

Abstract
Porphyrins such as protoporphyrin IX (PPIX) are known to occasionally cause conformational changes in proteins for which they are specific ligands. It has also been established that irradiation of porphyrins noncovalently intercalated between bases or bound to one of the grooves can cause conformational effects on DNA. Conversely, there is no evidence reported in the literature of conformational changes caused by noncovalently bound PPIX to globular proteins for which the porphyrin is not a specific ligand. This study shows that the irradiation of the porphyrin in the PPIX/lactoglobulin noncovalent complex indeed causes a local and limited (~7%) unfolding of the protein near the location of Trp19. This event causes the intrinsic fluorescence spectrum of the protein to shift to the red by 2 nm and the average decay lifetime to lengthen by approximately 0.5 ns. The unfolding of lactoglobulin occurs only at pH > 7 because of the increased instability of the protein at alkaline pH. The photoinduced unfolding does not depend on the presence of O2 in solution; therefore, it is not mediated by formation of singlet oxygen and is likely the result of electron transfer between the porphyrin and amino acid residues.
Introduction
The noncovalent interaction between porphyrins and proteins is an important, but often overlooked, aspect of the biomedical applications of these compounds. These interactions also represent an interesting biophysical model of the conformational effects induced by photoactive ligands on proteins.
In biomedical applications, porphyrins are used either as imaging contrast or as therapeutic agents. The binding of subcellular targets changes the photophysical and photochemical properties of porphyrins1,2 and affects their usefulness as fluorescence imaging probes, as well as their action as phototoxicity agents.1,3,4 The latter is a very intriguing aspect of the effects of porphyrins in vivo. Light-induced, porphyrin-mediated photoinduced structural effects that have been observed at the cellular level include the damage to microtubules, the formation of cross-links involving the membrane portion of Bcl-2 and conformational changes of Bcl-2.5–9 Despite this evidence, the porphyrin-induced conformational changes of proteins have not been investigated with a high level of mechanistic details. It is clear that porphyrins-photoinduced conformational changes in proteins occur, and with this study, we investigated their occurrence in a protein model which was not specific for porphyrin molecules. The light-induced, porphyrin-mediated conformational changes described in this study could also represent a model to study the unfolding mechanisms of globular proteins by using light as trigger. Other models have recently been developed that use light as a trigger of protein unfolding;10–12 however, our method would employ a more physiological molecular mediator of the photodamage such as protoporphyrin IX (PPIX).
Our group has been investigating the binding of porphyrins to globular proteins and established that PPIX binds both large and small globular proteins13–15 and that the binding causes protein-dependent changes in the optical and photochemical properties of the porphyrin.14 This evidence indicates the importance of understanding porphyrin/protein interactions in order to establish the direct structural effects produced on the polypeptides.
Binding of PPIX to β-lactoglobulin (BLG) is modulated by the pH of the solution;16 however, the location of the binding is still unclear. With this study, we provide a refined model of the binding site and characterize the effects that the irradiation of PPIX/BLG noncovalent complexes produce on the conformation of the protein.
Porphyrins are known to produce preferential photochemical reactions in the presence of specific free amino acids.17 Thus, we operated under the hypothesis that photochemical and/or photophysical mechanisms involving porphyrins and amino acids produce local conformational changes in the protein. Our results show that at pH > 7, irradiation of the complex with visible light causes BLG to partially unfold. Our experiments also show that the photoinduced unfolding does not depend on the presence of diffusing O2. This indicates that the photoinduced conformational effect is not mediated by singlet oxygen (1O2) sensitized by the triplet state of PPIX and might instead be produced by electron transfer between the porphyrin and amino acid residues in its proximity. This evidence may have important consequences for the biomedical application of PPIX (and other porphyrins) because an O2-indpendent mechanism of cellular photodamage may overcome some of the current limitations in the widespread use of porphyrins as photoactive drugs.
The results presented in this article represent the first evidence that PPIX noncovalently bound to a globular protein, for which the porphyrin is not specific, can produce conformational changes of the polypeptide.
Experimental Methods
Chemicals
TSPP (Frontier Scientific, Logan, UT), PPIX, and BLG (Sigma-Aldrich, St. Louis, MO) were used without further purification. Purity of BLG (>99%) was determined by HPLC and gel electrophoresis.
Buffers
Solutions were prepared in 10 mM phosphate buffer which was adjusted to the selected pH by adding small amounts of 0.5 N HCl or 0.1 M NaOH. The final pH values ranged from 5 to 9 at 1 unit increments. This range spans across the pH of the conformational transition of BLG.18
Sample Preparation
Solid BLG was dissolved directly into buffer and optically adjusted to 0.3 mg/mL (11 μM) by using ε280nm = 1.7 × 104 M−1 cm−1.16 PPIX solutions were prepared as 230 μM stocks in buffer. A small aliquot (100 μL) of this stock was added to 2 mL of the BLG solution to yield an equimolar BLG:PPIX solution. Solubility of PPIX in aqueous solutions is low; however, in the pH range of the study (5–9 units) and at our stock concentration, the porphyrin could be directly dissolved in buffer. This procedure was initially compared to previous protocols were the PPIX stock was prepared in dimethylsulfoxide (DMSO). The results yielded identical results; thus, we prepared the PPIX stock solution in buffer to eliminate the contribution of DMSO absorption in far-UV, CD spectra.
UV–Vis Absorption Spectroscopy
Spectra were recorded by using a dual beam spectrophotometer (Evolution 300, ThermoElectron, Verona, WI) at a speed of 240 nm/min and spectral resolution of 2 nm. All absorption spectra were recorded by using four clear-sides, 1 × 1 cm quartz cells (Starna Cells Inc., Atascadero, CA). Spectra were recorded between 220 and 350 nm for protein absorption and in the 350–700 nm range for the Soret band of porphyrin and corrected for the baseline.
Emission Spectroscopy
Steady state fluorescence was recorded by using a double monochromator fluorimeter (AB2, ThermoElectron, Verona, WI). For all emission spectra, the acquisition speed was set at 1 nm/s and 4 nm slit width in excitation and emission. Protein spectra were recorded in the 299–450 nm range upon excitation at 280 nm. Porphyrin spectra were recorded in the 580–750 nm range upon excitation at 405 nm (PPIX and PPIX/BLG complex). The optical density at the excitation wavelength was adjusted to <0.2 for all samples.
Emission intensity was calculated by integrating the area under the emission peak and normalized for absorbed radiation according to19
| (1) |
where Fraw is the area calculated from the raw data and Aex and Aem are the optical density of the sample at the excitation and maximum emission wavelength, respectively.
Fluorescence Lifetime
Fluorescence lifetime experiments were performed by using a time correlated single-photon counting instrument (5000 U, Horiba JobinYvon, Edison, NJ). Fluorescence decay of BLG was recorded upon excitation with a picosecond pulsed source at 280 nm (NanoLED-15, ~700 ps pulsewidth), whereas fluorescence decay of PPIX was recorded upon excitation with a picosecond 405 nm diode laser (NanoLED-07, ~150 ps pulsewidth) from Horiba JobinYvon. BLG fluorescence lifetime was recorded at 330 ± 4 nm, whereas the fluorescence lifetime of PPIX in the complex was recorded at 631 ± 8 nm.
Decay of the protein and PPIX were analyzed by using the deconvolution software DAS6.2 (IBH, Glasgow, U.K.). The software yields the value of the fluorescence lifetimes and their relative amplitude through a reconvolution between the time profile of the pulsed source (instrument response function or prompt, G(t)) and the theoretical fluorescence decay assumed to be19
| (2) |
The convolution of F(t) and G(t) is fitted, using conventional least-squares, to the experimental decay curve, I(t), by changing the parameters Ai and τi.19
The quality of the fitting is judged by (i) the value of the reduced χ2, which has to approach 1, (ii) the visual inspection of the residuals, and (iii) the value of the Durbin–Watson parameter (a measure of the autocorrelation of the residuals) that, for a good fitting with the number of data points employed in our analysis, ranges between 1.8 and 2.0.20 G(t) was recorded by using a 1 mg/mL scattering solution of glycogen in deionized water. Data analysis was carried out with one, two, or three lifetime components, and the best fitting that uses the fewest components was chosen. From the individual decay components, the average decay lifetime was calculated as
| (3) |
Circular Dichroism Spectroscopy (CD)
CD spectroscopy was carried out by using a Jasco J-815 (Jasco Inc., Easton, MD). Spectra were recorded in the 190–250 nm range, (far UV) which probes the protein secondary structure,21 and in the 250–450 nm range, which simultaneously probes the aromatic amino acids residues22 and the region of the porphyrin Soret band.23Experiment in the far UV nm region were carried out using 1 mm pathway quartz cells, whereas the experiments in the 250–450 nm range were carried out using 1 cm path length quartz cells. The 190–250 nm region of the spectrum was then analyzed by using CDPro,24 and the contribution of individual secondary structures was estimated. Analysis was carried out with all three fitting procedures offered by CDPro (SELCON 3, CONTINLL, and CDSSTR) to verify consistency in the retrieved secondary structures.
Irradiation
Irradiation was carried out by selecting the 405 ± 8 nm output of the fluorimeter 150 W Xe lamp. The incident power at the sample was measured at 0.35 mW, and the samples were exposed for increasing time intervals for a range of doses between 0 and 23 J. Irradiated samples included BLG alone and BLG/PPIX equimolar complexes. Controls were samples of BLG or BLG/PPIX complexes that were kept in solution for the same length of time as the irradiated samples but were not exposed to light until the recording of their absorption, fluorescence, or CD spectra.
Deoxygenated Samples
Oxygen was purged from the solution by flowing pure N2 (Praxair, Danbury, CT) into an airtight quartz cell (NSG Precision Cells Inc., Farmingdale, NY). Initially, the sample was purged with N2 for 30 min in the dark at which point absorption, emission, fluorescence lifetime, and CD experiments were carried out. Subsequently, the same sample was irradiated according to the procedure explained above, while under constant flow of N2. Thus, the sample was under N2 atmosphere before and during irradiation. This avoided the problem of having O2 re-entering the solution during irradiation. After irradiation under N2 atmosphere, the spectroscopic and fluorescence lifetime experiments were repeated.
Docking Simulations
In order to better determine the location of the binding site, molecular simulations of the docking of PPIX to BLG were carried out by using the software Arguslab 4.0.1 (Planaria Software LLC, Seattle, WA). The PPIX molecule was built by using ChemSketch (Advanced Chemistry Development Inc., Toronto, ON, Canada) and optimized by using Arguslab. The BLG dimer was downloaded from the Protein Data Bank (1BEB.pdb) and stripped of the water molecules contained in the PDB file. The docking was performed by approaching the ligand to different sites of the protein structure, including the aperture and the interior of the β-barrel, the external grove flanking the 3-turn helix,25 as well as the monomer/monomer interface.26,27 Calculations were carried out by using the AScore algorithm of the software which minimizes the interaction energy.
SDS-PAGE
In order to investigate whether the irradiation at 405 nm (i) produces the covalent attachment of PPIX to BLG and/or (ii) produces protein fragments, we used gel electrophoresis. Irradiated as well as nonirradiated samples were first subject to trichloroacetic acid precipitation. The precipitate was resuspended in buffer containing 15% sodium dodecyl sulfate (SDS) and, after a short incubation, run on a 12% SDS gel.
Results
Docking Simulations
Computational simulations of the docking of PPIX to the BLG dimer (1BEB) provide several scenarios for the binding of the porphyrin. BLG dimers provide several possible intradimeric sites located (i) on the side of the aperture of the β-barrel, (ii) on the opposite side between the two flanking 3-turn helices, or (iii) at the bottom of such helices.26,27 Computational docking of PPIX shows that all three sites produce stable binding configurations with different energy minima. The site between the apertures of the β-barrels produces an energy minimum of –7.07 ± 0.87 kcal/mol and places PPIX in close proximity (<12 Å) to the two Trp61 residues located near each CD loop.27 The site located between the two 3-turn helices yields an energy minimum of –8.33 ± 0.28 kcal/mol, whereas the site located at the bottom of the 3-turn helices yields an energy minimum of –9.54 ± 0.93 kcal/mol.
However, purely monomeric sites also exist at the surface of the protein. The one that yields the lowest energy configuration is the one located at the bottom of the barrel. Its energy is comparable to the lowest energy retrieved for binding to intradimeric sites (E = –9.25 ± 0.64 kcal/mol). This site is particularly significant because the porphyrin ring is located within 7 Å of the Trp19 indole ring (Figure 1). The energy minimization indicates that this configuration produces the possible formation of four hydrogen bonds: one between a N atom in the porphyrin macrocycle and Arg124, one between a carboxyl groups of PPIX and Lys14, and two between the other carboxyl groups of PPIX and amides of unordered protein structure. Attempts to dock PPIX inside or near the interior β-barrel of BLG did not yield energetically favorable conformations, neither did the binding to the groove formed between the outer wall of the barrel and the 3-turn α-helix.28
Figure 1.
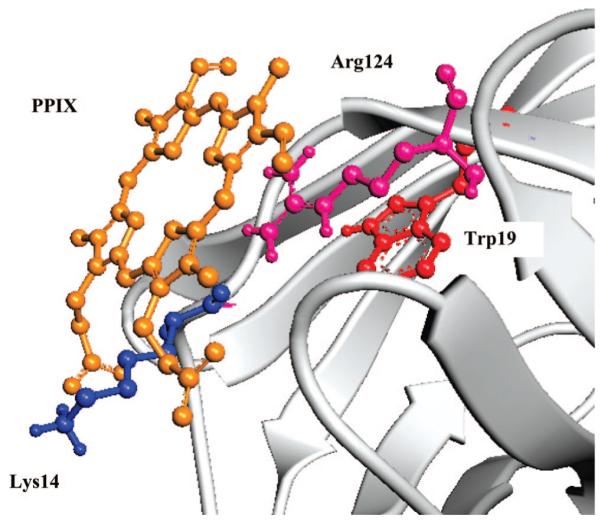
Superficial binding site of PPIX. The porphyrin is located within 7Å from Trp19 and forms hydrogen bonds with Lys14, Arg124, and amides of the unordered N terminal and FG loop. Notice that the porphyrin ring is not distorted.
The docking simulations do not show distortions of the porphyrin ring in any of the occupied binding sites, even though such distortion was allowed during the computations.
Absorption Spectroscopy
Difference absorption spectra were obtained by subtracting the spectra of irradiated samples from that of the initial (nonirradiated) spectra. At pH 7 and below, the comparison between nonirradiated and irradiated spectra of the PPIX/BLG complex does not show any effect of irradiation on the absorption spectrum of either the protein or the porphyrin (Figure 2A). The only effect being a small decrease of the protein absorption (negative difference peak near 280 nm) which is similar in irradiated and nonirradiated samples.
Figure 2.

Difference absorption spectra as a function of irradiation. Difference spectra were obtained by subtracting the irradiated spectra from the initial (nonirradiated) ones. (A) At pH 6, the spectra show a very small (<0.01) decrease in the region of the protein at increasing irradiation doses (solid lines). The spectra of the nonirradiated samples substantially superimpose to the ones of the irradiated ones (dotted lines). The spectra are representative of measurements carried out at pH 7 and below. (B) At pH 9, irradiation produces a large bleaching of PPIX absorption which produces a peak near 408 nm, which is where BLG-bound PPIX has its absorption maximum (solid lines). Nonirradiated samples show no change in the difference spectra (dotted lines). Spectra are representative of experiments at pH 8 and 9.
At pH 8 and 9, however, irradiation of the complex produces a decrease of the red-most region of the PPIX absorption spectrum compared with the nonirradiated samples (Figure 2B). The difference spectra (obtained by subtracting the irradiated spectra from the nonirradiated one) show a negative peak at 408 nm which increases in magnitude with increasing irradiation (Figure 2B). The same spectra also show a small positive band near 320 nm which could indicate the limited formation of Trp photoproducts.
Steady-State Fluorescence Spectroscopy
The irradiation of PPIX/BLG complexes is strongly pH dependent. At pH 5–7, the amount of PPIX bleaching is insignificant and nearly identical for irradiated and nonirradiated samples (Figure 3). Conversely, at pH 8 and 9, irradiation of the porphyrin/BLG complex leads to a marked bleaching of PPIX (Figure 3), the fluorescence of which decreases to <10% of the initial intensity. This strong photobleaching is not accompanied by either a shift of the emission maximum or the appearance of photoproducts as we observed with other proteins.13
Figure 3.
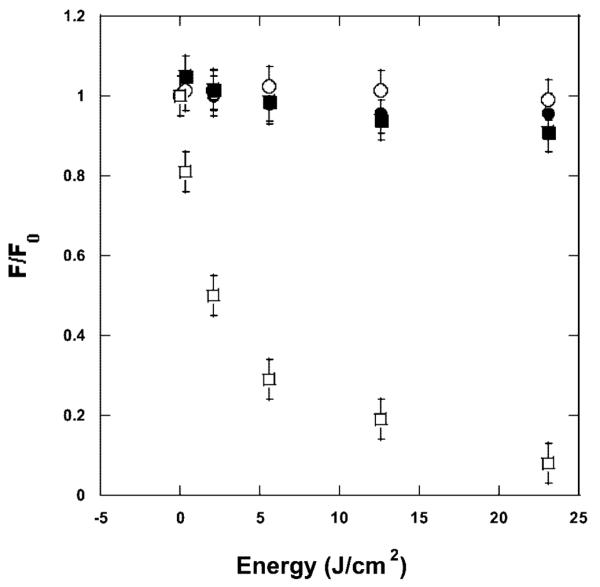
Photobleaching of PPIX upon irradiation of the porphyrin/BLG complex. At pH 6, there is insignificant difference between irradiated (○) and nonirradiated samples (●) (data are representative of experiments at pH 7 and below). At pH 9, irradiation (□) produces a large photobleaching ,whereas nonirradiated samples (■) do not photobleach (data are representative of experiments at pH 8 and above).
The intrinsic emission of BLG shows no substantial difference between spectra of irradiated versus nonirradiated complexes at pH 7 and below (Figure 4).
Figure 4.
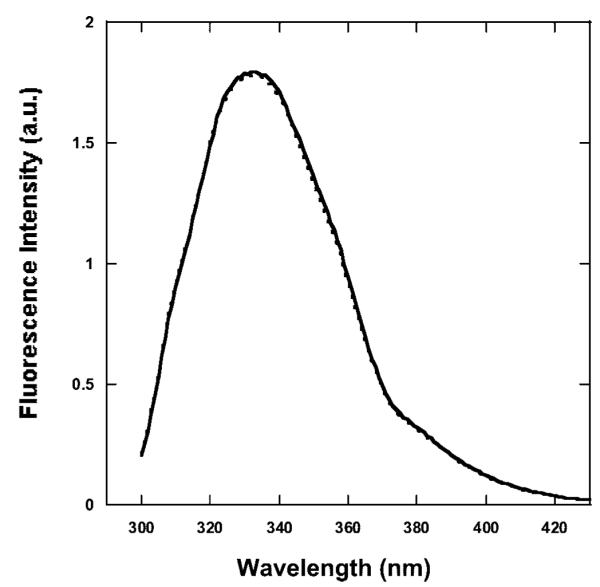
Emission spectra of BLG at pH 6 (λex ) 280 nm). The spectra are representative emission spectra of BLG/PPIX complexes at pH 7 and below for the initial nonirradiated complex (solid line), the final nonirradiated complex (dashed line), and the final irradiated (23 J/cm2) complex (dotted line). The emission of BLG in the complex remains unchanged upon irradiation.
At pH 8 and 9, the fluorescence of the protein in the irradiated complex increases by ~2%, whereas the spectrum of irradiated BLG/PPIX complexes shows a small (2 nm) but consistent shift to longer wavelengths compared to the nonirradiated complex (Figure 5).
Figure 5.
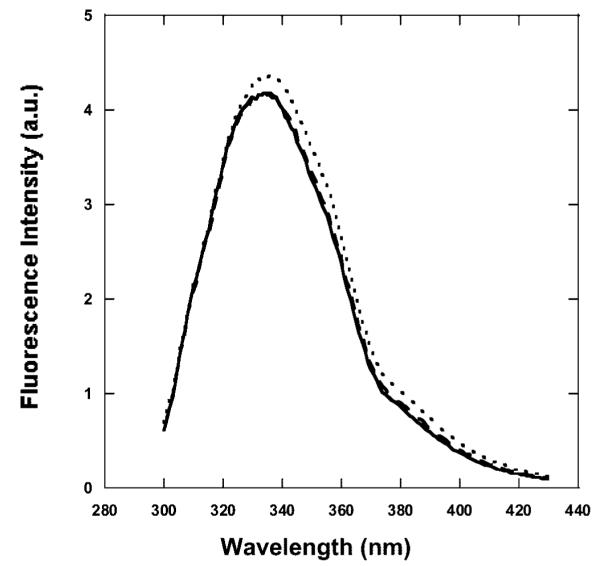
Emission spectra of BLG at pH 9 (λex ) 280 nm). The spectra are representative emission spectra of BLG/PPIX complexes at pH 8 and above for the initial nonirradiated complex (solid line), the final nonirradiated complex (dashed line), and the final (23 J/cm2) irradiated complex (dotted line). The emission of BLG in the irradiated complex increases and shifts to longer wavelengths.
Intrinsic Fluorescence Lifetime of BLG in the Presence of O2
Measurements of the fluorescence lifetime of BLG in the BLG/PPIX complex provide a better resolution of the effects of irradiation on the protein. At all pH values, the resolution of BLG decay into three lifetime components is in excellent agreement with our previous results that showed a pH-dependent fluorescence decay29 with the middle- and longer-lived components increasing in value between pH 5 and pH 9 .
In accord with the steady-state emission data, at pH 7 and below, there is no variation of the fluorescence decay as a function of irradiation (Table 1). The fluorescence decay of the protein in the BLG/PPIX complex before irradiation, after irradiation, and in the nonirradiated samples are almost exactly superimposed (Figure 6A).
TABLE 1.
Summary of Fluorescence Decay Parameters at pH 9 of Irradiated versus Nonirradiated BLG/PPIX Complex (λex = 280 nm and λem = 330 ± 4 nm, pH 9)
| 0 (J) | 1.8 (J) | 5.3 (J) | 12.3 (J) | 23 (J) | |
|---|---|---|---|---|---|
| α1 (irr.) | 0.29 ± 0.04 | 0.32 ± 0.05 | 0.31 ± 0.05 | 0.29 ± 0.04 | 0.28 ± 0.04 |
| α1 (non- irr.)a | 0.27 ± 0.03 | 0.32 ± 0.07 | 0.32 ± 0.07 | 0.30 ± 0.04 | 0.28 ± 0.06 |
| τ1 (irr. air) | 0.65 ± 0.06 | 0.65 ± 0.05 | 0.65 ± 0.07 | 0.63 ± 0.06 | 0.60 ± 0.05 |
| τ1 (irr. deoxy) | 0.35 ± 0.08 | 0.41 ± 0.05 | 0.37 ± 0.09 | 0.34 ± 0.07 | 0.28 ± 0.08 |
| τ1 (non- irr.) | 0.60 ± 0.01 | 0.62 ± 0.09 | 0.63 ± 0.08 | 0.61 ± 0.03 | 0.60 ± 0.02 |
| α2 (irr.) | 0.57 ± 0.02 | 0.54 ± 0.03 | 0.54 ± 0.03 | 0.54 ± 0.04 | 0.54 ± 0.03 |
| α2 (non- irr.)a | 0.59 ± 0.02 | 0.55 ± 0.04 | 0.55 ± 0.03 | 0.56 ± 0.02 | 0.57 ± 0.04 |
| τ2 (irr. air)b | 1.51 ± 0.05 | 1.53 ± 0.05 | 1.56 ± 0.11 | 1.60 ± 0.10 | 1.62 ± 0.11 |
| τ2 (irr. deoxy)b | 1.60 ± 0.07 | 1.62 ± 0.06 | 1.64 ± 0.09 | 1.71 ± 0.10 | 1.73 ± 0.12 |
| τ2 (non- irr.)a | 1.47 ± 0.06 | 1.53 ± 0.10 | 1.53 ± 0.12 | 1.50 ± 0.06 | 1.48 ± 0.01 |
| α3 (irr.)b | 0.14 ± 0.03 | 0.14 ± 0.02 | 0.15 ± 0.02 | 0.17 ± 0.02 | 0.18 ± 0.02 |
| α3 (non- irr.)a | 0.14 ± 0.03 | 0.13 ± 0.03 | 0.14 ± 0.03 | 0.15 ± 0.03 | 0.15 ± 0.03 |
| τ3 (irr. air)b | 4.30 ± 0.14 | 4.42 ± 0.16 | 4.49 ± 0.21 | 4.74 ± 0.21 | 4.82 ± 0.22 |
| τ3 (irr. deoxy)b | 4.71 ± 0.11 | 4.70 ± 0.13 | 4.85 ± 0.14 | 5.01 ± 0.24 | 5.40 ± 0.27 |
| τ3 (non- irr.)a | 4.32 ± 0.09 | 4.38 ± 0.13 | 4.41 ± 0.20 | 4.38 ± 0.20 | 4.36 ± 0.09 |
Summary.
The data are underlined in order to highlight the increase in the emission lifetime of the irradiated samples compared to the adjacent, nonirradiated samples.
Figure 6.

Fluorescence decay of BLG in the BLG/PPIX complexes (λex = 280 nm and λem = 330 ± 4 nm). (A) pH 6. The decay of the complex before irradiation (black), the final nonirradiated complex (blue), and the complex irradiated with 23 J/cm2 (red) are basically superimposed. (B) pH 9. The decay of the complex after irradiation with 23 J/cm2 (red) is clearly longer lived than the decay of the complex before irradiation (blue) and the nonirradiated complex (black). (C) The values of ⟨τ⟩ calculated from eq 3 show that at pH 7 and below, the decay of BLG in the complex does not change when comparing irradiated samples [(○) pH 5 and (□) pH 7] and non irradiated samples that were left in the dark for the same amount of time as that required for irradiation [(●) pH 5 and (■) pH 7]. At pH 8 and above ⟨τ⟩, the irradiated complex (Δ) increases with the dose compared to the nonirradiated samples (▲). (Note: the error bars for the data at pH 7 and below appear larger because of the difference between the left-hand side scale and the right-hand side scale of the plot). (D) Comparison of the BLG decay in air and under N2 saturation. The decay of the irradiated sample in air (blue) is longer than the decay of the nonirradiated sample in air (black). The decay of the nonirradiated sample under N2 (red) is slower than the corresponding one in air. After irradiation (green), the decay lengthens even further. In A, B, and D, the prompt is represented by □.
At pH 8 and 9, however, irradiated PPIX/BLG complexes produce substantial changes in the fluorescence decay of the protein. The increase of irradiation causes the fluorescence lifetime to lengthen (Figure 6B). By comparison, the nonirradiated samples maintained the same fluorescence decay as that of the sample before irradiation is carried out (Figure 6B). Deconvolution of the decay components shows that irradiation of the PPIX/BLG complex substantially lengthens the intermediate (τ2) and longer-lived (τ3) components of BLG fluorescence decay (Table 1) whereas the short component (τ1) as well as the relative amplitudes (α1, α2, and α3) of all components are left unaltered (within experimental uncertainty). At pH 8, τ2 and τ3 lengthen by approximately 100 and 250 ps, respectively, (from 0 to 23 mJ), whereas at pH 9, for the same range of irradiation energies, τ2 and τ3 increase by 110 and 500 ps, respectively. As a consequence, the average fluorescence lifetime (eq 3) also increases with increasing irradiation at alkaline pH (Figure 6C).
Intrinsic Fluorescence Lifetime of BLG in Deoxygenated Samples
The deoxygenation of solutions containing PPIX/BLG complexes resulted in the lengthening of the overall decay of the protein. This is due to the removal of O2, which is a well characterized quencher of protein intrinsic fluorescence.30 The lengthening of BLG fluorescence decay occurs at all pH values; however, we will limit the detailed analysis to alkaline pH because these are the only ones relevant to the mechanistic explanation of the photoinduced effects on the protein. At alkaline pH, the lifetime components of the protein in the PPIX/BLG complexes increased to τ1 = 0.3 ns, τ2 = 1.73 ns, and τ3 = 4.71 ns, whereas the relative amplitudes remained the same as those in the presence of O2 (Table 1). Upon irradiation, the fluorescence decay of the protein lengthened as in the presence of O2 (Figure 6D). The relative change of τ2 and τ3 was slightly more pronounced in the absence of O2 than in the presence of air. τ2 increased by 60 ps whereas τ3 increased by 690 ps (Table 1).
At pH 7 and below, there was no change of BLG fluorescence decay between irradiated and nonirradiated samples. Therefore, the effects of PPIX irradiation on BLG at alkaline pH occur regardless of the presence of O2.
At all pH values, irradiation of the protein alone (no PPIX bound) did not produce any change in the intrinsic fluorescence decay of either air-saturated or deoxygenated samples.
CD. 450–320 nm
The region of the Soret band of PPIX does not show any optical activity at any pH or as a result of irradiation. This is an indication that the ring of PPIX bound to BLG is not distorted by the binding site, in agreement with the docking simulations.
320–250 nm
At pH 7 and below, there is no substantial difference between irradiated and nonirradiated spectra of the PPIX/BLG complexes or between these and BLG alone. The spectra show the minima at 293 and 286 nm due to Trp residues and other minima between 280 and 260 nm due to Tyr residues.31,32 The ratio of 293/286 nm intensity remains constant for all samples. However, in irradiated BLG/PPIX complexes at pH 8 and 9, there is a slight decrease of the dichroic signal at 286 nm, indicating a small effect in the tertiary conformation of the protein (Figure 7A).
Figure 7.

CD spectra. (A) Near-UV CD spectra of the BLG/PPIX complex at pH 9. In comparison with the initial (black) and nonirradiated (blue) spectra, the irradiated complex (red) shows a near suppression of the Trp peak at 286 nm. (B) Far-UV CD spectra at pH 6. The spectra of BLG (black) superimposes with the spectrum of the BLG/PPIX complex before irradiation (blue), the spectrum of the nonirradiated complex (red), and the spectrum of the irradiated complex (green). (C) Far-UV CD spectra at pH 9. The spectra of BLG (black) superimposes with the spectrum of the BLG/PPIX complex before irradiation (orange), the spectrum of the nonirradiated complex (yellow), and BLG (green) after a time equivalent to the irradiation time. The spectrum of the irradiated complex in air (red) and deoxygenated (blue) are shifted to smaller wavelengths and show a slightly deeper trough.
250–196 nm (in the Presence of O2)
The experiments only yielded quality data between 250 and 196 nm. Although this range excludes a portion of the spectrum important for the analysis of the secondary structure,26 our analysis was comparable to the ones reported previously for CD spectra at λ > 200 nm.33 At pH 7 and below, there is no difference among the spectra of BLG alone before and after irradiation or between these and the spectra of BLG/PPIX complexes before and after irradiation (Figure 7B). According to this invariance, the analysis of the spectra with CDPro reveals a constant contribution of the secondary components (Table 2) which are in agreement with previous data.26
TABLE 2.
Analysis of the CD Spectra of BLG and PPIX/BLG Complexes by Using CDproa
| BLG initial | BLG final | BLG irradiated | complex initial | complex final | complex irradiated | |
|---|---|---|---|---|---|---|
| pH 9 | ||||||
| α | 0.060 ± 0.003 | 0.063 ± 0.008 | 0.059 ± 0.003 | 0.065 ± 0.006 | 0.065 ± 0.005 | 0.060 ± 0.002 |
| β | 0.375 ± 0.008 | 0.371 ± 0.005 | 0.371 ± 0.004 | 0.370 ± 0.003 | 0.375 ± 0.004 | 0.363 ± 0.004 |
| T | 0.200 ± 0.004 | 0.200 ± 0.001 | 0.200 ± 0.002 | 0.200 ± 0.002 | 0.200 ± 0.002 | 0.192 ± 0.002 |
| R | 0.365 ± 0.001 | 0.366 ± 0.004 | 0.370 ± 0.004 | 0.365 ± 0.005 | 0.360 ± 0.004 | 0.385 ± 0.002 |
| pH 6 | ||||||
| α | 0.047 ± 0.002 | 0.044 ± 0.001 | 0.043 ± 0.002 | 0.045 ± 0.007 | 0.046 ± 0.007 | 0.048 ± 0.003 |
| β | 0.373 ± 0.003 | 0.374 ± 0.001 | 0.375 ± 0.004 | 0.374 ± 0.008 | 0.373 ± 0.003 | 0.369 ± 0.003 |
| T | 0.203 ± 0.002 | 0.207 ± 0.002 | 0.205 ± 0.002 | 0.205 ± 0.002 | 0.204 ± 0.001 | 0.207 ± 0.006 |
| R | 0.377 ± 0.001 | 0.375 ± 0.003 | 0.377 ± 0.001 | 0.376 ± 0.001 | 0.377 ± 0.003 | 0.376 ± 0.006 |
Data are presented as percentages of each secondary structure.
At pH 8 and 9, however, the region of the secondary structure of BLG shows some intriguing changes associated with irradiation. Pre-irradiation spectra of BLG/PPIX complexes and BLG alone are identical (Figure 7C). The final nonirradiated samples of PPIX/BLG also overlap with the initial ones and yield contribution to the secondary structures similar to those mentioned above for pH 7 and below (Table 2). Conversely, the irradiated complex shows a 2–3 nm shift to lower wavelengths and a deepening of the negative peak (Figure 7C). This effect is similar to the one shown by Qi et al.26 for the temperature-induced unfolding of BLG. The analysis with CDPro reveals that the change can be attributed to a small decrease (<7%) in β-structure (sheet and turn) and a simultaneous increase in the contribution of unordered structures (Table 2).
250–196 nm (in Deoxygenated Samples)
Irradiation of PPIX/BLG complexes at alkaline pH in deoxygenated samples still produced the spectral shift observed in the presence of O2 (Figure 7C). The analysis with CDPro also yielded nearly identical percentages of the secondary structures compared to the samples irradiated in the presence of O2. Nonirradiated, deoxygenated samples did not show any change. Therefore, the photoinduced PPIX-mediated unfolding of BLG occurs to the same extent in the presence and in the absence of diffusing O2.
SDS-PAGE
Electrophoresis reveals an important result. At all pH investigated, irradiated and nonirradiated samples produce one single band near 18.5 kDa corresponding to the BLG monomer (as the dimer is separated by the presence of SDS). Thus, irradiation of BLG does not cause the protein to fragment. Electrophoresis and other methods failed, however, to determine whether irradiation of PPIX causes the porphyrin to covalently tag the protein.
Discussion
Refinement of the Binding Model
In order to interpret the conformational effects of the irradiation of PPIX/BLG complexes, we need to rationalize the location of the binding site. In a previous manuscript, we had established that binding of PPIX to BLG dimers was modulated by pH.16 Above the conformational transition of BLG, the affinity and the stoichiometry of PPIX binding increased.16 Modulation by pH appeared to suggest a binding site located inside or in proximity of the aperture of the β-barrel, which is the region of the protein that undergoes the major pH-induced conformational transition.25 The current study enabled us to refine the previous model. CD spectroscopy in the Soret band region shows that PPIX is not distorted by the binding. This virtually rules out that the porphyrin binds cavity sites such as the β-barrel or the groove between the exterior of the barrel and the 3-turn α-helix28 because they would likely require distortion of the porphyrin macrocycle. These sites are also ruled out by the docking simulations that does not produce any stable configuration of PPIX at these sites. The presence of intradimeric sites (BLG is a dimer under the conditions used in our experiments)27 is validated by our simulations but would not explain the strong quenching of BLG fluorescence,16 the major intrinsic chromophore of which, Trp19,29,34is distant from the monomer–monomer interface.27 The site of Figure 1, instead, produces an energy minimum comparable to the lower one obtained from intradimeric sites and places the porphyrin within 8 Å of Trp19. Thus, our results suggest that PPIX is located at the surface of the protein (as suggested by other studies)35 near the bottom of the β-barrel stabilized by hydrogen bonds with the C-terminal region and the loop between the G-strand and the 3-turn α-helix.
The nonlinear behavior of the binding curves determined previously16 however are consistent with the existence of at least another binding site which could be represented by an intradimeric site such as the ones discussed previously.
Photoinduced Conformational Changes
The possible location of the binding site described in the previous paragraph is paramount to interpret the structural changes induced by irradiation on the PPIX/BLG complex because the extent of the photodamage appears to be noncatastrophic, thus suggesting a localized effect. Our CD data demonstrate that PPIX causes localized unfolding of BLG only at alkaline pH. The fluorescence data suggests that the unfolding affects the environment of Trp19 as both the emission wavelength and the decay lifetime lengthen (Figures 5 and 6B). This experimental evidence can be interpreted with the simultaneous increased exposure of Trp19 and the increased separation from a proximal quencher in its surroundings. The CD spectra of Figure 7C and the retrieved secondary structures of Table 2 enable us to further refine the model. The 7% increase in unordered structure with consequent loss of β-structure (sheet and turn) is consistent with unfolding of the region of the binding site (Figure 1) localized near the Trp19 residue. The unfolding could involve the loss of structure of (i) the turn portion of the F-G loop (1BEB), (ii) the beginning of strand A, and (iii) part of strand G. These three structural features are part of PPIX binding site (Figure 1), and they enclose Trp19. A 5–7% unfolding of this region would increase exposure of Trp19 and decrease its interaction with proximal amino acid residues that could normally quench the fluorescence through hydrogen bonding or other mechanisms.36,37 This would explain the shift in emission, the lengthening of the decay lifetime, and the slight increase in intensity of BLG fluorescence upon irradiation. Moreover, because only the lifetimes and not the relative amplitudes of the decay components of Trp19 change upon irradiation, we suggest that the unfolding does not increase the mobility of Trp19, which would otherwise cause a substantial shift in the relative amplitude of the decays.38
Oxygen Independence
The evidence that the porphyrinmediated, photoinduced unfolding of BLG is O2-independent rules out the involvement of 1O2 as mediator of the protein damage. This is surprising given that the therapeutic action of PPIX is attributed to its ability to sensitize single oxygen.39 It is therefore likely that the mechanism of BLG unfolding is triggered by electron transfer (ET) between the porphyrin and proximal amino acid residues or even the peptide bond. Recent studies suggest the important role of the propionic moiety of PPIX in ET mechanisms.40,41 The residues in closer proximity to the propionic moiety of PPIX are Trp19 (~10Å), Tyr99 (~8.7Å), Pro48 (~7.6Å), Lys14 (~3.7Å), and Arg124 (~8.6Å). If one estimates the values of the reduction potential (versus SCE) of these amino acids42–44 and the oxidation potential of the porphyrins45–47 by using the Rehm–Weller equation,48 it is possible to establish that Trp19, Tyr99 and one of the proximal amides are the most likely residues for photoinduced ET from PPIX (ΔG = –1.95 = 0.26, –1.78 ± 0.26, and –1.83 ± 0.12eV, respectively). This estimate however is quite crude because it does not consider the difference in distance between donor and acceptor; moreover, the exact redox potentials in the region of the binding site is affected by the sequence of amino acids49,50 and is not trivial to estimate. It is conceivable however, on the basis of the free energy values, that ET to proximal residues or to an amide could trigger the localized unfolding. Additional experiments are being carried out to determine the ET acceptor and the effects on the protein conformation.
This finding is extremely exciting for both the mechanistic model and the potential biomedical applications of porphyrin/protein complexes. In fact, it demonstrates that under more controlled circumstances, PPIX is capable of molecular damage that does not depend on the intermediate formation of 1O2. This could have important consequences for the biomedical applications of PPIX because cellular phototoxicity could be induced in the absence of diffusing O2 (similar to type-I photosensitization).51
One question however remains to be addressed. Why does the unfolding occur only at pH 8 and 9? We suggest that because of the apparent location of PPIX, the photoinduced unfolding may not be related directly to the Tanford transition of BLG52 but rather to the combination of a decreased stability of the protein at alkaline pH53,54 and an increased photoactivity of PPIX at higher pH (Figure 3). A lesser stability (with weakening of intramolecular hydrogen bonds) of BLG at alkaline pH has been suggested,53 and it renders the protein more susceptible to exogenous modifications such as the ones produced by the irradiation of a ligand. On the other hand, PPIX photophysics and photochemistry is also pH-dependent as shown by the larger bleaching of PPIX at alkaline pH (Figure 3). However, to confirm this, we are currently undertaking studies of the photochemical intermediates formed upon irradiation of the PPIX/BLG complex.
Conclusions
Our results show that porphyrins such as PPIX are indeed capable of modifying the structure of nonspecific proteins upon irradiation of the Soret band. Although more detailed mechanistic studies are necessary, these results suggest that (i) phototoxicity of porphyrins may proceed through direct reaction with target proteins and (ii) porphyrins may be used to trigger conformational changes useful for the investigation of protein folding/unfolding mechanisms. The unfolding mechanism is not O2-dependent; therefore, a direct charge transfer between the porphyrin and one amino acid in the binding site is the likely cause of the conformational change rather than a more trivial oxidation produced by PPIX-sensitized singlet oxygen. The fact that the mechanism of photoinduced conformational change does not depend on O2 is extremely important for future development of porphyrin-induced conformational changes of proteins in biomedical applications and in proteomics.
Acknowledgment
The study was funded in part by a Faculty Research Award to L.B. from the University of Texas at San Antonio. For the duration of the project, N.F.F. received MARC U*STAR support (NIH/NIGMS GM07717). The authors would like to thank Dr. Donald Kurtz and Dr. David Wampler for their assistance with SDS-PAGE experiments.
References and Notes
- (1).Lang K, Mosinger J, Wagnerova DM. Coord. Chem. Rev. 2004;248:321. [Google Scholar]
- (2).Berg K, Selbo PK, Weyergang A, Dietze A, Prasmickaite L, Bonsted A, Engesaeter BO, Angell-Petersen E, Warloe T, Frandsen N, Hogset AJ. Microsc. 2005;218:133. doi: 10.1111/j.1365-2818.2005.01471.x. [DOI] [PubMed] [Google Scholar]
- (3).Aveline BM, Hasan T, Redmond RW. J. Photochem. Photobiol. B. 1995;30:161. doi: 10.1016/1011-1344(95)07174-z. [DOI] [PubMed] [Google Scholar]
- (4).Horsey BE, Whitten DG. J. Am. Chem. Soc. 1978;100:1293. [Google Scholar]
- (5).Boekelheide K, Eveleth J, Tatum AH, Winkelman JW. Photochem. Photobiol. 1987;46:657. doi: 10.1111/j.1751-1097.1987.tb04828.x. [DOI] [PubMed] [Google Scholar]
- (6).Berg C, Moan J. Photochem. Photobiol. 1997;65:403. doi: 10.1111/j.1751-1097.1997.tb08578.x. [DOI] [PubMed] [Google Scholar]
- (7).Kessel D, Castelli M. Photochem. Photobiol. 2001;74:318. doi: 10.1562/0031-8655(2001)074<0318:etbitt>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- (8).Usuda J, Chiu SM, Murphy ES, Lam M, Nieminen AL, Oleinick NL. J. Biol. Chem. 2003;278:2021. doi: 10.1074/jbc.M205219200. [DOI] [PubMed] [Google Scholar]
- (9).Xue LY, Chiu SM, Fiebig A, Andrews DW, Oleinick NL. Oncogene. 2003;22:9197. doi: 10.1038/sj.onc.1207019. [DOI] [PubMed] [Google Scholar]
- (10).Lee CT, Jr, Smith KA, Hatton TA. Biochemistry. 2005;44:524. doi: 10.1021/bi048556c. [DOI] [PubMed] [Google Scholar]
- (11).Wang SC, Lee CT., Jr J Phys Chem B. 2006;110:16117. doi: 10.1021/jp060981n. [DOI] [PubMed] [Google Scholar]
- (12).Abbruzzetti S, Sottini S, Viappiani C, Corrie JE. Photochem. Photobiol. Sci. 2006;5:621. doi: 10.1039/b516533d. [DOI] [PubMed] [Google Scholar]
- (13).Brancaleon L, Moseley H. Biophys. Chem. 2002;96:77. doi: 10.1016/s0301-4622(02)00035-2. [DOI] [PubMed] [Google Scholar]
- (14).Brancaleon L, Magennis SW, Samuel IDW, Namdas E, Lesar A, Moseley H. Biophys. Chem. 2004;109:351. doi: 10.1016/j.bpc.2003.12.008. [DOI] [PubMed] [Google Scholar]
- (15).Tian F, Johnson EM, Zamarripa M, Sansone S, Brancaleon L. Biomacromol. 2007;8:3767. doi: 10.1021/bm700687x. [DOI] [PubMed] [Google Scholar]
- (16).Tian F, Johnson K, Lesar AE, Moseley H, Ferguson J, Samuel IDW, Mazzini A, Brancaleon L. Biochim. Biophys. Acta. 2005;1760:38. doi: 10.1016/j.bbagen.2005.09.005. [DOI] [PubMed] [Google Scholar]
- (17).Krieg M, Whitten DG. J. Am. Chem. Soc. 1984;106:2477. [Google Scholar]
- (18).Tanford C, Bunville LG, Nozaki Y. J. Am. Chem. Soc. 1959;81:4032. [Google Scholar]
- (19).Lakowicz JR. Principles of Fluorescence Spectroscopy. 3rd ed Springer; New York: 2006. [Google Scholar]
- (20).Durbin J, Watson GS. Biometrika. 1951;38:159. [PubMed] [Google Scholar]
- (21).Johnson WC. Annu. Rev. Biophys. Biophys. Chem. 1988;17:145. doi: 10.1146/annurev.bb.17.060188.001045. [DOI] [PubMed] [Google Scholar]
- (22).Albinsson B, Kubista M, Norden B, Thulstrup EW. J. Phys. Chem. 1989;93:6646. [Google Scholar]
- (23).Huang X, Nakanishi K, Berova N. Chirality. 2000;12:237. doi: 10.1002/(SICI)1520-636X(2000)12:4<237::AID-CHIR10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- (24).Sreerama N, Woody RW. Anal. Biochem. 2000;287:252. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- (25).Brownlow S, Morais Cabral JH, Cooper R, Flower DR, Yewdall SJ, Polikarpov I, North ACT, Sawyer L. Structure. 1997;5:481. doi: 10.1016/s0969-2126(97)00205-0. [DOI] [PubMed] [Google Scholar]
- (26).Qi XL, Holt C, McNulty D, Clarke DT, Brownlow S, Jones GR. Biochem. J. 1997;324:341. doi: 10.1042/bj3240341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Fessas D, Iametti S, Schiraldi A, Bonomi F. Eur. J. Biochem. 2001;268:5439. doi: 10.1046/j.0014-2956.2001.02484.x. [DOI] [PubMed] [Google Scholar]
- (28).Monaco HL, Zanotti G, Spadon P, Bolognesi M, Sawyer L, Eliopoulos EE. J. Mol. Biol. 1987;197:695. doi: 10.1016/0022-2836(87)90476-1. [DOI] [PubMed] [Google Scholar]
- (29).Harvey BJ, Bell E, Brancaleon LJ. Phys. Chem. B. 2007;111:2610. doi: 10.1021/jp065783a. [DOI] [PubMed] [Google Scholar]
- (30).Eftink MR, Ghiron CA. Anal. Biochem. 1981;114:189. doi: 10.1016/0003-2697(81)90474-7. [DOI] [PubMed] [Google Scholar]
- (31).Rasmussen P, Barbiroli A, Bonomi F, Faoro F, Ferranti P, Iriti M, Picariello G, Iametti S. Biopolymers. 2007;86:57. doi: 10.1002/bip.20704. [DOI] [PubMed] [Google Scholar]
- (32).Zhang X, Keiderling TA. Biochemistry. 2006;45:8444. doi: 10.1021/bi0602967. [DOI] [PubMed] [Google Scholar]
- (33).Crougennec T, Molle D, Mehra R, Bouhallab S. Protein Sci. 2004;13:1340. doi: 10.1110/ps.03513204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cho Y, Batt CA, Sawyer L. J. Biol. Chem. 1994;269:11102. [PubMed] [Google Scholar]
- (35).Dufour E, Marden MC, Haertle T. FEBS Lett. 1990;277:223. doi: 10.1016/0014-5793(90)80850-i. [DOI] [PubMed] [Google Scholar]
- (36).Vivian JT, Callis PR. Biophys. J. 2001;80:2093. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Callis PR, Petrenko A, Muino PL, Tusell JR. J. Phys. Chem. B. 2007;111:10335. doi: 10.1021/jp0744883. [DOI] [PubMed] [Google Scholar]
- (38).Eftink MR. Biophys. J. 1994;66:482. doi: 10.1016/s0006-3495(94)80799-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kennedy JC, Pottier RH. J. Photochem. Photobiol. B. 1992;14:275. doi: 10.1016/1011-1344(92)85108-7. [DOI] [PubMed] [Google Scholar]
- (40).Tsukahara K, Okazawa T, Takahashi H, Yamamoto Y. Inorg. Chem. 1986;25:4756. [Google Scholar]
- (41).Rosales-Hernandez MC, Correa-Basurto J, Flores-Sandoval C, Marin-Cruz J, Torres E, Trujillo-Ferrara J. J. Mol. Struct. (THEOCHEM) 2007;804:81–88. [Google Scholar]
- (42).Jonsson M, Kraatz HB. J. Chem. Soc., Perkin Trans. 1997;2:2673. [Google Scholar]
- (43).Milligan JR, Aguilera JA, Ly A, Tran NQ, Hoang O, Ward JF. Nucleic Acid Res. 2003;31:6258. doi: 10.1093/nar/gkg816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Bumber AA, Kornienko IV, Profatilova IA, Vnukov VV, Kornienko IE, Garnovskii AD, Russian J. Gen. Chem. 2001;71:1311. [Google Scholar]
- (45).Chen FC, Ho JH, Chen CY, Su YO, Ho TI. J. Electroanal. Chem. 2001;499:17. [Google Scholar]
- (46).D’Souza F, Deviprasad GR, Hsieh YY. Chem. Comm. 1997;1997:533. [Google Scholar]
- (47).Bensasson RV, Land EJ, Truscott TG. Excited States and Free Radicals in Biology and Medicine. Oxford University Press; Oxford: 1993. [Google Scholar]
- (48).Rehm D, Weller A. Isr. J. Chem. 1970;8:259–271. [Google Scholar]
- (49).Eugster N, Jensen H, Fermin DJ, Girault HH. J. Electroanal. Chem. 2003;560:143. [Google Scholar]
- (50).Moffeta DA, Foleyb J, Hechta MH. Biophys. Chem. 2003;105:231. [Google Scholar]
- (51).Girotti AW. J. Photochem. Photobiol. 1992;13:105. doi: 10.1016/1011-1344(92)85050-5. [DOI] [PubMed] [Google Scholar]
- (52).Qin BY, Bewley MC, Creamer LK, Baker HM, Baker EN, Jameson GB. Biochemistry. 1998;37:14014. doi: 10.1021/bi981016t. [DOI] [PubMed] [Google Scholar]
- (53).Taulier N, Chalikian TV. J. Mol. Biol. 2001;314:873. doi: 10.1006/jmbi.2001.5188. [DOI] [PubMed] [Google Scholar]
- (54).Fujiwara K, Ikeguchi M, Sugai S. J. Prot. Chem. 2001;20:131. doi: 10.1023/a:1011029524100. [DOI] [PubMed] [Google Scholar]