

Abstract
Objective
To report a 3-generation white family clinically diagnosed variably with Wagner, Stickler, and Jansen syndromes and screened for sequence variants in the COL2A1 and CSPG2 genes. Wagner syndrome is an autosomal dominant vitreoretinopathy with a predisposition to retinal detachment and cataracts. It has significant phenotypic overlap with allelic Jansen syndrome and ocular Stickler syndrome type 1. Sticker syndrome type 1 maps to chromosome 12q13.11-q13.2, with associated COL2A1 gene mutations. Wagner syndrome maps to chromosome 5q13-q14 and is associated with mutations in CSPG2 encoding versican, a proteoglycan present in human vitreous.
Methods
Genomic DNA samples derived from venous blood were collected from all family members. Complete sequencing of COL2A1 was performed on a proband. Primers for polymerase chain reaction and sequencing were designed to cover all exon and intronexon boundaries. Direct sequencing of CSPG2 was performed on all family member samples.
Results
No detectable COL2A1 mutations were noted, making the diagnosis of ocular Stickler syndrome highly unlikely for this family. A unique base pair substitution (c.9265+1G>T) in intron 8 of the CSPG2 gene cosegregating with disease status was identified. This mutation occurred in a highly conserved previously reported splice site with a similar base pair substitution(G>A). Direct sequencing of this splice site mutation in 107 unrelated external controls revealed no variants, supporting the rarity of this base pair change and its causation in Wagner syndrome. This novel base pair substitution is thought to cause the deletion of exon 8 and formation of a truncated protein product.
Conclusion
Mutation screening of CSPG2 in autosomal dominant vitreoretinopathy families is important for accurate diagnosis.
Clinical Relevance
This study underscores the importance of obtaining extensive pedigree information and comparative ophthalmologic clinical information, as the phenotypic findings may vary greatly among independent family members. The study also affirms the paradigm shift from diagnosis assignment based on eponyms to that based on gene mutation type.
Wagner First Described the ophthalmic clinical features of his eponymous syndrome (OMIM 14200) with a 16-member Swiss kindred in 1938.1 The ophthalmic features consist of an optically empty vitreous with avascular vitreous strands and veils, moderate myopia, presenile cataracts, and retinal degeneration with atrophy. In 1995, clinical reexamination of the expanded original Wagner pedigree was performed, incorporating an additional 44 relatives.2 An optically empty vitreous with avascular strands or fibrillary condensation, chorioretinal atrophy, and cataracts were a consistent finding in all affected individuals. By age 45 years, 87% of syndromic individuals had electrophysiologic testing abnormalities, and 55% had tractional retinal detachments.2
Families with Wagner syndrome demonstrate an autosomal dominant inheritance pattern with near complete penetrance.3 The prevalence estimate of Wagner syndrome is less than 1:1 000 000.3 The condition was mapped to chromosome 5q, and in 2005, a mutation of the chondroitin sulfate proteoglycan 2 gene (CSPG2) encoding for the protein versican was found to cosegregate with the disease in a Japanese pedigree.4 Versican is a large proteoglycan found in many tissues and is a major constituent of the extracellular matrix of human vitreous.4,5 Since 2005, several groups have identified similar mutations of the CSPG2 gene in families with Wagner syndrome.4–7
Two ocular-only syndromes share clinical and allelic features with Wagner syndrome. Jansen syndrome has a predominance of retinal detachments.4,8 Jansen syndrome maps to the same chromosome 5q region as Wagner syndrome.9 Erosive vitreoretinopathy syndrome (OMIM 143200) is a vitreoretinal degeneration first reported in 1994.10 Affected individuals also have night blindness, visual field defects, and chorioretinal atrophy. Erosive vitreoretinopathy syndrome is also allelic with Wagner syndrome.7
Ophthalmic features associated with Stickler syndrome are similar to those found in Wagner syndrome. Stickler syndrome involves myopia, presenile cataract, vitreous degeneration, radial perivascular retinal degeneration, and tractional retinal detachments.11 The nonocular features of mid-face hypoplasia, cleft palate, bifid uvula, hearing loss, and skeletal abnormalities help to differentiate these 2 syndromes.12 Vitreous phenotypes can help to distinguish subtypes of Stickler syndrome and Wagner syndrome. Stickler syndrome demonstrates a membranous anterior vitreous (type 1 vitreous) or a fibrillar or beaded vitreous (type 2 vitreous.)11 The vitreous phenotype in Wagner syndrome is described as having an optically empty vitreous with avascular veils or a fibrillary condensation.5 A variant of Stickler syndrome devoid of systemic findings, the so-called ocular-only Stickler syndrome, apart from the vitreous phenotypes can be particularly difficult to distinguish from Wagner syndrome.13,14
Stickler syndrome is genetically distinct from Wagner syndrome. Three forms of autosomal dominant Stickler syndrome are each associated with an extracellular matrix collagen gene.12 Type I (OMIM 108300) is associated with COL2A1, while type II (OMIM 604841) is associated with COL11A1.12 Both types have ocular and systemic manifestations. The vitreous phenotype is a distinguishing feature between type I and type II. Type I Stickler syndrome is associated with retrolental membranous vitreous while type II Stickler syndrome is described as having a fibrillar or beaded vitreous phenotype. Type III (OMIM 184840) is associated with COL11A2 and involves only systemic manifestations.12 The ocular-only variant of Stickler syndrome is a subgroup of type I (COL2A1) and shares its membranous vitreous phenotype.13
In this report, we clinically and genetically characterize a family with autosomal dominant vitreoretinal degeneration demonstrating a wide ocular phenotypic spectrum. Affected individuals of this family had tentative initial clinical diagnoses of Stickler syndrome and Wagner syndrome/Jansen syndrome as well as retinitis pigmentosa. This study underscores the importance of obtaining consolidated family history and clinical data as well as seeking genetic testing in the setting of highly variable clinical presentations of a mutual disorder.
METHODS
STUDY SUBJECTS
The study family was identified after a referral of 2 different family members from a community retinal specialist (Claxton Baer, MD) to the Duke University Eye Center. Consenting family members were recruited under an approved human subject research institutional review board protocol for the clinical and molecular analysis of genetic eye disorders to include molecular genetic testing.
All participating family members were offered full ophthalmic examinations. In addition to the standard ophthalmic history, health histories included questions regarding hearing loss, previous repair of hard or soft cleft palate, other midline defects, skeletal or joint abnormalities, and early-onset arthritis. The clinical evaluation included assessment tests of Early Treatment Diabetic Retinopathy Study visual acuity (Snellen equivalent) and intraocular pressure, slit lamp inspection of the anterior segment, and indirect ophthalmoscopy to inspect the fundus.15,16 Ancillary tests included fundus photographs, axial length measurements, keratometry measurements, and ocular coherence tomography. Goldman visual field tests were performed in affected individuals.
Venous blood was collected from participating family members. Genomic DNA was extracted from venous blood using Auto Pure LS DNA extractor and PUREGENE reagents (Gentra Systems Inc, Minneapolis, Minnesota) and stored.
GENE SCREENING
COL2A1
Genomic DNA from 1 affected individual was screened for sequence variations of the COL2A1 gene by a commercial laboratory (Matrix DNA Diagnostics, New Orleans, Louisiana). All 54 exons, including intronexon boundaries, were examined. The resulting sequence was compared with DNA of normal controls and available published sequences.
CSPG2
Primers for polymerase chain reaction and sequencing were designed to cover coding and untranslated gene regions, including intronexon boundaries, using the ExonPrimer and Primer3 programs (Helmholtz Center Munich, Munich, Germany) (http://ihg2.helmholtz-muenchen.de/ihg/ExonPrimer.html). Primers were selected to produce amplification product sizes not to exceed 600 base pairs (bp) for optimal sequence output and reading. Large exons or untranslated gene regions were covered with overlapping amplicons, with a minimal 50 bp of overlapped sequence. Table 1 displays the optimized primer sequences used in this study.
Table 1.
Primers for Polymerase Chain Reaction and Sequencing of CSPG2
| Exon | Forward Primer | Reverse Primer | Approximate Size, Base Pairs |
|---|---|---|---|
| 1 | GTGAATGAACcctcctc | TTTCTTAACTTCGCCTTAC | 517 |
| 2 | ACCCTTATTACATA-CAATGC | ATCAATCTTTTATTC-CAGTG | 270 |
| 3 | CAAACTATTATAAAGGC-TGC | CAAACAACAGTTACTTCT-GAG | 562 |
| 4/5 | CTAAGTTGATACATT-GCTCC | ACAAGCACGAAGACTTG | 569 |
| 6.1 | AAAGTATTACAT-GCTCCTCC | TGTGGTTTTAACCATAT-CAC | 578 |
| 6.2 | CATTGATGTTCAACTAGCC | AGTAAACCTAACTAAA-GATACTCTG | 581 |
| 7.1 | TCCCTACAAAATACTCAGG | ATTTTCTTTTCAGTTTTGG | 657 |
| 7.2 | CCTTTGGTAACATCTATGG | GTATCTGTTCTCATCTC-TGG | 605 |
| 7.3 | AGAATTGTTTCCT-TATTCTG | AATAAGAGTAAATTCATC-TGC | 613 |
| 7.4 | TACAGAACCTTCAGCCTC | AGTTTCTG-GAGAAATAGTCG | 610 |
| 7.5 | TTAGTACCTTCTGTTC-CATC | AATAATCCT-GAGCCTAAATC | 599 |
| 7.6 | TTTACATCATCTTT-GAGTCC | TCTAATAGCAGCATTTGG | 658 |
| 8.1 UTR | GGAATTTGTCTTGG-TAGTTC | TCGAAAAGTATGTATCAT-CAG | 635 |
| 8.2 UTR | TTTGCTGATGTATTTCTG | aaaattGAGGCCATATC | 584 |
| 8.2 UTR | GCTCTAGAACAGAT-TATAAAGG | TGGATCTGTTTCTTCAC-TAC | 644 |
| 8.1 | GATTTGAATCCTTCTTT-GTG | CTCATGGAATGGGACTG | 633 |
| 8.2 | TTACATGGAAGCCTGAG | GTGATTATCCTGCTAGT-GTC | 598 |
| 8.3 | AATTACAGAAGGCTCTGG | CCCTGCTCCATAAAGAC | 597 |
| 8.4 | TACATTAGAAAATTTGGGG | CTACTGGGTCCTTCTGAG | 582 |
| 8.5 | AGAAGAAACGGTAAT-GATG | TTCAGTAAATGTCTGT-GAATC | 592 |
| 8.6 | ATTGAAAGTGAAACAA-CATC | CCACATTTTCCATTCTG | 595 |
| 8.7 | AAGTGGAACAAAT-CAATAAC | ATCCATCAGCAGTAACAG | 621 |
| 8.8 | GAAGTGGATATTGTT-GATTC | TCAGTTAAAGAAAGGTT-GAC | 596 |
| 8.9 | TGGAATGCAAACA-GATATAG | AGTAGCAATACTTAG-ATAGGG | 580 |
| 8.10 | AAACATGCTGGTCCTTC | AAACATCTTGATT-GCTTCTC | 582 |
| 8.11 | GAAGATGATGG-TAAACCTG | AATAATATGAAACTCTC-CATTG | 680 |
| 9 | CTAAATTGCTATGG-TAAAGC | TTGAAATCTCTGATT-GATTC | 322 |
| 10 | aatttaactggctgtcttg | GAAATGTGAATTTC-CACTG | 318 |
| 11 | GACCAAATTTTATGAAT-CAG | ttaagtaattcacagatgagg | 433 |
| 12 | AATCCAGCCAGTAAAGAG | agtctaatgcaccctctg | 287 |
| 13 | GAAATTGTTGAGTC-TATTCC | GTCTCAAACAAT-GAATTTG | 342 |
| 14 | TTGGTAC-CATAAAGAAAGAG | CACACAATATTACTT-GCTCC | 387 |
| 15.1 | CTAGACACCTTCATTT-TACG | AATCATCTTATTTA-CATGGC | 576 |
| 15.2 | TTTAGTTTTCTATTTGCCTC | TATGAAATGCATTGATCG | 593 |
| 15.3 | AGGCCTGAATGGAG-GACTTT | ACAGGAAGAAATGCCCA-CAC | 587 |
| 15.4 | TCCTCACACAATTTG-GAATCA | TCCTTCTAAGCCAAAG-GAGGT | 475 |
Abbreviation: UTR, untranslated region.
Polymerase Chain Reaction and Sequence Analysis
Genomic DNA of 2 affected and 2 unaffected family members was initially screened. The DNA of remaining family members was screened if any sequence variants followed the affection status. Polymerase chain reactions were performed and amplicons were visualized by agarose gel electrophoresis by standard procedures. Polymerase chain reaction amplicon purification was conducted using the Quickstep 2 SOPE Resin (Edge BioSystems, Gaithersburg, Maryland). BigDye Terminator 3.1 (Applied Biosystems Inc, Foster City, California) was used to perform sequencing reactions, and ABI3730 robotics (Applied Bio-systems Inc) was used to process the DNA fragments. Base pair calls were made using the Sequencher 4.8 software (Gene Codes, Ann Arbor, Michigan). Sequences of affected and unaffected individuals were aligned to a known reference genomic sequence (UCSC Genome Browser, http://genome.ucsc.edu) and compared for sequence variation.
RESULTS
CLINICAL FEATURES
The study family consisted of 3 generations with 6 affected and 3 unaffected participating individuals (Figure 1). The 6 affected individuals provided blood samples for genomic DNA isolation and underwent complete clinical evaluations. Of the 3 unaffected individuals, 2 provided blood samples and underwent clinical examinations (individuals 2 and 9), and 1 provided a blood sample and ophthalmic history but was unavailable for clinical inspection (individual 7).
Figure 1.

Study family pedigree. The study family consisted of 11 individuals in 3 generations with 6 affected and 3 unaffected participants.
The participant demographic and clinical examination information is summarized in Table 2. None of the affected family members had historical or systemic clinical features of the Stickler syndromes. The funduscopic examinations revealed significant phenotypic variation and are described next.
Table 2.
Subject Demographics and Ocular Clinical Features
| Subject/ Sex/Age, y | Eye | Best-Corrected VA (Snellen Equivalent) | Recent Refractive Error Spherical Equivalent,a D | Past Refractive Error (Presurgical) | Axial Length, mm | Lens Status | Retinal Detachment Age, y | Surgery |
|---|---|---|---|---|---|---|---|---|
| 1/M/60 | ODb | HM | −0.875 | NA | NA | ACIOL pupillary membrane | NA | CE/IOL tube shunt |
| OS | 20/25 | 0.25 | NA | 23.44 | ACIOL | NA | CE/IOL | |
| 2/F/59.5 | OD | 20/20 | 0.50 | +0.25+0.50×126 | NA | NSC | NA | None |
| OS | 20/20 | 0.625 | +0.25+0.75×40 | NA | NSC | NA | None | |
| 4/F/37 | OD | 20/20 | −2.00 | NA | 24.69 | PCIOL | NA | CE/IOL |
| OS | 20/25 | −0.25 | NA | 24.46 | PCIOL | NA | CE/IOL | |
| 6/F/40 | ODb | NLP | NA | NA | NA | Dense NSC | 5 | SB/PPV |
| OS | 20/400 | Plano | NA | 22.38 | PCIOL | NA | None | |
| 8/M/16 | OD | 20/20 | NA | (age 14 y) −1.00+1.00×90 | 24.56 | Clear | NA | None |
| OSc | 20/32 | −1.00 | (age 14 y) −1.00+2.00×90 | 25.07 | Clear | 16 | SB/PPV | |
| 9/F/8 | OD | 20/20 | Plano | Plano | NA | Clear | NA | None |
| OS | 20/20 | Plano | Plano | NA | Clear | NA | None | |
| 10/M/16 | ODb | NLP | NA | (age 4 y) + 2.50 sph | NA | Dense NSC | 9 | SB/PPV |
| OS | 20/160 | −0.25 | (age 4 y) + 2.50 sph | 22.18 | Clear | NA | None | |
| 11/F/11 | OD | 20/200 | −3.25 | (age 2.5 y) 1.25+3.00×90 | 23.62 | PSC | 8 | SB/PPV |
| OS | 20/50 | 0.75 | (age 2.5 y) −1.50+4.25×95 | 22.85 | Clear | NA | None |
| Subject/ Sex/Age, y | Eye | Optically Empty Vitreous | Vitreal Avascular Membranes | Retinal Traction | Chorioretinal Atrophy | Retinal Pigmentary Changes | Ocular Alignment | Visual Field |
|---|---|---|---|---|---|---|---|---|
| 1/M/60 | ODb | NA | NA | NA | NA | NA | Ortho | NA |
| OS | Yes | Yes | None | Mild | Mild | Ortho | Paracentral scotoma | |
| 2/F/59.5 | OD | None | None | None | None | None | NA | NA |
| OS | None | None | None | None | None | NA | NA | |
| 4/F/37 | OD | Yes | Yes | Moderate | Moderate | Moderate | XT | Ring scotoma |
| OS | Yes | Yes | Moderate | Moderate | Moderate | XT | Ring scotoma | |
| 6/F/40 | ODb | NA | NA | NA | NA | NA | XT | NA |
| OS | Yes | Yes | Mild | Severe | Severe | XT | Severe constriction | |
| 8/M/16 | OD | Yes | Yes | Severe | None | None | Ortho | Enlarged blind spot |
| OSc | Yes | Yes | Severe | None | None | Ortho | Paracentral scotoma | |
| 9/F/8 | OD | None | None | None | None | None | NA | NA |
| OS | None | None | None | None | None | NA | NA | |
| 10/M/16 | ODb | NA | NA | NA | NA | NA | Ortho | NA |
| OS | Yes | Yes | Moderate | Severe | Severe | Ortho | Moderate constriction | |
| 11/F/11 | OD | NAd | NAd | None | Moderate | None | XT | Nasal scotoma |
| OS | Yes | Yes | None | Moderate | None | XT | Superior scotoma |
Abbreviations: ACIOL, anterior chamber intraocular lens; CE/IOL, cataract extraction/intraocular lens; D, diopters; HM, hand motions; NA, not available; NLP, no light perception; NSC, nuclear sclerotic cataract; Ortho, orthophoria; PCIOL, posterior chamber intraocular lens; PPV, pars plana vitrectomy; PSC, posterior subcapsular cataract; SB, scleral buckle; sph, sphere; XT, exotropia; VA, visual acuity.
Spherical equivalent=sphere+cylinder/2.
No view of fundus because of opacity of anterior ocular media.
Patient examined both previtrectomy and postvitrectomy.
Unable to determine secondary to postvitrectomy status.
INDIVIDUAL 8 (PROBAND)
The proband was a 16-year-old boy who presented with complaints of acute onset of floaters in his right eye. Tentative diagnoses of Wagner/Jansen syndrome and Stickler syndrome had been made in the past based on his family history of retinal detachments. He had no history of cataract or retinal detachment. His distance visual acuity was 20/20 OD and 20/32 OS by Early Treatment Diabetic Retinopathy Study testing. On examination, his lenses were clear bilaterally. He had an optically empty vitreous, with preretinal vitreous condensation in the mid-periphery and avascular vitreous sheets in the far periphery bilaterally. There was severe retinal traction in both eyes, with nasal dragging of the arcades and fovea as seen in Figure 2A. The optic nerve was inverted nasally in the left eye and was detected in the corresponding ocular coherence tomography (Figure 2B). No chorioretinal atrophy was noted; however, both eyes had multiple areas of peripheral hyperpigmentation, lattice degeneration, and cystic tufts. Multiple round retinal holes and a localized tractional detachment were identified in the periphery of the left eye. The detachment and holes were successfully treated with encircling scleral buckle, pars plana vitrectomy, gas tamponade, and laser. Bilateral Goldman visual field testing demonstrated an enlarged blind spot in the right eye and a paracentral scotoma in the left.
Figure 2.
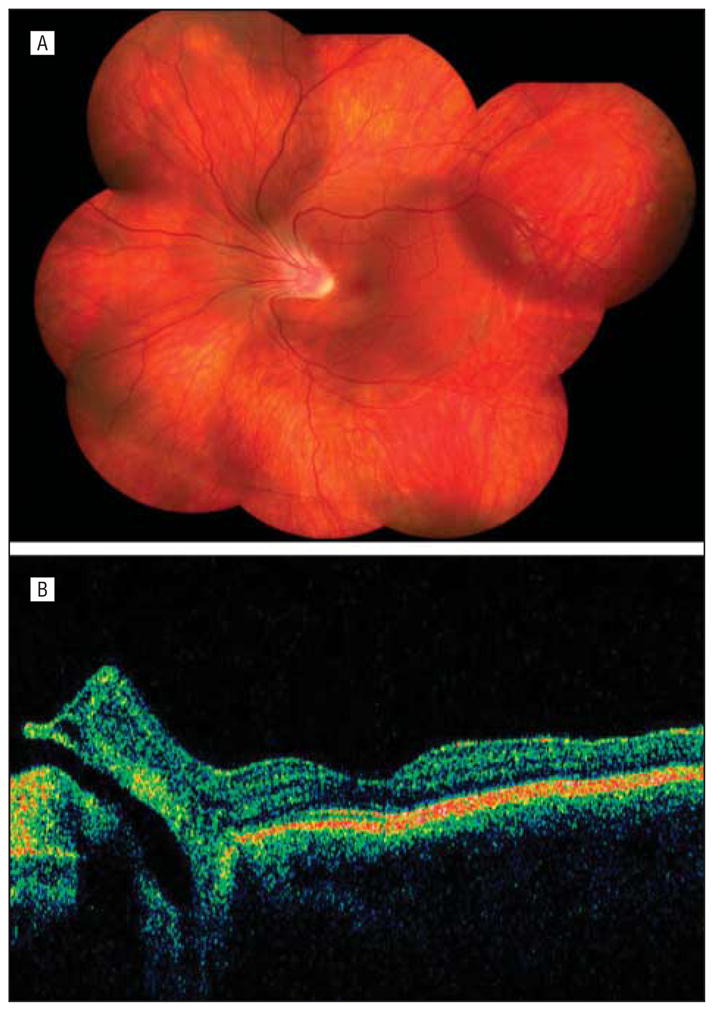
Individual 8. A, Composite fundus photograph of the left eye shows an optically empty vitreous with preretinal vitreous condensation in the midperiphery, avascular vitreous sheets in the far periphery, and nasal dragging of the arcades and fovea. B, An optical coherence tomogram of the left eye shows the optic nerve inverted nasally.
INDIVIDUAL 4
The proband’s mother was 37 years of age at the time of examination and had a history of bilateral presenile cataracts requiring surgical extraction at 18 years of age for the left eye and 30 years of age for the right eye. She had a long-standing history of a large exotropia. She had no history of retinal detachment. Her Snellen distance visual acuity was 20/20 OD and 20/25 OS. A composite fundus photograph of her right eye is shown in Figure 3A. There was an optically empty vitreous except for a vascular vitreous sheets in the midperiphery and far periphery. Both the temporal and nasal retinal vascular arcades were straightened, suggesting moderate retinal traction. There was a focal area of chorioretinal atrophy and pigmentary changes. There were also perivascular pigmentary changes. Visual field testing demonstrated ring scotomata of both eyes (Figure 3B).
Figure 3.

Individual 4. A, Composite fundus photograph of the right eye shows an optically empty vitreous except for avascular vitreous sheets in the midperiphery and far periphery, straightening of the temporal and nasal retinal vascular arcades, an area of chorioretinal atrophy, and perivascular pigmentary changes. B, Goldmann visual field test of the left eye shows a ring scotoma.
INDIVIDUAL 1
The proband’s maternal grandfather was 60 years of age at the time of examination. He had primary open-angle glaucoma requiring tube shunt placement in the right eye, with subsequent development of a pupillary membrane. He had bilateral presenile cataracts with a history of surgical extraction at approximately 30 years of age. He had no history of retinal detachment. His distance visual acuity was hand motions OD and 20/25 OS. The view of the right fundus was obscured by anterior ocular media opacification. His vitreous cavity on the left was optically empty except for an avascular vitreous membrane inferiorly, as demonstrated in Figure 4. There were retinal pigmentary changes underlying the area of the vitreous membrane. A few patchy areas of chorioretinal atrophy could also be seen. Goldman visual field testing of the left eye revealed a paracentral scotoma.
Figure 4.
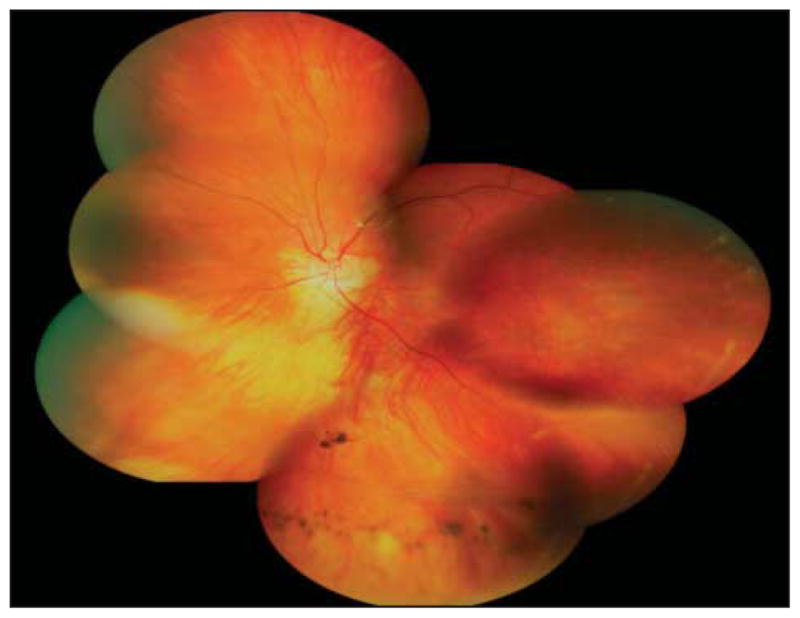
Individual 1. Composite fundus photograph of the left eye shows an optically empty vitreous except for an avascular vitreous membrane inferiorly with underlying retinal pigmentary changes and patchy areas of chorioretinal atrophy.
INDIVIDUAL 6
The proband’s maternal aunt was 40 years of age, with an ocular history significant for a chronic retinal detachment of the right eye requiring 3 surgical repairs starting at the age of 5 years. She had a significant cataract of the left eye requiring surgical extraction at 25 years of age. She had an initial diagnosis of retinitis pig-mentosa. She described slow degradation of left eye visual acuity since childhood. She had a long-standing small-angle right exotropia. Her visual acuity was no light perception OD and 20/400 OS. There was no view of the fundus of the right eye because of a dense cataract. The left eye vitreous cavity was optically empty except for avascular vitreous sheets in the far periphery. The left fundus had notable diffuse chorioretinal atrophy with dense pigment in the midperiphery to far retinal periphery (Figure 5A). The left eye retinal vascular arcades were straightened, suggesting mild retinal traction. The corresponding Goldman visual field test of the left eye showed severe constriction with a few scattered small islands (Figure 5B).
Figure 5.

Individual 6. A, Composite fundus photograph of the left eye shows an optically empty vitreous cavity except for avascular vitreous sheets in the far periphery, diffuse chorioretinal atrophy with dense pigment in the midperiphery to far retinal periphery, and straightening of the vascular arcades. B, Goldmann visual field test of the left eye shows severe constriction with a few scattered small islands.
INDIVIDUAL 10
The proband’s maternal cousin (and son to individual 6) was 16 years of age at the time of evaluation. He had a history of a retinal detachment in the right eye at a young age, leading to a poor visual outcome. He had not had cataract surgery. He described slow worsening visual acuity of his left eye. His distance visual acuity was no light perception OD and 20/160 OS. There was a dense cataract in the right eye, obscuring the view of the fundus. The left vitreous cavity was optically empty except for a vascular preretinal membranes in the far periphery. The left fundus demonstrated findings similar to that of his mother, with diffuse retinal pigmentary changes, patchy chorioretinal atrophy, and straightened vascular arcades (Figure 6A). The corresponding left visual field showed moderate constriction (Figure 6B).
Figure 6.

Individual 10. A, Composite fundus photograph of the left eye shows an optically empty vitreous except for avascular preretinal membranes in the far periphery, diffuse retinal pigmentary changes, patchy chorioretinal atrophy, and straightened vascular arcades. B, Goldmann visual field test of the left eye shows moderate constriction.
INDIVIDUAL 11
A second maternal cousin and daughter to individual 6 had a history of a retinal detachment in the right eye at the age of 8 years. She was treated for a macula-off retinal detachment at Duke University Eye Center. The detachment originated from a posterior retinal break and was associated with rings of dense vitreous bands at the equator. The retina was successfully reattached with vitrectomy, scleral buckling, and silicone oil tamponade with subsequent oil removal. She had a moderate-angle exotropia. Her distance visual acuity was 20/200 OD and 20/50 OS. She had a significant posterior subcapsular cataract of the right eye that had developed after retinal detachment repair. The vitreous cavity of the left eye was optically empty except for a subtle midperipheral a vascular vitreous membrane (Figure 7A). Retinal pigmentary changes were conspicuously absent except for mild peripapillary pigmentation. There was straightening of the vascular arcades. She had a blonde fundus with subtle inferior chorioretinal atrophy, which corresponded to early superior scotomatous changes with visual field testing (Figure 7B).
Figure 7.

Individual 11. A, Composite fundus photograph of the left eye shows an optically empty vitreous except for a subtle midperipheral avascular vitreous membrane, mild peripapillary pigmentation, straightening of the vascular arcades, and subtle inferior chorioretinal atrophy. B, Goldmann visual field test of the left eye shows early superior scotomatous changes that correspond to the inferior chorioretinal atrophy seen on fundus photography.
MOLECULAR GENETIC EVALUATION SCREENING OF CANDIDATE GENES
Proband (individual 8) genomic DNA was commercially screened for sequence variation of the COL2A1 gene. No sequence changes were identified with comparison to internal and external control DNA and to the published sequence for the COL2A1 gene.
Initial sequence screening of the CSPG2 gene uncovered 17 single-nucleotide polymorphic (SNP) variations: 10 exonic SNPs, 2 untranslated gene region SNPs, 4 in-tronic SNPs, and 1 SNP at a splice site (Table 3). Only the splice site SNP cosegregated with affection status in the initial 4 DNA samples screened. The splice site sequence change was a single base pair substitution of a guanine for a thymine at the 5′ end of intron 8 at position c.9265+1G>T (Figure 8). For confirmation, all family member DNA was sequenced at the mutation site. The mutation cosegregated with all affected individuals (n=6). The splice site mutation did not appear with sequence screening of the DNA of the 3 unaffected family members or in 107 external control DNA samples.
Table 3.
Sequence Variants Identified in CSPG2
| Location | SNP | Position | Function | Heterozygous Sequence Variant Present |
|---|---|---|---|---|
| Exon 3 | rs12332199 | c.348T>C | Coding synonymous | U2 and U9 |
| Exon 5 | rs4470745 | c.645A>G | Coding synonymous | U2 |
| Exon 8 | rs309559 | c.4547A>G | Coding nonsynonymous | U2 |
| Exon 8 | rs188703 | c.5477G>A | Coding nonsynonymous | U2 |
| Exon 8 | rs309557 | c.5808T>C | Coding synonymous | U2 |
| Exon 8 | rs160279 | c.6723A>G | Coding synonymous | U2 |
| Exon 8 | rs160278 | c.6902T>A | Coding nonsynonymous | U2 |
| Exon 8 | rs160277 | c.8809G>T | Coding nonsynonymous | U2 |
| Exon 8 | rs16900532 | c.9003C>A | Coding nonsynonymous | U2 |
| Exon 14 | rs308365 | c.9882C>T | Coding synonymous | A6 (T/T), A8 (T/T), U2 (T/T), U9 (T/T) |
| UTR 8 | Novel | c.4004–1284C>G | UTR | U9 |
| UTR 8 | rs309560 | c.4004–687C>G | UTR | U2 |
| Intron 8 | Novel | c.9265+1G>T | Splice site | A6 and A8 |
| Intron 9 | rs695103 | c.9379+7T>C | Intronic | U9 |
| Intron 10 | rs6873404 | c.9494–63T>A | Intronic | U2 |
| Intron 13 | Novel | c.9880+70G>A | Intronic | U9 |
| Intron 13 | Novel | c.9880+73T>C | Intronic | A6 (C/C) |
Abbreviations: A6, affected individual 6; A8, affected individual 8; U2, unaffected individual 2; U9, unaffected individual 9; UTR, untranslated region.
Figure 8.

Sequence chromatogram of CSPG2 splice site mutation in intron 8 CSPG2 splice donor mutation (c.9265+1G>A) from 4 individuals (2 affected, 2 unaffected) who were initially screened. The N in the sequence depicts the heterozygous mutation present. DNA analysis of all other affected family members showed cosegregation with this mutation. One hundred seven control DNA samples were negative for this mutation.
COMMENT
We have identified a novel mutation with a single base pair substitution of a guanine for a thymine at the 5′ end of intron 8 at position c.9265+1G>T. This mutation co-segregates with the disease state in our study family with clinical manifestations of Wagner syndrome. This splice site has previously been associated with Wagner syndrome. In 2006, Kloeckener-Gruissem et al5 identified a guanine to adenine substitution at the 9265+1 position in the original Swiss family described by Wagner. With messenger RNA transcript analysis, this group found a21-bp deletion causing an in-frame deletion of 7 amino acids. This was likely caused by the disruption of the usual splice site sequence, allowing activation of a subsequent cryptic splice site, although none was identified in that study. In 2007, Meredith et al6 reported a father-daughter pair with the same c.9265+1G>A mutation. We believe that our mutation c.9265+1G>T leads to the same 21-bp deletion described by Kloeckener-Gruissem et al. We were unable to confirm this with transcript analysis, however, because the cell lines for the family samples were not viable. No control DNA samples had sequence variants at this site, which strongly suggests that this is a highly conserved splice site consensus sequence and that the base substitution does not represent a common polymorphism.
The wide ocular phenotypic spectrum demonstrated in this relatively small family is notable. In the original Wagner syndrome report, the term situs inversus was used to describe the nasal displacement of the temporal arcades presumably due to tractional effects.1 To our knowledge, the current report is the first to describe tractional forces leading to the inversion of the papilla in Wagner syndrome. Figure 2B is an ocular coherence tomography photograph showing the “inverted papilla” of individual 8 (proband) and also demonstrating nasal foveal dragging. This is in contrast to the proband’s mother (individual 4) with a temporally displaced fovea (Figure 3A). She had a large-angle exotropia on examination, which likely represents a positive angle kappa. A large angle kappa is an infrequent feature of Wagner syndrome.10
Retinal detachments were not thought to be a significant finding in Wagner syndrome until the reexamination of the expanded original family in 1995.2 In that study, rhegmatogenous retinal detachments were rare (4%), while tractional retinal detachments were a common finding after the age of 45 years (55%). A majority of the affected individuals in our pedigree sustained retinal detachments, suggesting that this is a more common feature than the rate previously reported. More interestingly, the average age of retinal detachment occurrence and detection in our family was 9.5 years. This finding refutes retinal detachment as a middle-age issue and has significant implications for how patients with Wagner syndrome should be monitored clinically.
Our studied pedigree contains a mother-son pair with unusual fundus features that are not necessarily associated with age. The diffuse retinal atrophy and pigmentary changes seen in individuals 6 and 10 could readily be diagnosed as a distinct disease entity if they had not been evaluated as members of a larger kindred. This clinical phenotype closely resembles erosive vitreoretinopathy syndrome with diffuse chorioretinal degeneration and constriction of the visual fields.7 This report, as well as that by Meredith et al, demonstrates that a single kindred can have members with the more traditional Wagner syndrome phenotype features along with individuals with an erosive vitreoretinopathy syndrome phenotype, all caused by a single CSPG2 mutation.10 As a progressive degeneration, the severity of the clinical presentation cannot be fully explained as late changes since both individuals are relatively young. In our kindred, the oldest affected family member (individual 1) had the mildest phenotype and had an initial diagnosis of retinitis pigmentosa.
Given the broad phenotypic variation seen in Wagner syndrome, as is evident in the family we present herein, making a definitive diagnosis clinically can be extremely challenging. This may be especially difficult in families with limited family member availability because of adoption or estrangement or in questions of paternity. Affected individuals from this kindred were given different diagnoses that changed over time with disease progression. Confirming a molecular diagnosis is helpful in determining a clinical care plan and prognosis and also allows the patient and family to seek same-condition larger-family support networks.
Acknowledgments
Funding/Support: This work was supported by National Institutes of Health grant EY014685, Research to Prevent Blindness, Inc, a Lew R. Wasserman Award (Dr Young), and Alice and John Haynes.
Footnotes
Additional Contributions: We thank Claxton Baer, MD, for bringing this family to our attention. We are thankful to all patient subjects who participated in this study.
Financial Disclosure: Dr Toth is a consultant to Genentech and Alcon and has patents pending related to ophthalmic ocular coherence tomography imaging. She receives research support from Genentech, Sirion, Bioptigen, North Carolina Biotechnology Center, Duke Translational Research Institute, and the National Institutes of Health.
References
- 1.Wagner H. Ein bisher unbekanntes des auges (degeneration hyaloideoretinalis hereditaria), beobachtet im Kanton Zurich. Klin Monatsbl Augenheilkd. 1938;100:840–857. [Google Scholar]
- 2.Graemiger RA, Niemeyer G, Schneeberger SA, Messmer EP. Wagner vitreoretinal degeneration—follow-up of the original pedigree. Ophthalmology. 1995;102 (12):1830–1839. doi: 10.1016/s0161-6420(95)30787-7. [DOI] [PubMed] [Google Scholar]
- 3.Hinton DR. Basic Clinical Science and Inherited Retinal Diseases. Philadelphia, PA: Elsevier Mosby; 2006. pp. 519–538. [Google Scholar]
- 4.Miyamoto T, Inoue H, Sakamoto Y, et al. Identification of a novel splice site mutation of the CSPG2 gene in a Japanese family with Wagner syndrome. Invest Ophthalmol Vis Sci. 2005;46(8):2726–2735. doi: 10.1167/iovs.05-0057. [DOI] [PubMed] [Google Scholar]
- 5.Kloeckener-Gruissem B, Bartholdi D, Abdou MT, Zimmermann DR, Berger W. Identification of the genetic defect in the original Wagner syndrome family. Mol Vis. 2006;12:350–355. [PubMed] [Google Scholar]
- 6.Meredith SP, Richards AJ, Flanagan DW, Scott JD, Poulson AV, Snead MP. Clinical characterisation and molecular analysis of Wagner syndrome. Br J Ophthalmol. 2007;91(5):655–659. doi: 10.1136/bjo.2006.104406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukhopadhyay A, Nikopoulos K, Maugeri A, et al. Erosive vitreoretinopathy and Wagner disease are caused by intronic mutations in CSPG2/Versican that result in an imbalance of splice variants. Invest Ophthalmol Vis Sci. 2006;47(8):3565–3572. doi: 10.1167/iovs.06-0141. [DOI] [PubMed] [Google Scholar]
- 8.Jansen LM. Degeratio hyaloideoretinalis herditaria. Ophthalmologica. 1962;144:348–363. [Google Scholar]
- 9.Perveen R, Hart-Holden N, Dixon MJ, et al. Refined genetic and physical localization of the Wagner disease (WGN1) locus and the genes CRTL1 and CSPG2 to a 2- to 2.5-cM region of chromosome 5q14.3. Genomics. 1999;57(2):219–226. doi: 10.1006/geno.1999.5766. [DOI] [PubMed] [Google Scholar]
- 10.Brown DM, Kimura AE, Weingeist TA, Stone EM. Erosive vitreoretinopathy: a new clinical entity. Ophthalmology. 1994;101(4):694–704. doi: 10.1016/s0161-6420(94)31276-0. [DOI] [PubMed] [Google Scholar]
- 11.Edwards AO. Clinical features of the congenital vitreoretinopathies. Eye. 2008;22 (10):1233–1242. doi: 10.1038/eye.2008.38. [DOI] [PubMed] [Google Scholar]
- 12.Snead MP, Yates JR. Clinical and molecular genetics of Stickler syndrome. J Med Genet. 1999;36(5):353–359. [PMC free article] [PubMed] [Google Scholar]
- 13.McAlinden A, Majava M, Bishop PN, et al. Missense and nonsense mutations in the alternatively-spliced exon 2 of COL2A1 cause the ocular variant of Stickler syndrome. Hum Mutat. 2008;29(1):83–90. doi: 10.1002/humu.20603. [DOI] [PubMed] [Google Scholar]
- 14.Richards AJ, Martin S, Yates JR, et al. COL2A1 exon 2 mutations: relevance to the Stickler and Wagner syndromes. Br J Ophthalmol. 2000;84(4):364–371. doi: 10.1136/bjo.84.4.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck RW, Moke PS, Turpin AH, et al. A computerized method of visual acuity testing: adaptation of the early treatment of diabetic retinopathy study testing protocol. Am J Ophthalmol. 2003;135(2):194–205. doi: 10.1016/s0002-9394(02)01825-1. [DOI] [PubMed] [Google Scholar]
- 16.Ferris FL, III, Kassoff A, Bresnick GH, Bailey I. New visual acuity charts for clinical research. Am J Ophthalmol. 1982;94(1):91–96. [PubMed] [Google Scholar]