10.1073/pnas.95.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith D., Yarus M. tRNA-tRNA interactions within cellular ribosomes. Proc. Natl. Acad. Sci. USA. 1989;86:4397–401. doi: 10.1073/pnas.86.12.4397. doi:10.1073/pnas.86.12.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pruitt K.D., Tatusova T., Maglott D.R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–5. doi: 10.1093/nar/gkl842. doi:10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crick F.H. Codon–anticodon pairing: the wobble hypothesis. J. Mol. Biol. 1966;19:548–55. doi: 10.1016/s0022-2836(66)80022-0. doi:10.1016/S0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- 25.Rosner B. Fundamentals of Biostatistics(7th Edition) Brooks/Cole Publishing Company, Cengage Learning, Inc., Independence, Kentucky, USA; 2010. [Google Scholar]
- 26.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. doi:10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larkin M.A., Blackshields G., Brown N.P., et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. doi:10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 28.Sharp P.M., Li W.H. The codon Adaptation Index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–95. doi: 10.1093/nar/15.3.1281. doi:10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharp P.M., Tuohy T.M., Mosurski K.R. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986;14:5125–43. doi: 10.1093/nar/14.13.5125. doi:10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das S., Ghosh S., Pan A., Dutta C. Compositional variation in bacterial genes and proteins with potential expression level. FEBS Lett. 2005;579:5205–10. doi: 10.1016/j.febslet.2005.08.042. doi:10.1016/j.febslet.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 31.Das S., Paul S., Chatterjee S., Dutta C. Codon and amino acid usage in two major human pathogens of genus Bartonella—optimization between replicational-transcriptional selection, translational control and cost minimization. DNA Res. 2005;12:91–102. doi: 10.1093/dnares/12.2.91. doi:10.1093/dnares/12.2.91. [DOI] [PubMed] [Google Scholar]
- 32.Das S., Paul S., Dutta C. Synonymous codon usage in adenoviruses: influence of mutation, selection and protein hydropathy. Virus Res. 2006;117:227–36. doi: 10.1016/j.virusres.2005.10.007. doi:10.1016/j.virusres.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 33.Das S., Roymondal U., Chottopadhyay B., Sahoo S. Gene expression profile of the cyanobacterium Synechocystis genome. Gene. 2012;497:344–52. doi: 10.1016/j.gene.2012.01.023. doi:10.1016/j.gene.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 34.Das S., Roymondal U., Sahoo S. Analyzing gene expression from relative codon usage bias in yeast genome: a statistical significance and biological relevance. Gene. 2009;443:121–31. doi: 10.1016/j.gene.2009.04.022. doi:10.1016/j.gene.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 35.Roymondal U., Das S., Sahoo S. Predicting gene expression level from relative codon usage bias: an application to Escherichia coli genome. DNA Res. 2009;16:13–30. doi: 10.1093/dnares/dsn029. doi:10.1093/dnares/dsn029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shields D.C., Sharp P.M. Synonymous codon usage in Bacillus subtilis reflects both translational selection and mutational biases. Nucleic Acids Res. 1987;15:8023–40. doi: 10.1093/nar/15.19.8023. doi:10.1093/nar/15.19.8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharp P.M., Cowe E., Higgins D.G., Shields D.C., Wolfe K.H., Wright F. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens: a review of the considerable within-species diversity. Nucleic Acids Res. 1988;16:8207–11. doi: 10.1093/nar/16.17.8207. doi:10.1093/nar/16.17.8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rocha E.P. Codon usage bias from tRNA's point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004;14:2279–86. doi: 10.1101/gr.2896904. doi:10.1101/gr.2896904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.dos Reis M., Savva R., Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004;32:5036–44. doi: 10.1093/nar/gkh834. doi:10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vieira-Silva S., Rocha E.P. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 2010;6:e1000808. doi: 10.1371/journal.pgen.1000808. doi:10.1371/journal.pgen.1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.