

Abstract
Selenium is an essential micronutrient in the diet of humans and other mammals. Based largely on animal studies and epidemiological evidence, selenium is purported to be a promising cancer chemopreventive agent. However, the biological mechanisms by which chemopreventive activity takes place are poorly understood. It remains unclear whether selenium acts in its elemental form, through incorporation into organic compounds, through selenoproteins or any combination of these. The purpose of this study was to determine whether selenoproteins mitigate the risk of developing chemically induced mammary cancer. Selenoprotein expression was ablated in mouse mammary epithelial cells through genetic deletion of the selenocysteine (Sec) tRNA gene (Trsp), whose product, designated selenocysteine tRNA, is required for selenoprotein translation. Trsp floxed and mouse mammary tumor virus (MMTV)-cre mice were crossed to achieve tissue-specific excision of Trsp in targeted mammary glands. Eight- to twelve-week-old second generation Trspfl/+;wt, Trspfl/+;MMTV-cre, Trsp fl/fl;wt and Trspfl/fl;MMTV-cre female mice were administered standard doses of the carcinogen, 7,12-dimethylbenzylbenz[a]antracene. Our results revealed that heterozygous, Trspfl/+;MMTV-cre mice showed no difference in tumor incidence, tumor rate and survival compared with the Trspfl/+;wt mice. However, 54.8% of homozygous Trspfl/f l;MMTV-cre mice developed mammary tumors and exhibited significantly shorter survival than the corresponding Trspfl/fl;wt mice, where only 36.4% developed tumors. Loss of the homozygous Trsp alleles was associated with the reduction of selenoprotein expression. The results suggest that mice with reduced selenoprotein expression have increased susceptibility to developing carcinogen-induced mammary tumors and that a major protective mechanism against carcinogen-induced mammary cancer requires the expression of these selenoproteins.
Introduction
Selenium is an essential micronutrient in the diet of many life forms including humans and other mammals. Numerous health benefits have been attributed to this element including roles as a chemopreventive agent in cancer, heart disease and other cardiovascular and muscle disorders and roles in inhibiting viral expression, the onset of AIDS in HIV-positive patients, slowing the aging process, mammalian development and boosting the immune system (1). Among these beneficial effects, the one that has received the most attention is selenium’s role in preventing cancer (see reviews in 2–14).
The anticarcinogenic properties of selenium, small molecular weight selenium-containing compounds (smw selenocompounds) and selenium-containing proteins (selenoproteins) have been reported in numerous in vitro, animal and epidemiological studies (1,11–14) However, despite these many studies, little is known about the underlying metabolic mechanisms of how selenium acts in preventing cancer; and there has been considerable debate in the selenium field whether small molecular weight selenium-containing compounds (4,5,6,7,9) or selenoproteins (11–13), or both (8,10), are the responsible agents. More recently, the emphasis appears to have shifted in favor of selenoproteins as the more likely selenium-containing components involved in cancer prevention (1,2,3,11–14). In addition, two recent studies have shown that selenoproteins are directly involved in preventing colon (15) and prostate cancers (16) and several other studies implicate specific selenoproteins in cancer prevention (1,11,12,14,17–19).
Selenocysteine (Sec) is the selenium-containing amino acid that is incorporated into protein in response to the codon, UGA, and is the 21st amino acid in the genetic code (20–22). Interestingly, Sec is biosynthesized, unlike any other known amino acid in eukaryotes, on its tRNA which is designated selenocysteine tRNA (Sec tRNA[Ser]Sec) (23). Sec tRNA[Ser]Sec is a single-copy gene in the mammalian genome, is designated as Trsp and its expression is essential for selenoprotein synthesis (24). As a consequence, any modulation of tRNA[Ser]Sec expression has a dramatic impact on selenoprotein expression. This feature of regulating selenoprotein expression has provided us with a means of elucidating the function of this protein class by generating various transgenic, standard knockout and conditional knockout mouse models involving wild type and mutant Sec tRNA[Ser]Sec transgenes and the loss, or targeted loss, of Trsp (reviewed in 24,25).
In an initial study, the targeted removal of Trsp in mammary tissues using loxP-cre technology was examined (26). Only a slight loss in most selenoproteins was found, since the promoter-cre-recombinase was specific to epithelial cells and epithelial cells represent only a small proportion of the cell population of mammary tissue in comparison with other cell types. Although mammary tissue consists of relatively few epithelial cells, the mammary gland remains an ideal tissue to examine the role of selenoproteins in cancer since both chemically induced and relevant genetic mouse mammary cancer models have been developed. Furthermore, this tissue is a major focus of cancer occurrence in women with known alterations in several genes that are involved in breast cancer development. For instance, BRCA1 and p53 tumor suppressor genes which are frequently altered in familial breast cancers play a central role in maintaining the genetic integrity of the cell (27–30). Most importantly, BRCA1 and p53 expression have been shown to be altered in the Trsp knockout model (26). Therefore, in the present study, we examined whether the reduction of selenoprotein expression in mammary epithelium would affect the incidence of mammary cancer induced through chemical carcinogenesis by targeting the removal of Trsp using loxP-cre technology (26). We found that homozygous deletion of Trsp in mammary epithelial cells resulted in a shortened time to tumor formation and shortened survival time compared with control animals. The reduction in Sec tRNA[Ser]Sec was accompanied by reduced expression of selenoproteins. Therefore, these results demonstrate that selenoproteins play a critical role in preventing mammary cancer and that therapies to enhance the production of selenoproteins may be a useful strategy for chemoprevention.
Materials and methods
Chemicals
Paraformaldehyde, glutaraldehyde, NP-40, X-gal, K3Fe (CN)6, K4Fe(CN)6·3H2O, MgCl2 and Na-deoxycholate were purchased from the Sigma (St Louis, MO).
Animals
Mice were handled in accordance with protocols approved by the NCI Animal Care and Use Committee and housed in microisolator cages on a 12 h light/dark cycle with food and water provided ad libitum. The mice were fed the Charles River Rat and Mouse 18% protein (Autoclavable) 5L79 diet (PMI Nutrition International, St Louis, MO). Mice exhibited normal weight gain, and no differences in weight were observed between the cohorts of mice. Generation of Trsp floxed (Trspfl/fl) mice in a C57Bl/6 background (26), mouse mammary tumor virus (MMTV)-cre mice in an FVB/N background (31) and Rosa26R mice in a B6/129 background (32) have been described previously. Crosses of these mice to generate the animals used in the present study, their genotypes and strain backgrounds are summarized in the scheme shown in Table I. Briefly, F1 generation control mice were produced by crossing homozygous Trspfl/fl mice with mice heterozygous for the MMTV-cre transgene resulting in Trsp fl/+;wt and Trsp fl/+;MMTV-cre offspring. The F2 generation was produced by crossing Trsp fl/+;MMTV-cre with Trspfl/fl;wt mice to generate Trsp fl/+;MMTV-cre, Trspfl/+;wt Trsp fl/fl;MMTV-cre and Trspfl/fl;wt offspring. In addition, control animals were generated from MMTV-cre and Rosa26R crosses in order to determine whether recombination of an unrelated allele (Rosa26) by cre recombinase could influence carcinogen-induced tumor development. Rosa26R contain a flox-STOP-flox cassette upstream of the lacZ gene inserted into the Rosa26 locus. Upon cre-induced excision of the STOP cassette, lacZ is expressed. Wild-type FVB and C57Bl/6 mice were also treated with 7,12-dimethylbenz[a] anthracene (DMBA) to determine differences in tumor susceptibilities of these individual strains which have been reported to be different (31).
Table I.
Mouse genotypes, background strains, crosses, and resulting offspring used in the study
| Mouse genotypesa | Crossed with | Mouse genotypes | Offspringb |
| F1 generation | |||
| MMTV-cre (FVB/N) | X | Wt (C57Bl/6) | MMTV-cre wt |
| Rosa26R (B6/129) | X | MMTV-cre (FVB/N) | Rosa26R;MMTV-cre Rosa26R; wt |
| Trspfl/fl (C57Bl/6) | X | MMTV-cre (FVB/N) | Trspfl/+;MMTV-cre Trspfl/+;wt |
| F2 generation | |||
| Trspfl/+;MMTV-cre (C57Bl/6; FVB/N) | X | Trspfl/fl;wt (C57Bl/6) | Trspfl/+;MMTV-cre |
| Trspfl/+;wt | |||
| Trspfl/fl;MMTV-cre | |||
| Trspfl/fl;wt |
Background strain in parentheses.
Represent the offspring that were dosed with DMBA.
Genotyping
DNA was extracted from mouse tissues using the Genomic DNA Purification Kit (Promega, Madison, WI) according to the manufacturer’s instructions to determine the presence of floxed Trsp and the cre transgene by PCR as described (26). In addition, we also determined the presence of Rosa26R by PCR as described previously (32). Moreover, mammary tissue was randomly selected from Rosa26R mice to determine cre recombination through the expression of β-galactosidase. Tissues were fixed for 1–2 h in 2% paraformaldehyde, 0.25% glutaraldehyde, 0.01% NP-40 in PBS and stained for β-galactosidase activity (1 mg/ml X-gal, 30 mM K3Fe (CN)6, 30 mM K4Fe(CN)6·3H2O, 2 mM MgCl2, 0.01% Na-deoxycholate, 0.02% NP-40, 1x PBS) overnight at 30°C. Stained and unstained tissues were viewed under an inverted microscope.
Tumor induction by DMBA
7,12-dimethylbenz[a] anthracene was purchased from Sigma. Numbers of female mice with the following genotypes (8–12 week old) produced in the F2 generation received 0.1 ml of 10 mg/ml DMBA dissolved in sesame oil by gavage once a week for 6 weeks: 49 Trspfl/ +;MMTV-cre, 34 Trspfl/+;wt, 31 Trspfl/fl;MMTV-cre, and 33 Trspfl/fl-;wt. In addition, thirty 8- to 12-week-old female MMTV-cre and thirty wild-type mice and thirty-six 8- to 12-week-old female Rosa26R; MMTV-cre and 20 Rosa26R;wt mice received 0.1 ml of 10 mg/ml DMBA dissolved in sesame oil by gavage once a week for 6 weeks. Forty 8- to 12-week-old female mice from each FVB and C57Bl/6 group also received 0.1 ml of 10 mg/ml DMBA as described above. Mice were observed twice a week for the development of tumors that were measured by caliper. Mice were euthanized for humane reasons when a tumor reached 2 cm in diameter.
Pathology
All mice were necropsied. The #4 mammary glands, mammary tumors, all tissues with tumors, lungs and liver were fixed in 4% paraformaldehyde (Fisher Scientific, Carlsbad, CA) overnight and then placed in 70% ethanol and embedded in paraffin. Sections were prepared and stained with hematoxylin and eosin (H&E) and subjected to a blind review by a single pathologist (M.H.).
Western blot
Protein extracts were prepared from mammary glands by homogenizing the tissue in cold lysis buffer [50 mM Tris; pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% Igepal and Complete Mini Protease Inhibitor (Roche)]. Protein was electrophoresed through 10% polyacrylamide gels, transferred onto PVDF membranes and immunoblotted with antibodies against glutathione peroxidase 1 (GPx1) (Abcam, Burlingame, CA; 1:1000 dilution), glutathione peroxidase 4 (GPx4) (Epitomics, Cambridge, MA; 1:1000 dilution) or 15 kDa selenoprotein (Sep15) (Epitomics; 1:1000 dilution). Following incubation of the primary antibody, membranes were washed with tris-buffered saline (20 mM Tris/HCl, pH 7.5 and 150 mM NaCl) containing 0.1% Tween 20 and incubated in anti-rabbit horseradish peroxidase–conjugated secondary antibody (Thermo Scientific, Rockford, IL; 1:10 000). Following secondary antibody incubation, membranes were washed with tris-buffered saline, incubated in SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and exposed to X-ray film.
Statistical analysis
Differences in tumor incidence between groups were tested using Fisher’s Exact Test. Differences in median number of tumors were tested using the Wilcoxon or Kruskal–Wallis test as appropriate. Differences in time to tumor or survival time were tested using Kaplan–Meier methods and the Wilcoxon chi-square statistics.
Results
Generation of mammary-specific, Trsp-deficient mice
F1 and F2 generation mice were generated as shown in Table I to obtain animals with the desired genotypes for use in these studies. Table II indicates the number of mice used in each experimental group and the number of mammary tumors that developed in each group. Genotyping of mice was performed using PCR of DNA extracted from tail snips. The presence of the MMTV-cre transgene was confirmed by a 280 bp PCR fragment (Figure 1, upper panel). Confirmation of recombination of the floxed Trsp alleles in the mammary glands was performed by PCR on DNA extracted from mammary glands. The wild-type Trsp allele is detected as a 900 bp band, the Trsp floxed allele as a 1.1 kb band and the Trsp knockout allele as a 450 bp band (Figure 1, lower panel). Recombination of the floxed Trsp allele was detected only in mice carrying the MMTV-cre transgene. The 450 bp knockout allele appears relatively weak compared with the other allele bands that is due to the fact that only a relatively small percentage of cells in the mouse mammary gland are epithelial cells that undergo cre-recombination in this model system (26). Body weights of the groups of mice in the F2 generation (Trspfl/fl;MMTV-cre; 25.7 gm ± 3.47, Trspfl/fl;wt; 25.5 gm ± 3.41, Trspfl/+;MMTV-cre; 24.8 gm ± 1.67, p = 0.705) were not significantly different from one another suggesting that the loss of Trsp in the mammary gland did not affect the overall health of the mice. Our previous study of selenoprotein loss in the normal mammary gland using Trsp fl/fl;MMTV-cre mice demonstrated strong cre expression in the mammary epithelial cells with evidence of moderate expression in the skin and spleen and virtually undetectable expression in the kidney (26).
Table II.
Listing of experimental groups, number of mice and number of mice developing mammary tumors in each group
| Experimental groups | Number/group | Number with tumors (%) |
| 1) wt FVB/N + DMBA | 40 | 17 (42.5) |
| 2) wt C57/Bl6 + DMBA | 40 | 9 (22.5) |
| 3) Rosa26R; wt + DMBA | 20 | 6 (30.0) |
| 4) Rosa26R;MMTV-cre + DMBA | 36 | 11 (30.5) |
| 5) Trspfl/fl;wt + DMBA | 33 | 12 (36.4) |
| 6) Trspfl/fl;MMTV-cre + DMBA | 31 | 17 (54.8) |
| 7) Trspfl/+;wt + DMBA | 34 | 21 (61.8) |
| 8) Trspfl/+;MMTV-cre + DMBA | 49 | 26 (53.1) |
Fig. 1.

Excision of floxed-Trsp alleles in the mammary glands of Trspfl/fl;MMTV-cre mice. Upper panel shows a 280 bp band for the MMTV-cre transgene in lanes 1, 3 and 5 as demonstrated by PCR. Lower panels show heterozygous and homozygous floxed Trsp mice wherein Trspfl/+ is represented by a 1.1 kb band, Trspwt by a 900 bp band and Trspfl/fl by a 450 bp band. F1 and F2 generations are shown in lanes 1 and 2 and in lanes 3, 4, 5 and 6, respectively.
DMBA-induced tumor susceptibility in FVB, C57Bl/6, MMTV-cre and Rosa26R mice
Since mice used in this study were hybrids between different strain backgrounds, we compared the response of FVB and C57Bl/6 wild-type mice with the induction of tumors by DMBA (Figure 2). There was no statistically significant difference in the mammary tumor incidence [FVB, 42.5% (17/40) versus C57Bl/6; 22.5% (9/40), p = 0.09] (Table II) or median tumor number (1.0 versus 1.0, p = 0.99) comparing FVB and C57Bl/6 mice. However, following exposure to DMBA, FVB mice developed mammary tumors in a significantly shorter period of time compared with C57B/6 mice (166 versus 273 median days, respectively; p = 0.0001) (Figure 2A and Table III) and exhibited a significantly reduced survival rate (about 132 versus 180 median days, respectively; p = 0.004) (Figure 2B and Table III). These data are consistent with previous reports demonstrating that DMBA-induced mammary tumors arise earlier and are more aggressive in FVB than in C57Bl/6 mice (31). Since the floxed model system that we utilized in this study introduced a recombination event in the mouse genome, we examined the possibility that such an event could modify the risk of DMBA-induced carcinogenesis unrelated to the Trsp allele. Therefore, to control for cre recombination, we determined whether tumor incidence following exposure to DMBA differed between Rosa26R; MMTV-cre mice and Rosa26R; wt mice in the same genetic background. No significant differences in mammary tumor incidence (30.5% versus 31.6%; respectively; p = 0.739) (Table II), median tumor number (1.0 versus 1.0; respectively; p = 0.363) or survival (about 238 versus 249 median days; respectively; p = 0.845) were observed between these groups (Table III). The results indicate that cre-mediated recombination of an allele unrelated to tumorigenesis does not increase tumor susceptibility (see Supplementary Figure 2A and B is available at Carcinogenesis Online).
Fig. 2.

Mammary tumor development and survival rates in FVB and C57Bl/6 control mice. (A) Time to mammary tumor formation following DMBA exposure in control FVB (solid line) and C57Bl/6 mice (dotted line). Represents the percent of mice that develop mammary tumors over time. (B) Time to death of control FVB (solid line) and C57Bl/6 mice (dotted line) from all causes.
Table III.
Median time to death and time to mammary tumor onset
| Confidence interval | ||||
| Group | Median | Lower 95% | Upper 95% | P-value |
| Time to death | ||||
| FVB | 132 | 119 | 146 | 0.004 |
| C57Bl/6 | 180 | 138 | 210 | |
| Trspfl/fl;MMTV-cre | 146 | 126 | 216 | 0.018* |
| Trspfl/+;MMTV-cre | 264 | 234 | 322 | 0.21* |
| Trspfl/fl;wt | 214 | 168 | 266 | 0.068# |
| Trsp fl/+;wt | 241 | 196 | 286 | |
| Rosa26R; MMTV-cre | 238 | 168 | 287 | 0.845 |
| Roas26R; wt | 249 | 133 | 295 | |
| Time to mammary tumor onset | ||||
| FVB | 166 | 133 | 197 | 0.0001+ |
| C57Bl/6 | 273 | 238 | 306 | |
| Trspfl/fl;MMTV-cre | 236 | 154 | 261 | 0.03* |
| Trspfl/+;MMTV-cre | 300 | 265 | 412 | 0.18* |
| Trspfl/fl;wt | 356 | 294 | 377 | |
| Trspfl/+wt | 294 | 188 | 384 | |
*p-value when compared with Trspfl/fl;wt. #p-value comparing Trspfl/fl;wt versus Trspfl/+;wt. *p-value when compared with Trspfl/fl;wt. +p-value comparing FVB versus C57Bl/6.
Loss of Sec-tRNA[Ser]Sec increases susceptibility to mammary cancer
DMBA is known to cause multiple pathologies in mice leading to death including a significant incidence of lymphomas (33). We determined overall survival curves for each group of mice (Figure 3). Mammary tumor incidence was calculated as the number of mice developing a mammary tumor (confirmed histologically)/total number of mice treated in each group (Table III). The median time of tumor onset was determined for the mice in each group that developed mammary tumors (Table III). We compared the time with mammary tumor onset and survival between Trspfl/fl;wt, Trspfl/+;MMTV-cre and Trspfl/fl;MMTV-cre mice from the F2 generation in response to DMBA exposure as described in the Materials and methods (Figure 3A and B). There was no significant difference between Trspfl/fl;MMTV-cre and Trspfl/fl;wt mice in overall mammary tumor incidence [54.8% (17/31) versus 36.4% (12/33), respectively; p = 0.21] (Table II) and median tumor number (1.0 versus 1.0; p = 0.30). However, Trspfl/fl;MMTV-cre mice developed mammary tumors more rapidly than Trspfl/fl;wt mice (154 versus 356 median days, respectively; p = 0.03) and had a shorter survival period compared with Trspfl/fl;wt mice (146 versus 214 median days; p = 0.018) (Figure 3A and Table III). Compared with Trspfl/+;wt mice, however, Trspfl/+;MMTV-cre mice showed no differences in mammary tumor incidence [61.8% (21/34) versus 53.1% (26/49), respectively; p = 0.50] (Table III), median tumor number (1.0 versus 1.0; p = 0.94), time to tumor development (294 versus 300 median days, respectively; p = 0.18) and survival (242 versus 264 median days, respectively; p = 0.21) (Table III). This is consistent with our previous study showing that Trspfl/ +MMTV-cre mice produce sufficient levels of selenoproteins to provide adequate selenoprotein function (26). The histology of the mammary tumors generated by DMBA were mostly of the adenosquamous type, although other phenotypes included tubloacinar adenocarcinoma, solid carcinoma, carcinosacrcomas, spindle cell sarcoma and keratoacanthoma (see Supplementary Table I and Figure 1 is available at Carcinogenesis Online).
Fig. 3.

Tumor development and survival rates in Trsp knockout and control mice. (A) Time to tumor formation in Trspfl/fl;MMTV-cre (solid line), Trspfl/+;MMTV-cre (dashed line) and Trspfl/fl;wt mice (blue dashed line). Represents the percent of mice that develop mammary tumors over time. (B) Time to death of Trspfl/fl;MMTV-cre (solid line), Trspfl/+;MMTV-cre (dashed line) and Trspfl/fl;wt mice (blue dashed line) from all causes. P values are compared with Trspfl/fl;wt and Trspfl/+;wt mice. There was no significant differences between Trspfl/fl;wt and Trspfl/+;wt mice. Only the Trspfl/fl;wt mice are graphed.
Loss of Sec-tRNA is associated with reduced GPx1, GPx4 and Sep15
Since loss of Sec-tRNA results in the loss of selenoprotein expression, we evaluated the expression levels of representative selenoproteins, Sep15, GPx4 and GPx1 from DMBA-induced mammary tumors generated in Trspfl/fl;MMTV-cre and Trspfl/fl;wt mice (Figure 4). Western blot analyses demonstrated that mammary tumors arising in Trspfl/fl;MMTV-cre mice expressed reduced levels of Sep15, GPx4 and GPx1 selenoproteins as compared with tumors from Trspfl/fl;wt mice. Since tumors are composed of a mixture of epithelial cells in which the Trsp knockout has occurred as well as stromal cells containing wt Trsp, we did not observe a complete loss of these proteins by western blotting. Although Sec tRNA[Ser]Sec expression was reduced in Trspfl/fl;MMTV-cre mice in our earlier study (26), it was apparent from the level of expression that there was substantially more cell types in addition to epithelial cells in mammary tissue used for tRNA analysis. Unfortunately, no reagents are available to perform immunohistochemistry on the tumor samples to demonstrate loss of the proteins within the epithelial cells.
Fig. 4.
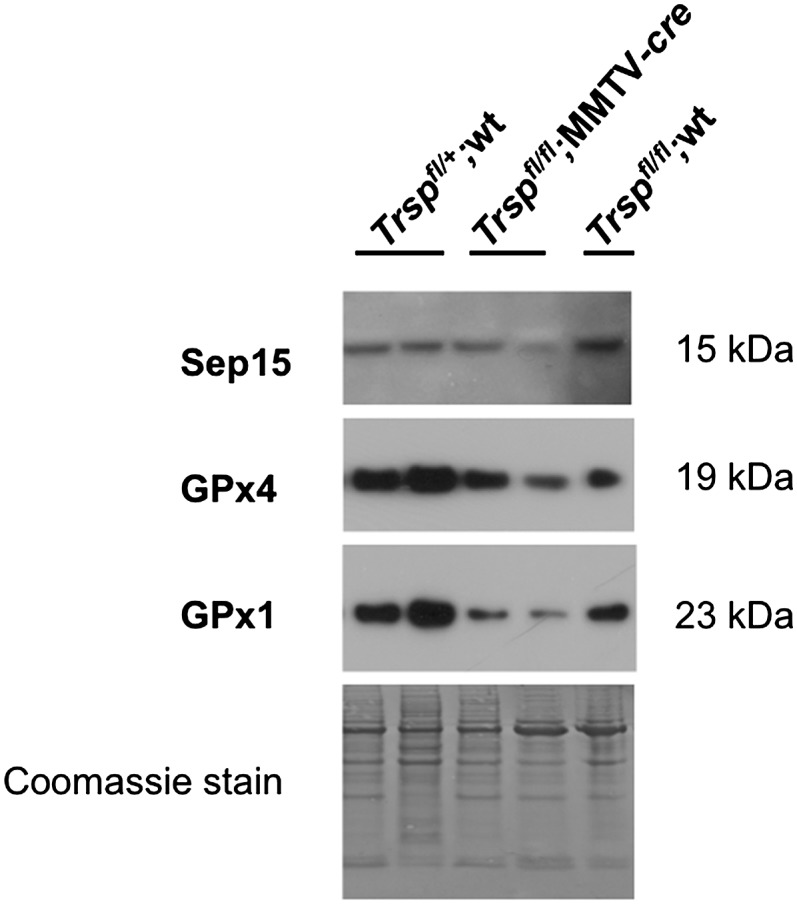
Selenoprotein expression in DMBA-induced mammary tumors. Western blot analyses of protein extracts from carcinogen-induced mouse mammary tumors showing Sep15, GPx4 and GPx1 protein levels are reduced in Trspfl/fl;MMTV-cre tumors compared with Trspfl/+; wt and Trspfl/fl;wt tumors. Genotypes of tumors are indicated at the top of the figure.
Discussion
Although epidemiological data have associated a protective effect of selenium in the prevention of prostate and lung cancer in humans, the relationship between selenium and breast cancer risk is less clear (1,11,12). Since selenium may exist in various forms within an organism, recent studies have attempted to elucidate what role each form of selenium may play in cancer chemoprevention. This element may exist as elemental selenium, small molecular weight selenium-containing compounds and selenium incorporated into selenoproteins as the amino acid, Sec. All three forms of selenium appear to have positive effects on preventing cancer (2–14) (1,3,15). The current study has demonstrated, by using a genetic approach, that selenoproteins are protective against chemical carcinogen-induced mammary cancer in mice.
Our previous study in which Trsp was knocked out in wild-type mouse mammary glands revealed a reduction in Sec tRNA[Ser]Sec and selenoprotein levels but the reduction was only partially observed due to the low percentage of epithelial cells among other cells in mammary tissue (26). Herein, we expanded the earlier study and explored whether the loss of Trsp and, therefore, the loss of selenoprotein expression increased the risk of development of mammary tumors induced by DMBA using Trspfl/fl;MMTV-cre, Trspfl/+;MMTV-cre and Trspfl/+;wt mice. Previous studies have shown that different strains of mice respond differently to DMBA. Hennings et al. (31) showed that FVB mice were more susceptible to developing DMBA tumors than C57Bl6 mice. Therefore, we carefully controlled for genetic background of the mice in these experiments to exclude genetic background as a confounding variable in influencing the outcome of our results.
Furthermore, we observed that genomic recombination alone did not increase the incidence of DMBA-induced mammary carcinogenesis. Genomic recombination by cre has been shown to cause DNA damage in mammalian cells (34), and thus, we evaluated whether cre recombination in the mammary gland in an unrelated allele contributes to a higher risk of developing DMBA-induced mammary tumors. Rosa26R; MMTV-cre mice undergo homologous recombination at the Rosa26 locus in the mammary epithelium resulting in the loss of Rosa26 protein expression which is replaced by expression of lacZ. No differences in tumorigenesis were observed in Rosa26R; MMTV-cre mice exposed to DMBA compared with Rosa26R; wt mice. Therefore, we conclude that the differences in mammary tumor development observed in Trsp knockout mice compared with wild type is not the result of nonspecific cre recombination.
Hu et al. showed that loss of heterozygosity at the GPx1 locus is a common event in cancer of head and neck, breast, lung and colon (18,35–37). GPx1 was shown to protect against hypoxia in human breast cancer cells (38). In addition, a gene variant of GPx1 was shown to be associated with breast cancer (18). Furthermore, loss of heterozygosity of Sep15 was shown to be associated with beast cancer (3). Our results are consistent with these studies wherein we found that reduction in the expression of selenoproteins (see Figure 4) is associated with accelerated mammary tumor development. Unfortunately, we were not able to directly compare activities of these selenoproteins in normal mammary tissues (due to the low numbers of mammary epithelial cells in the gland) or tumor tissue since the other cell types present have not lost the Trsp alleles. However, our earlier study of Trsp loss in normal mammary epithelium has demonstrated that this model system does knock-out selenoprotein expression in the mammary gland (26). We demonstrated a reduction in levels of the selenoproteins GPx1, GPx4 and Sep15 in the present study.
Clearly, selenoproteins are not expressed in mammary epithelial cells due to the dependence of their synthesis on the presence of Sec tRNA[Ser]Sec which is absent by the targeted removal of Trsp (26). Interestingly, loss of both Trsp alleles and lack of selenoprotein expression in mammary epithelium resulted in a significantly shortened time to tumor onset and reduced survival time but did not reduce overall tumor incidence or significantly impact tumor phenotype. The shortened time to tumor onset and reduced survival time might be explained by the ability of selenoproteins to delay tumor promoting events induced by the carcinogen, but the inhibition is eventually overcome. As cancer cells suffer from oxidative stress and the major role of most characterized selenoproteins is to serve as antioxidants (39), such a proposal would seem to be a logical possibility. Our results appear to be significant since they suggest that chemoprevention may be improved by identifying mechanisms that increase selenoprotein expression, but it should also be noted that other prevention targets may also be necessary in order to ultimately prevent the development of mammary cancer. If translatable to preventing human breast cancer, then combinations of chemopreventive agents along with enhanced selenoprotein production may be critical in reducing the development of breast cancer.
Two well-characterized selenoproteins that serve as antioxidants are TR1 and GPx1 and the loss of their antioxidant properties most certainly has an impact on the role that this selenium-containing protein class has in maintaining reduced oxidative, cellular stress in normal cells (40). TR1 is known to have a role in protecting normal cells from cancer as it is one of the major redox regulators in mammals having roles in cell proliferation, transcription and angiogenesis, contributing to the antioxidant defense and acting as a redox regulator of cell signaling (reviewed in 41). One of the principal functions of TR1 in normal cells is to maintain thioredoxin in the reduced state (42) and thus its loss in mammary epithelium ablates the thioredoxin system. As noted above, cancer cells suffer from oxidative stress and must depend on a strong antioxidant system to maintain their malignant properties. The interrelationship between the glutathione and thioredoxin systems and the upregulation of the glutathione system in malignant cells following the downregulation of the thioredoxin system has been thoroughly reviewed recently (43–45).
It should also be noted that GPx1 has a role in serving as an antioxidant in the glutathione system (reviewed in 46,47), but, the targeted loss of the GPx1 gene in mice resulted in no observable phenotype unless the mouse was subjected to stress (46,47). It would appear, therefore, that the loss of GPx1 expression contributes to the shortened time to tumor onset and reduced survival time observed in the present study. It should also be noted that although selenoproteins play an important role as antioxidants, they have other functions which may also be critical in chemoprevention. Further studies will be required to address this latter point.
Our results may have important translational implications in light of the failure of the Selenium and Vitamin E Cancer Prevention Trial (SELECT) (48). The SELECT Trial was based upon observations made during the Nutritional Prevention of Cancer Trial wherein an association was made between increased prostate cancer risk and low baseline levels of serum selenium (49). In the prospectively designed SELECT Trial, L-selenomethionine, vitamin E (all rac-alpha-tocopheryl acetate) or a combination of both were administered to men to determine whether these compounds could reduce the risk of prostate cancer. The study concluded that there was no reduction in prostate cancer for these compounds at the doses administered. Thus, the chemopreventive properties of selenium may be related to the levels of selenoprotein expression and not simply the ingestion of selenium-containing compounds. Our results suggest that an important strategy for cancer chemoprevention may be to identify mechanisms that increase selenoprotein expression. Combinations of chemopreventive agents along with enhanced selenoprotein production may be critical in reducing the development of breast cancer.
There are at least two other studies that have shown a direct connection between selenoprotein deficiency and enhanced carcinogenesis. Irons et al. (15) and Diwadkar-Navsariwala et al. (16) demonstrated that selenoprotein deficiency generated by using a specific transgenic mouse model that causes a reduction in certain selenoproteins resulted in enhanced colon cancer and prostate cancer, respectively. The current study provides another example of a direct role of selenoproteins, and likely their role as antioxidants, in affecting cancer development, and more specifically, in the onset and degree of intensity in breast cancer.
While this study supports the role of selenoproteins in delaying mammary tumor progression in this model system, our results indicate that mammary tumors ultimately develop. This indicates that while selenoproteins are protective against chemically induced carcinogenesis in the mammary glands, they may be insufficient as a completely chemopreventive agent. Other strategies may need to be combined with enhanced selenoprotein expression to provide full chemoprevention. Alternatively, it may be possible that further mutations occur in lesions that are initially suppressed by selenoproteins leading to the eventual development of mammary tumors. Further work will be required to dissect more detailed mechanisms of how selenoproteins retard mammary tumor formation and whether the protective effect is more pronounced in certain subtypes of human breast cancer.
In summary, the current study demonstrates that loss of Trsp expression in mammary epithelial cells accelerates the development of carcinogen-induced tumorigenesis associated with reduced expression of selenopreoteins and supports a chemopreventive mechanism through the enhanced expression of selenoproteins.
Funding
This study was funded by the intramural research program of NIH, National Cancer Institute, Center for Cancer Research.
Supplementary material
Supplementary Table I and Figures 1–2 can be found at http://carcin.oxfordjournals.org/
Acknowledgments
We would like to acknowledge Lisa Riffle, Darlene Reaver and Christina Hernandez for their animal caretaking assistance. J.E.G., D.L.H., B.A.C., T.S.H. conceived and designed the experiments; T.S.H., B.A.C., L.S. performed the experiments; H.A.Y., J.E.G., D.L.H., M.H., L.S. analyzed the data; and W.M., L.S., D.L.H., B.A.C. contributed reagents, materials, analysis tools and animals.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- DMBA
7,12-dimethylbenzylbenz[a]antracene
- GPx1
glutathione peroxidase 1
- GPx4
glutathione peroxidase 4
- MMTV
mouse mammary tumor virus
- Sec
selenocysteine
- Sec tRNA[Ser]Sec
selenocysteine tRNA
References
- 1.Hatfield DL, et al. Selenium: its molecular biology and role in human health. NYC: Springer; 2011. [Google Scholar]
- 2.Davis CD, et al. Are selenoproteins important for the cancer protective effects of selenium. Curr. Nutr. Food Sci. 2005;1:201–214. [Google Scholar]
- 3.Diwadkar-Navsariwala V, et al. The link between selenium and chemoprevention: a case for selenoproteins. J. Nutr. 2004;134:2899–2902. doi: 10.1093/jn/134.11.2899. [DOI] [PubMed] [Google Scholar]
- 4.Ganther HE. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: complexities with thioredoxin reductase. Carcinogenesis. 1999;20:1657–1666. doi: 10.1093/carcin/20.9.1657. [DOI] [PubMed] [Google Scholar]
- 5.Ip C. Lessons from basic research in selenium and cancer prevention. J. Nutr. 1998;128:1845–1854. doi: 10.1093/jn/128.11.1845. [DOI] [PubMed] [Google Scholar]
- 6.Ip C, et al. New concepts in selenium chemoprevention. Cancer Metastasis Rev. 2002;21:281–289. doi: 10.1023/a:1021263027659. [DOI] [PubMed] [Google Scholar]
- 7.Jung HJ, et al. Current issues of selenium in cancer chemoprevention. Biofactors. 2010;36:153–158. doi: 10.1002/biof.81. [DOI] [PubMed] [Google Scholar]
- 8.Lu J, et al. Selenium and cancer chemoprevention: hypotheses integrating the actions of selenoproteins and selenium metabolites in epithelial and non-epithelial target cells. Antioxid. Redox Signal. 2005;7:1715–1727. doi: 10.1089/ars.2005.7.1715. [DOI] [PubMed] [Google Scholar]
- 9.Medina D, et al. Se-methylselenocysteine: a new compound for chemoprevention of breast cancer. Nutr. Cancer. 2001;40:12–17. doi: 10.1207/S15327914NC401_5. [DOI] [PubMed] [Google Scholar]
- 10.Rayman MP. Selenium in cancer prevention: a review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005;64:527–542. doi: 10.1079/pns2005467. [DOI] [PubMed] [Google Scholar]
- 11.Rayman MP. Selenoproteins and human health: insights from epidemiological data. Biochim. Biophys. Acta. 2009;1790:1533–1540. doi: 10.1016/j.bbagen.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Zhuo P, et al. Molecular mechanisms by which selenoproteins affect cancer risk and progression. Biochim. Biophys. Acta. 2009;1790:1546–1554. doi: 10.1016/j.bbagen.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson MI, et al. Selenium and anticarcinogenesis: underlying mechanisms. Curr. Opin. Clin. Nutr. Metab. Care. 2008;11:718–726. doi: 10.1097/MCO.0b013e3283139674. [DOI] [PubMed] [Google Scholar]
- 14.Brigelius-Flohe R. Selenium compounds and selenoproteins in cancer. Chem. Biodivers. 2008;5:389–395. doi: 10.1002/cbdv.200890039. [DOI] [PubMed] [Google Scholar]
- 15.Irons R, et al. Both selenoproteins and low molecular weight selenocompounds reduce colon cancer risk in mice with genetically impaired selenoprotein expression. J. Nutr. 2006;136:1311–1317. doi: 10.1093/jn/136.5.1311. [DOI] [PubMed] [Google Scholar]
- 16.Diwadkar-Navsariwala V, et al. Selenoprotein deficiency accelerates prostate carcinogenesis in a transgenic model. Proc. Natl. Acad. Sci. USA. 2006;103:8179–8184. doi: 10.1073/pnas.0508218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu J, et al. GPX1 Pro198Leu polymorphism and breast cancer risk: a meta-analysis. Breast Cancer Res. Treat. 2010;124:425–431. doi: 10.1007/s10549-010-0841-z. [DOI] [PubMed] [Google Scholar]
- 18.Hu YJ, et al. Role of glutathione peroxidase 1 in breast cancer: loss of heterozygosity and allelic differences in the response to selenium. Cancer Res. 2003;63:3347–3351. [PubMed] [Google Scholar]
- 19.Maia AT, et al. Extent of differential allelic expression of candidate breast cancer genes is similar in blood and breast. Breast Cancer Res. 2009;11:R88. doi: 10.1186/bcr2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birringer M, et al. Trends in selenium biochemistry. Nat. Prod. Rep. 2002;19:693–718. doi: 10.1039/b205802m. [DOI] [PubMed] [Google Scholar]
- 21.Hatfield DL, et al. How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 2002;22:3565–3576. doi: 10.1128/MCB.22.11.3565-3576.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadtman TC. Discoveries of vitamin B12 and selenium enzymes. Annu. Rev. Biochem. 2002;71:1–16. doi: 10.1146/annurev.biochem.71.083101.134224. [DOI] [PubMed] [Google Scholar]
- 23.Xu XM, et al. New developments in selenium biochemistry: selenocysteine biosynthesis in eukaryotes and archaea. Biol. Trace Elem. Res. 2007;119:234–241. doi: 10.1007/s12011-007-8003-9. [DOI] [PubMed] [Google Scholar]
- 24.Hatfield DL, et al. Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Prog. Nucleic Acid Res. Mol. Biol. 2006;81:97–142. doi: 10.1016/S0079-6603(06)81003-2. [DOI] [PubMed] [Google Scholar]
- 25.Carlson BA, et al. Mouse models targeting selenocysteine tRNA expression for elucidating the role of selenoproteins in health and development. Molecules. 2009;14:3509–3527. doi: 10.3390/molecules14093509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumaraswamy E, et al. Selective removal of the selenocysteine tRNA [Ser]Sec gene (Trsp) in mouse mammary epithelium. Mol. Cell. Biol. 2003;23:1477–1488. doi: 10.1128/MCB.23.5.1477-1488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng CX, et al. Roles of BRCA1 and its interacting proteins. Bioessays. 2000;22:728–737. doi: 10.1002/1521-1878(200008)22:8<728::AID-BIES6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 28.Evan G, et al. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- 29.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, et al. BRCA1 physically associates with p.53 and stimulates its transcriptional activity. Oncogene. 1998;16:1713–1721. doi: 10.1038/sj.onc.1201932. [DOI] [PubMed] [Google Scholar]
- 31.Hennings H, et al. FVB/N mice: an inbred strain sensitive to the chemical induction of squamous cell carcinomas in the skin. Carcinogenesis. 1993;14:2353–2358. doi: 10.1093/carcin/14.11.2353. [DOI] [PubMed] [Google Scholar]
- 32.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 33.Nicol CJ, et al. PPARgamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004;25:1747–1755. doi: 10.1093/carcin/bgh160. [DOI] [PubMed] [Google Scholar]
- 34.Loonstra A, et al. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc. Natl. Acad. Sci. USA. 2001;98:9209–9214. doi: 10.1073/pnas.161269798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayashi T, et al. Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J. Biol. Chem. 1993;268:11380–11388. [PubMed] [Google Scholar]
- 36.Hu Y, et al. Allelic loss of the gene for the GPX1 selenium-containing protein is a common event in cancer. J. Nutr. 2005;135:3021S–3024S. doi: 10.1093/jn/135.12.3021S. [DOI] [PubMed] [Google Scholar]
- 37.Hu YJ, et al. Allelic loss at the GPx-1 locus in cancer of the head and neck. Biol. Trace Elem. Res. 2004;101:97–106. doi: 10.1385/BTER:101:2:097. [DOI] [PubMed] [Google Scholar]
- 38.Bilodeau JF, et al. Glutathione peroxidase-1 expression enhances recovery of human breast carcinoma cells from hyperoxic cell cycle arrest. Free Radic. Biol. Med. 2002;33:1279–1289. doi: 10.1016/s0891-5849(02)01013-4. [DOI] [PubMed] [Google Scholar]
- 39.Kryukov GV, et al. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 40.Reeves MA, et al. The human selenoproteome: recent insights into functions and regulation. Cell Mol. Life Sci. 2009;66:2457–2478. doi: 10.1007/s00018-009-0032-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arner ES. Focus on mammalian thioredoxin reductases—important selenoproteins with versatile functions. Biochim. Biophys. Acta. 2009;1790:495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 42.Turanov AA, et al. Mammalian thioredoxin reductase 1: roles in redox homoeostasis and characterization of cellular targets. Biochem. J. 2010;430:285–293. doi: 10.1042/BJ20091378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franco R, et al. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009;16:1303–1314. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 44.Montero AJ, et al. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs. 2011;71:1385–1396. doi: 10.2165/11592590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 45.Corti A, et al. Gamma-glutamyltransferase of cancer cells at the crossroads of tumor progression, drug resistance and drug targeting. Anticancer Res. 2010;30:1169–1181. [PubMed] [Google Scholar]
- 46.Lei XG, et al. Metabolic regulation and function of glutathione peroxidase-1. Annu. Rev. Nutr. 2007;27:41–61. doi: 10.1146/annurev.nutr.27.061406.093716. [DOI] [PubMed] [Google Scholar]
- 47.Lubos E, et al. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox. Signal. 2011;15:1957–1997. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lippman SM, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duffield-Lillico AJ, et al. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: an analysis of the complete treatment period of the Nutritional Prevention of Cancer Trial. BJU Int. 2003;91:608–612. doi: 10.1046/j.1464-410x.2003.04167.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.