

Abstract
Despite continuous research efforts directed at early diagnosis and treatment of pancreatic cancer (PC), the status of patients affected by this deadly malignancy remains dismal. Its notoriety with regard to lack of early diagnosis and resistance to the current chemotherapeutics is due to accumulating signaling abnormalities. Hoarding experimental and epidemiological evidences have established a direct correlation between cigarette smoking and PC risk. The cancer initiating/promoting nature of cigarette smoke can be attributed to its various constituents including nicotine, which is the major psychoactive component, and several other toxic constituents, such as nitrosamines, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, and polycyclic aromatic hydrocarbons. These predominant smoke-constituents initiate a series of oncogenic events facilitating epigenetic alterations, self-sufficiency in growth signals, evasion of apoptosis, sustained angiogenesis, and metastasis. A better understanding of the molecular mechanisms underpinning these events is crucial for the prevention and therapeutic intervention against PC. This review presents various interconnected signal transduction cascades, the smoking-mediated genotoxicity, and genetic polymorphisms influencing the susceptibility for smoking-mediated PC development by modulating pivotal biological aspects such as cell defense/tumor suppression, inflammation, DNA repair, as well as tobacco-carcinogen metabolization. Additionally, it provides a large perspective toward tumor biology and the therapeutic approaches against PC by targeting one or several steps of smoking-mediated signaling cascades.
Introduction
Pancreatic cancer (PC) is a devastating disease due to lack of early diagnosis and its unresponsiveness to conventional therapeutic regimens, resulting in 5-year survival rate of less than 5% and a mortality rate of nearly 100% (1,2). Till date, tumor resection is the only potentially curative approach. But unfortunately, >80% patients are presented with unresectable tumor with distant organ metastasis at the time of diagnosis (3). Furthermore, the mortality rate for PC has remained unchanged over the past few decades (4). As there are no tests for early screening, and once detected, therapeutic options are limited, the only prospect for reducing mortality from PC is prevention. Understanding the etiology and identifying the risk factors associated with PC are essential for the prevention of this deadly disease.
Although several risk factors, such as higher body mass index, alcohol consumption, and history of diabetes, have been associated with PC, cigarette smoking is the only unequivocal risk factor that has been identified (5). Cigarette smoking is also an independent risk factor for developing chronic pancreatitis, a disease with a subsequent high risk of progression to PC (6). In addition, the cigarette smoke (CS) causes pancreatic damage by inducing alteration in pancreatic enzyme secretions and leading to acinar cell destruction by aggravating the ongoing pancreatic inflammatory events (7). CS-induced pancreatic damage is a multifactorial event caused by various tobacco-derived components, including nicotine and various other carcinogenic components, such as polycyclic aromatic hydrocarbons (PAHs), nitrosamines, aromatic amines (AA), and heterocyclic amines (HCA). Herein, we discuss the effects of these tobacco-derived components on the pancreas through the cross-talk between the CS-mediated signaling pathways and gene alterations and thus decipher the direct causal relationship between cigarette smoking and PC.
Correlation between Cigarette Smoking and PC Incidence: Epidemiological Evidence
Smoking is the most common method of consuming tobacco, and tobacco is the most common substance smoked. There are no international definitions of light/heavy smoking. However, smoking <20 pack years is considered light/moderate smoking, where one pack-year is smoking one pack per day for 1 year. Any amount above this value is considered heavy smoking and smoking >30 pack years significantly increases the risk of developing all smoking-related diseases. The association of long-term smoking and lung cancer is well established through various studies, and it has been validated that tobacco smoking is responsible for 90% of all lung cancers (8). However, smoking also increases the risk of developing cancers of the esophagus, uterine cervix, kidney, bladder, stomach, and pancreas. Cigarette smoking is the only environmental factor that is strongly associated with risk of developing PC, and it is estimated to account for approximately 25–30% of all pancreatic tumors (9).
An association between smoking and PC was first noted through several studies during the 1960s and 1970s. These survey-based studies showed that cigarette smokers had a 70% greater risk of developing PC in comparison to non-smokers (10,11). Also, a recent meta-analysis of 82 studies published between 1950 and 2007 on smoking and PC found that current smokers have a 1.74-fold (95% confidence interval, 1.61–1.87) increased risk of developing PC (12). An ongoing multicenter prospective cohort study, European Prospective Investigation into Cancer and Nutrition (EPIC), undertaken by Vrieling et al. including 10 European countries, revealed that both active cigarette smoking as well as exposure to environmental tobacco smoke is associated with increased risk of PC and that risk is reduced to levels of never smokers within 5 years of quitting (13). A recent hospital-based case-control study by Talamini et al. on PC in Northern Italy, between 1991 and 2008, found that tobacco smoking may be responsible for approximately one-third of PC cases (14).
Smoking has also been associated with earlier onset of PC (9,15,16). In an international study by Lowenfels et al., hereditary pancreatitis patients were shown to have a greatly elevated risk of PC, and if they were active smokers, they were noted to develop PC on an average of 20 years earlier than non-smokers (17). A plethora of studies have also developed race-/sex-dependent trends in cancer incidence. A cohort study of the US population reflected a higher incidence of tobacco-mediated PC among men, specifically in Black and non-Hispanic populations (18). Between 2001 and 2005, Blacks were diagnosed with PC with a 33% higher incidence compared with Whites. Moreover, the PC mortality rates were 27% greater in Black men and 38% greater in Black women as compared to their White counterparts (19).
Although various studies have established smoking as undeniable risk factor for development of PC, detailed studies on impact of smoking intensity and smoking duration on PC initiation, progression, and development are still limited. Also, data on passive smoking and the use of smokeless tobacco are inconsistent, proving an insufficient evidence for increased risk of PC, hence demanding further examination. Further studies are required to find the direct causal relationship between cigarette-smoking and PC. It is a well-established fact that CS is a cocktail of many toxic constituents (20–22) and it contributes to PC pathogenesis due to its complexity.
Components of CS: their Contribution to Pancreatic Damage
More than 4000 chemicals are generated during cigarette combustion (20–22) that include butadiene, aldehydes, nicotine, bacterial endotoxin, a large amount of free radicals such as alkyl, alkoxyl, peroxyl, and guinone/hydroquinone, PAHs such as benzo[a]pyrene (BaP), tobacco-specific N-nitrosamine such as 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and nitrosonornicotine, and a large amount of nitric oxide (NO). Carcinogenic AA and tobacco-specific nitrosamines detected in CS are one of the best examined factors in the pathology of various cancers (23). Table I briefly covers the chemical nature and structure of the major CS components discussed in the review and provides information about their doses per cigarette.
Table I.
The major components of CS involved in PC pathogenesis
| Components of CS | Lowest delivery dose/cigarettea | Highest delivery dose/cigarettea | Description | Structure |
|---|---|---|---|---|
| Nicotine | 1.0 mg | 1.5 mg | An alkaloid (the major component of tobacco smoke) |
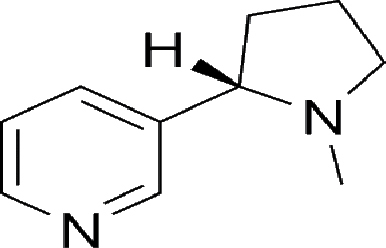
|
| Carbon monooxide (CO) | 14 mg | 20 mg | Gas, which is highly toxic, released during burning and charring of tobacco |

|
| Polycyclic aromatic hydrocarbons (PAHs) | 0.1 µg | 0.2 µg | Highly carcinogenic constituent of tobacco smoke. They are also the known ligands of the aryl hydrocarbon receptor, a cellular xenobiotic sensor responsible for activating the metabolic machinery |

|
| 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone(NNK) | 0.3 µg | 2 µg | Nitrosamine, a highly carcinogenic derivative of nicotine and related compounds, formed by a nitrosation reaction that occurs during the curing and processing of tobacco |

|
| Nitrogen oxides | 0.1 mg | 0.4 mg | Chemical compounds of nitrogen produced as a by-product of combustion of tobacco |

|
aThe values for the lowest dose/cigarette and highest dose/cigarette are listed for the individual components (dose variation depends upon the brand and source of cigarette).
Abundant evidence has been provided both through experimental and epidemiological studies for the CS-mediated pancreatic injury. Nicotine is the major psychoactive component of tobacco and CS. It is an addictive agent and has been characterized as a drug of abuse by the US Surgeon General (24). Although it is not carcinogenic by itself, it has been shown to play a key role in PC pathogenesis by leading to an uncontrolled increase of pancreatic protein synthesis in isolated acini (25). A study using the experimental mouse model of intraepithelial lesions induced by 7,12-dimethylbenzanthracene (DMBA) showed that nicotine promotes pancreatic ductal adenocarcinoma in these mice (26). Thus, smoking not only exhibits the regulatory effect on the pancreatic acinar cells but also influences the ductal cell function. A recent study has shown an upregulation of pro-collagen type 1 in pancreatic tissue with an induction of morphologic changes due to high dose of smoke exposure, which is an indicator for fibrotic tissue replacement (27). Chowdhury et al. showed that nicotine itself induces cytoplasmic vacuolation and cellular edema in the exocrine pancreas and increases total cellular amylase content (28). It was observed that the increase in pancreatic enzymes in nicotine-treated rats may be the causative factor in nicotine-induced pancreatic cell pathology (6). Studies have shown that the exposure to nicotine causes a significant decrease in secretion of duodenal bicarbonate in rabbits and affects the composition of the pancreatic secretions (29). On the subcellular level, nicotine exposure also activates multiple signaling pathways in cells resulting in high levels of intracellular calcium release that is paralleled by increased enzyme release, including lipase (30) and amylase (31).
CS contains various toxic chemicals, including the nitrosamine and NNK, which is a derivative of nicotine formed by nitrosation during the processing of tobacco plants into cigarettes (32). Prokopczyk et al. showed higher NNK levels in the pancreatic juice of smokers as compared with the non-smokers (33). Experimental data using female hamsters revealed that 65% of offspring develop PC by 1 year of age when injected with an intra-tracheal dose of NNK 1 day before the delivery of the pups (34). In another study, when administered in the drinking water for F344 rats, NNK induced pancreatic tumors of both the exocrine and the ductal/endocrine type (23,35). These studies suggest that smoking during pregnancy adds risk of developing PC in early stage of life. To elicit its effects, NNK undergoes an organ-specific metabolization into its stereo-selective metabolites, (R, S)-4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (R, S-NNAL), by human pancreatic microsomes and cytosols (36). Similar to NNK, NNAL is a also a potent pancreatic carcinogen (37) and the stereo-selective differences in metabolism and/or tissue distribution contributes to the difference in carcinogenicity between its enantiomers. Although (R)-NNAL is excreted into the urine, (S)-NNAL is extensively retained and sequestered in the target tissues of NNK-treated rats, which is subsequently re-oxidized to NNK contributing to carcinogenicity (38). Thus, (S)-NNAL is a more potent carcinogen in the A/J mouse than the (R)-NNAL. All these facts support a possible role for NNK in the etiology of PC and demonstrate that NNK contributes to tobacco-induced carcinogenesis.
Cigarette smoking is one of the most common sources of PAHs such as anthracene, phenanthrene, naphthalene, and pyrene. The most abundant PAHs in CS are the low molecular weight compounds such as methylated anthracenes and phenanthrenes that are present in CS at approximately 62-fold higher concentration levels than BaP and benzo(e)pyrene (39). 1-methylanthracene, with an angular pocket formed with a bay-like structure, are active in the induction of arachidonic acid release, inhibition of gap-junctions, and the activation of mitogen-activated protein kinase (MAPK) pathways, whereas the 2-methylanthracene lacking the bay-like structures are inactive (40). These studies showing differences between the linear versus the bay-like isomers of methylanthracene speculated an involvement of some unknown receptor requiring further validation. DMBA, a laboratory carcinogen and yet another PAH component, although not present in human environment causes pancreatic tumor in rodents due to DMBA–DNA adducts (41). To measure its metabolism, DMBA was injected into the pancreas of male rat and oddly a high concentration of metabolites of DMBA such as 5, 6-epoxy-7-hydroxymethyl-12-MBA was observed in pancreatic tissues.
The active metabolites of the CS constituents, once inhaled, react with all classes of biomolecules including carbohydrates, lipids, proteins, and nucleic acids in organs that can be reached directly, such as the lung. In addition, these harmful substances are taken up by the bloodstream and also reach other organs that are not directly in contact with tobacco smoke such as the pancreas, hence causing genetic and signaling abnormalities.
CS-induced Genotoxicity
A very important comprehensive genetic analysis was performed by Jones et al. on 24 cancers with sequencing of more than 20 000 protein-coding genes. The study identified core signaling pathways that were altered by genetic mutations or chromosomal loss in human PC and suggested that, on average, at least 63 genetic alterations linked to 12 core cellular signaling pathways were in PC, and each of these pathways were altered in 67–100% of pancreatic tumors (42).
Smoking is associated with cancer of 11 organs (as reported by International Agency for Research on Cancer, through monographs on the evaluation of the carcinogenic risks of chemicals to humans, vol. 83), and mutations in some of these smoking-associated tumors have been identified in both oncogenes and tumor-suppressor genes. Hruban et al. observed significantly high frequency of K-ras mutations in the pancreatic carcinomas harbored from smokers than from non-smokers (43,44). Berger et al. also demonstrated an aggravated PC risk with KRAS mutational activation in cigarette smokers (45). Many investigators have observed a strong “fingerprint” of tobacco carcinogens in the DNA from PC patients. The tobacco carcinogen NNK has been implicated in the pancreatic carcinogenesis by its ability to form DNA adducts, which has been associated with activating KRAS mutations (46). Studies have revealed higher proportion of G to A transition due to the involvement of nitrosamines (47,48). These mechanisms have been extensively studied in laboratory animals where the hydroxylation of NNK by CYP isoenzyme induces DNA methylation (32). The level of DNA adducts, in response to another component of CS, PAHs, has also been interpreted as being positively correlated with the spectrum of KRAS mutations in PC (49). Studies have also established an association between G to T transversion hotspots and preferential sites of formation of PAHs (50,51).
Human pancreatic tissues have many carcinogen-metabolizing enzymes (52), and individual variations in these genes modify the risk of smoking-induced PC. The metabolism takes place by a two-phase process. Phase I involves the activation or oxidative modification of the carcinogen by enzymes encoded by the cytochrome P450 (CYP) gene superfamily (53). During the phase II process, carcinogens are conjugated to cofactors by the family of glutathione S-transferases, transforming them into more polar metabolites that cannot diffuse across membranes thereby facilitating excretion. CYP2A6 (54,55), CYP2A13 (34,56), and CYP1A2 are the major isoforms of the phase I hepatic CYP enzymes responsible for metabolizing nicotine to cotinine, nitrosamines to reactive electrophiles (57), and N-hydroxylation-induced activation of AA and HCA (58), respectively. Two other important enzymes involved in the metabolism of AA and HCA carcinogens are N-acetyltransferase 1 (NAT1) and N-acetyltransferase 2 (NAT2) (59). Exposure of hamsters to CS induces the expression of CYP1A2, which metabolizes HCA and enhances the ability of their liver to convert HCA to mutagens, suggesting that CS can itself induce functioning of carcinogen-metabolizing genes (60).
Various smoke-induced mutations in the genes such as p16 and p53 have been detected mainly in PC tissues (61,62). The tumor-suppressive p53 gene is seen to be frequently mutated in cigarette smokers, causing an uncontrolled cellular growth and tumor formation (63,64). Additionally, numerous studies have provided evidence for CS-induced DNA-strand breaks in mammalian cells (65,66) and genotoxicity through sister chromatid exchanges in CS-exposed bone marrow and lung cells (67). Interestingly, in humans, newborns of smoking mothers have higher frequencies of hypoxanthine–guanine phosphoribosyltransferase mutants, translocations, and DNA strand breaks. Moreover, sperm of smokers have been shown to have higher frequencies of aneuploidy, DNA adducts, strand breaks, and oxidative damage (68). CS components exert their carcinogenicity not only through direct covalent binding to DNA but also through indirect mechanisms, for example, chronic or low-level inflammation, causing reactive oxygen species (ROS) production and lipid peroxidation products.
Collectively, these studies suggest that CS may be a potential mutagen in pancreatic carcinogenesis. However, most of these studies associating carcinogen exposure, DNA adduct formation, and gene mutations have been demonstrated mainly in the animal models and the human evidences are limited. It is a known fact that not all smokers develop cancer and even when they develop cancer, the proportion is relatively small. The mechanism for this selectivity is not clear, but it is speculated that the components contained in the CS might manifest their casual relations with PC at a greater extent when the smokers carry the susceptible genotypes/polymorphisms.
Gene-polymorphisms-influenced Susceptibility to Smoking-induced PC
Individuals have inherent genetic differences in their ability to metabolize the CS components that may influence their response to smoking initiation, dependence, addiction, and cessation. Interestingly, studies have found that individuals with genetic loss of CYP2A6 have a reduced risk of becoming smokers, and those who smoke tend to smoke fewer cigarettes per day and have a higher smoking cessation success rate (69,70).
The polymorphisms in the well-defined tumor suppressor and/or oncogenes are potential hallmarks of smoke-associated PC (71). Smoking can increase the risk of PC in BRCA2 germ-line mutation carriers who have a known genetic predisposition for PC (72). In a recent study by Li et al., the somatostatin receptor (SSTR) gene polymorphisms were seen to be inversely related to increased PC risk due to its ability to inhibit cell proliferation and negatively regulate the release of growth hormones in response to somatostatin, a polypeptide hormone (73). Among participants in four United States prospective cohort studies, low plasma levels of insulin-like growth factor binding protein-1 (IGFBP-1) significantly predicted an increased risk of PC (74). IGFBP-1 is a downstream target of insulin that inhibits the growth-promoting effects of IGF1. A case-control study by Suzuki et al. suggests that polymorphic variants of the IGF genes such as the IGF1 haplotype and the IGF2 Ex4-233 C>T TT genotype may increase susceptibility for developing PC (75). Interestingly, Brian et al. observed that the influence of IGFBP-1 on the relative risk for PC was stronger among participants who never smoked cigarettes (74). Future studies are warranted to explore polymorphisms in IGF pathways and their association with the smoking-mediated PC pathogenesis.
Patients with pancreatitis have a marked increased risk of developing PC as compared with the general population, and this risk is further increased upon smoking (17). A recent study has shown that nicotine induces oxidative stress and cholecystokinin (CCK)-stimulated amylase release in pancreatic acinar cells (56). Wheatley et al. demonstrated that the (myeloperoxidase) MPO2 G463A polymorphism and the (superoxide dismutase) SOD2 A16V polymorphism have an impact on the capacity to regulate ROS, thereby affecting the risk of PC (76). Studies by Wittel et al. showed that the exposure to high-dose CS caused pancreatic inflammation and overexpression of many chronic pancreatitis-associated genes, such as interleukin-1β (IL-1β) and transforming growth factor-β (TGF-β) (6). A meta-analysis of seven case-control studies of various cancer types presented that carriers for the variant of TGFβR1 (TGFβR1*6A) had a 26% increased risk of developing PC (77). The role of TGFβR1*6A in PC still remains unclear and is a subject of future study. Thus, polymorphisms in inflammatory molecules can act as susceptibility markers for smoking-induced PC.
There is mounting epidemiologic evidence that polymorphisms in the DNA repair genes in combination with heavy tobacco smoking increases the risk for PC (78,79). XRCC2 and XRCC3 are key components of the homologous recombination machinery that repairs DNA double-strand breaks, often resulting from radiation- and smoking-induced DNA damage. Duell et al. identified the polymorphisms in the genes in base excision repair (8-oxoguanine DNA glycosylase; OGG1), nucleotide excision repair (xeroderma pigmentosum group D, A, C; XPD, XPA, XPC), and double-strand break repair (XRCC3) in combinations with cigarette smoking, as potential risk factors for PC (80). Jiao et al. reported an interaction between the XPD codon 312 variant (D312N) and smoking in relation to the risk of PC (81).
Existence of gender-specific susceptibility to tobacco carcinogen for development of PC has been observed in various recent studies. The presence of the CYP1A2 and NAT1 genotypes has an additive effect on increased risk of PC among smoking women but not in men (82). Sonoyama et al. found that the p53 Pro/Pro genotype compared with the Arg/Arg genotype had a profound effect on PC risk among males, particularly among heavy smokers (83). Miyasaka et al. analyzed the possible associations between the polymorphism in the aldehyde dehydrogenase 2 (ALDH2) genes and incidence of PC in the Japanese population. They observed that male smokers with inactive ALDH2 are prone to increasing risk of PC, a trend not observed in female subjects (84). The exact reason for these gender-specific differences are unknown, but may possibly be related either to differences in smoking practices or to differences in the ability to detoxify carcinogenic substances contained within tobacco smoke.
The susceptible genotypes/polymorphisms diminish the ability of pancreatic cells to protect themselves from environmental or metabolic stressors, resulting in reactive molecules that lead to inflammation/cell injury and DNA damage with a series of events resulting in knocking out of key tumor-suppressor genes and/or activation of oncogenes (Table II). Although much has been explored in this field, further studies with a larger sample size and adequate study design are warranted for better conclusive results. Also, the impact of these events on PC incidences and initiation of PC are still obscure and in-depth studies are needed to fill these lacunas.
Table II.
List of gene polymorphism affecting the susceptibility to cigarette-smoke-induced pancreatic cancer
| Gene | Function | Observed polymorphism(s) and associated genotypes/alleles | Polymorphism affecting the susceptibility to PC (Refs) |
|---|---|---|---|
| Tobacco-carcinogen metabolizing genes | |||
| Cytochrome P450 (CYP2A6) | Metabolization of nicotine to cotinine | CYP2A6*2, CYP2A6*4, CYP2A6*9, CYP2A6*12 | CYP2A6*2 and CYP2A6*4 genotypes: slow metabolism of nicotine in Caucasian population. No association in Japanese population (54,55,69,70). |
| Cytochrome P450 (CYP2A13) | Metabolization of nicotine to cotinine, metabolic activation of nitrosamines | CYP2A13*1/*7 (Arg101Stop) | CYP2A13*1/*7 (Arg101Stop): Inactive enzyme, no significant association with PC risk was observed due to lack of carrier of CYP2A13*7 among the PC cases (158). |
| N-acetyl-transferase 1 (NAT1) | O-acetylation of aromatic and HCA | C1095A (NAT1*10, 3′UTR, C97T, Arg33Stop, NAT1*19), C190T (Arg64Trp, NAT1*17), G445A (Val149lIe, NAT1*11) | NAT1*10: Rapid acetylator (4-fold higher risk of PC in female smokers in comparison to never smokers who did not carry the NAT1*10 allele (59,82)). |
| N-acetyl-transferase 2 (NAT2) | O-acetylation of aromatic and HCA | G191A (Arg64Gln) C282T, T341C (Ile114Thr), C481T, G590A (Arg197Gln), A803G (Lys268Arg), G857A (Gly286Thr) | Slow acetylator increases the susceptibility to PC in smokers (82,159). |
| Glutathione S-transferase T1 (GSTT1) | Adding reduced glutathione | Wild type/heterozygous del, Null/homozygous del (deletion) | GSTT1-null genotype: Increases the susceptibility to PC in smokers (160) |
| Tumor suppressors genes | |||
| p21 | Critical mediator of G1-phase cell cycle arrest preventing the G1/S transition by inhibiting various CDK–cyclin complexes | p21 exon 2 (C/A) Ser31Arg | p21 C-to-A SNP: Increases the susceptibility to PC among homozygous wild-type carrier of p27 especially among non-smokers (71) |
| Methylene-tetrahydro- folate reductase (MTHFR) | Catalyzes conversion of THF to 5-methyl THF during folate metabolism | MTHFR C677T (exon 4) (Ala/Val) MTHFR A1298C (exon 7) Glu429Ala | MTHFR 677T: Increases susceptibility to PC in ever-smokersMTHFR 1298C: Significantly reduced risk for PC (161,162). |
| Somatostatin receptor 5 (SSTR5) | Inhibits proliferation of normal and neoplastic cells | Pro109Ser (CC,CT) Leu48Met (CC, AC) Pro335Leu (TT, TC) | Leu48Met: Increases the risk for PC in combination with smoking (73) |
| Proliferator-activated receptor gamma (PPARG) | Regulate the adipose cell differentiation and inhibits the invasive behavior of PC cells in vitro | PPARG exon 2(C/G) Pro12Ala | Pro12Ala: Increases the risk for PC among high-risk smokers in vitamin-administered subject in comparison to placebo-administered subjects (163) |
| Tumor protein p53 (TP53) | Protection from DNA damage | TP53 exon 4 (G/C), Arg 72 Pro, Arg/Arg, Arg/Pro and Pro/Pro | TP53 Pro/Pro: Profound effect on PC risk heavy smoker males (83) |
| Pro-inflammatory genes | |||
| Cyclooxygenase-2 (COX-2) | Converts free arachidonic acid into PGH2 (precursor of prostaglandins and thromboxanes) | Promoter region—765G/C—1195G/A—1290A/G | 765C: Increased COX2 promoter activity upon cigarette smoking in comparison to −765G allele (164). |
| DNA repair genes | |||
| X-ray repair cross-complementing group 2 (XRCC2) | Double-strand break repair gene | XRCC2 exon 3 (G/A) (Arg 188His), Arg/Arg, Arg/His, His/His | XRCC2 Arg188Arga: Increased risk of PC among ever-smokers in comparison to never-smokers (79,165). |
| XRCC3 | Double-strand break repair gene | XRCC3 exon 7 (C/T) Thr(241)Thra, Thr(241)Met, Met (241) Meta, XRCC3.241C/T | Combination of smoking and XRCC3 variant (XRCC3.241* and XRCC3 Thr241Met) associated with increased risk of PC (80) |
| Xeroderma pigmentosum group D (XPD)/(ERCC2) | Member of the human transcriptional initiation factor TFIIH with ATP-dependent helicase activity | XPD exon 10 (G/A) Asp(312)Asn and exon 23 (A/C) Lys(751)Gln | Asn(312)Asna: Reduced risk of PC in ever smokers in comparison to carriers of Asp(312)Aspa allele (81,166). |
| Aldehyde dehydrogenase 2 (ALDH2) | Catalyzes the chemical transformation of acetaldehyde to carboxylic acid in mitochondria | Glu504 homozygotes (ALDH2*1/1), Glu504/Lys504 (active enzyme) heterozygotes (ALDH2*1/2) (inactive enzyme) | The OR of smoking patients with ALDH2*1/2*2 polymorphism was more than 7-fold higher than that of non-smoking patients with the active form of ALDH2 (84). |
| ABO | A, B, and O glycosyl-transferases transfer GalNAc, Gal, and no sugar residue, respectively, to H histo-blood group antigen expressed by red blood cells, endothelial cells, and epithelial cells | Genotype derived from ABO, O, A, B, AB | In a joint model with smoking, current smokers with non-O blood type had an adjusted OR of 2.68 (95% CI, 2.03–3.54) compared with non-smokers of blood type O (167, 168). |
CS, Cigarette smoke; THF, 5,6,7,8-tetrahydrofolate; PGH2, prostaglandin H2; ERCC2, excision repair cross-complementing repair deficiency; GalNAc, N-acetyl-D-galactosamine; Gal, D-galactose; OR, odd ratio;
PC, pancreatic cancer.
*The variant alleles of the gene.
aNo amino acid change due to the alteration in the wobble nucleotide.
CS-induced Altered Signaling
Cancer research has generated a rich and complex body of knowledge, revealing cancer to be a disease involving dynamic changes in the signaling cascades. At least six major pathways must be disrupted for a transition of a normal cell into a tumor cell (85). These include self-sufficiency in growth signals, insensitivity to antigrowth signals, limitless replicative potential, evasion of apoptosis, sustained angiogenesis, tissue invasion, and metastasis. Despite the strong evidences gleaned from various epidemiological studies and genotoxic analysis for the correlation between PC incidence and cigarette smoking, the exact components responsible and the signal-transduction cascades involved require further examination. A better understanding of molecular events occurring during the CS-mediated initiation/development of PC may improve the management of patients, enabling early diagnosis in high-risk individuals and permitting the development of improved therapeutic approaches targeting specific genes and key molecules of specific pathways. Initiating events alone are not sufficient for the development of cancer. Therefore, understanding whether mixtures or specific compounds found in CS can contribute in promoting the progression phase of cancer is also very important.
Inflammatory Pathways
In experimental models, nicotine has been observed to incite an acute inflammatory reaction in the pancreas without the changes that are characteristic of chronic inflammation (86). However, frequent sessions of smoking-induced acute pancreatic inflammation may progress to chronic inflammation and might even cause chronic pancreatitis. Chronic inflammation is characterized by a shift in the cell types at the site of inflammation that can cause lasting and detrimental health effects (87,88). Thus, active and consistent cigarette smoking may eventually lead to chronic intra-pancreatic inflammation and the development of cancer within the inflamed tissue (89,90).
There is evidence that proinflammatory cytokines, chemokines, and their receptors are expressed in pancreatic cells eventually leading to the infiltration of immune cells within inflamed pancreatic tissues (91). The chemokine, CCL5 (C-C motif ligand 5) also known as RANTES (Regulated upon Activation, Normal T-cell Expressed, and Secreted), and one of its receptors, CCR5 (Chemokine (C-C motif) receptor 5), are believed to play a role in antitumor immunity through immune cell recruitment (92). Studies by Goecke et al. showed that CCR5 was significantly expressed by macrophages (91). Duell et al. investigated for the possible interaction between current active smoking and CCR5-32bp deletion. The studies suggested that intact CCR5 may offer pancreatic cells protection from the damaging effects of tobacco smoking (93).
The link between cigarette smoking and pancreatitis is supported by the results of various animal studies (28). Smoke enhances ethanol-induced pancreatic injury and accelerates the development and progression of chronic pancreatitis, which might further lead to the development of PC (94). The article by Wittel et al. sheds further light on this by providing experimental evidence that tobacco smoke leads to focal chronic inflammatory changes in the pancreas, increased turnover of the pancreatic digestive enzymes, and reduction of the antiprotease activity (6). A recent study by Song et al. examined the carcinogenic effects of an aqueous extract of CS (tobacco smoke, TS) and Snus (the Swedish variant of oral smokeless tobacco) in an elastase-IL-1β transgenic mouse model of chronic pancreatitis. The studies showed that both TS- and Snus-treated elastase-IL-1β mice developed significant pancreatic ductal epithelial flattening and severe glandular atrophy. Also, TS-elastase-IL-1β mice had an earlier onset and a greater extent of phenotypic changes, which were associated with upregulation of tumor necrosis factor-α, IL-6, and TGF-β (95).
Inflammation can be oncogenic through multiple molecular mechanisms (Figure 1). Inflamed tissues characteristically generate NO via inducible NO synthase (iNOS) and various other free radicals that carry the oncogenic potential of causing direct DNA and protein damage, promoting angiogenesis, inhibiting apoptosis and cellular repair functions (96). Various studies have shown an increased expression of iNOS and increased protein tyrosine nitration during the development and progression of PC (97,98). Also, an increased expression of iNOS and CS-induced pathogenesis in various cancers are significantly correlated (99,100). A recent study has shown that nicotine is able to enhance oxidative stress and CCK-stimulated amylase release in AR42J pancreatic acinar cells via the XOD pathway and that these events trigger pathophysiological changes in the pancreas (56). Studies by Tai et al. in a pluripotent rat liver epithelial stem cell line indicate that a distinct structural configuration of the methylanthracenes could be a potential etiological agent contributing to the epigenetic events of PC, such as an induced release of arachidonic acid (49). Cyclooxygenase enzymes catalyze a critical step in the conversion of arachidonic acid to prostaglandins, which are important mediators of acute and chronic inflammation (101). A recent study by Lazar et al. showed that cigarette smoking contribute to PC inflammation by inducing monocyte chemoattractant protein-1 and provided evidence for osteopontin (OPN) being a downstream effector of nicotine, capable of mediating these pro-inflammatory effects in PC cells (102).
Fig. 1.

Interconnected signaling cascades for CS-mediated pancreatic injury. CS produces various components, including nicotine, nitrosamines (NNK), and PAH that cause the pancreatic injury. (A) Both nicotine and NNK are selective agonists for α7nAChR, which upon binding causes the cell depolarization and lead to an enhanced influx of cations including calcium ions through voltage-dependent channels. Calcium ions mediate the nicotine entry into the pancreatic cells inducing altered exocrine pancreatic secretions associated with pancreatic injury. Enhanced calcium influx triggers the release and synthesis of excitatory neurotransmitters (adrenalin and noradrenalin) that activates the adenylyl cyclase downstream of Gαs-coupled receptors. GABA normally balances these effects by inhibiting adenylyl cyclase downstream of the Gαi-coupled GABA receptor through a α4β2nAChR-dependent mechanism. Both nicotine and NNK desensitizes the α4β2nAChR, hence inhibiting the release and synthesis of GABA and virtually shutting down all inhibitory GABA signaling. (B) NNK and nicotine leads to the stimulation of the cell proliferation, migration, and angiogenesis either by the direct activation of the β-adrenoreceptor-mediated signaling via cAMP/PKA/p-CREB or indirectly by regulating the release and synthesis of EGF, VEGF, or arachidonic acid and PKA-mediated transactivation of EGFR. This leads to the nicotine/NNK-mediated indirect induction of the Ras-Raf-MEK ERK pathway. (C) MAPK pathway and nicotine/α7nAChR-mediated JAK2/STAT3 pathway further stimulates the upregulation of the genes such as MYC, CYP, KRAS, FOS, and JUN in the nucleus, inhibiting the apoptosis of the cells and causing cells to proliferate and grow. (D) Nicotine elicits a prometastatic response in pancreatic cells by stimulation of osteopontin production through α7 nAChR-dependent mechanism. Nicotine can also induce through EGFR/AKT/NFκB-mediated pathways, changes in gene expression consistent with epithelial to mesenchymal transition (EMT). PAH also contributes to the cell metastasis by inducing the release of arachidonic acid and inhibiting the gap junctional intercellular communication. (E) The carcinogens NNK and PAH can be metabolically activated to intermediates that react with DNA, forming DNA adducts resulting in the mutation of crucial genes such as KRAS gene. If the DNA adducts are repaired by cellular repair enzymes, DNA is returned to its normal undamaged state. Nicotine is able to enhance the production of ROS, which plays an important role in inhibiting the DNA repair mechanism of the cell. Also, increase in the ROS has been directly linked to the lipid peroxidation leading to the pancreatic injury. Cells with damaged or mutated DNA can be removed by apoptosis, which is inhibited by nicotine as it regulates EGFR leading to activation of the serine threonine kinase, AKT, and other factors such as NFκB and Bcl2, causing decreased apoptosis. The red arrows merge the various pathways defining their specific role in modulating the functional properties of PC cells such as proliferation, angiogenesis, migration, EMT, and reduced apoptosis. AA, aromatic amines; AKT, cAMP, Cyclic adenosine monophosphate; COX-2, cyclooxygenase-2; CREB, “http://en.wikipedia.org/wiki/Cyclic_adenosine_monophosphate” \o “Cyclic adenosine monophosphate” cAMP response element-binding; EGFR, epidermal growth factor receptor; EMT, epithelial-mesenchymal transition; GABA, γ-aminobutyric acid; GPCRs, G‑protein coupled receptors; JAK2, Janus kinase 2; MAPK, mitogen activated protein kinase; nAChR, nicotinic acetylcholine receptor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone;OPN, osteopontin; PAH’s, polycyclic aromatic hydrocarbons; STAT3, signal transducer and activator of transcription 3; TGF-β, transforming growth factor- β; VEGF, Vascular endothelial growth factor.
Nicotinic Acetylcholine Receptor-mediated Pathways
The nicotinic acetylcholine receptor (nAChR), first characterized in 1970 as a membrane spanning protein and a ligand-gated ion channel, is known to be localized both in the neuro-muscular junctions as well as in a large variety of non-neuronal cells where they serve diverse functions (103). A very recent study by Sullivan et al. demonstrated the expression and regulation of nAChRs in PC (104). These receptors are usually activated by the neurotransmitter acetylcholine, but significant evidence exists in the literature suggesting that they are the primary site of nicotine action in the central nervous system (105,106). Several laboratories have demonstrated that prolonged exposure of mice and rats to nicotine and other nicotinic agonists produce a significant increase in the number of agonist binding sites in many brain regions (107,108). The binding of nicotine to nAChR causes a conformational change that either opens or closes the receptor ion channels, thereby changing the receptors functional state. If the nicotine remains bound for a longer time, a second conformational change occurs termed as desensitization in which the channel is closed. Each nAChR is made up of five subunits, arranged symmetrically around a central pore. These subunits belong to a multigene family and the different combinations of subunits results in a great functional diversity of the receptors.
The nAChRs have been demonstrated to play a key role in smoking-induced pathogenesis in various types of human cancers. Recent studies have shown that treatment of several PC and colon cancer cell lines with nicotine induces proliferation and metastasis in a receptor-dependent manner and that α7 is the main nAChR subunit that mediates this effect (109–111). Studies by Chen et al. have shown that nicotine causes α7nAChRs-mediated rapid activation of STAT3 and Erk1/2 leading to cell proliferation in human bladder cancer cells (112). Our recent studies have also demonstrated that α7nAChR modulate JAK2/STAT3 pathway in PC both in vitro and in vivo (unpublished data). The α7nAChR, being a pivotal subunit involved for the nicotine-mediated effects, has received profound interest as a valuable molecular target for cancer therapeutics (113,114). Recently, multiple single-nucleotide polymorphisms were identified in the gene clusters encoding for α3, α5, and β4nAChR subunits, which are associated with an increased risk for nicotine dependence and lung cancer (115). Also, studies by Dasgupta et al. have shown antiapoptotic effects of nicotine in non-small cell lung cancer (NSCLC) mediated by α3-containing nAChRs that imparts resistance against various chemotherapeutic agents (116).
Various in vitro and in vivo animal studies show that homo-pentameric α7nAChR-specific inhibitors, such as methyllycaconitine and α-bungarotoxin, can attenuate nicotine-induced proliferative, angiogenic, and metastatic effects on lung, colon, and bladder cancer cells. Studies have demonstrated that α9nAChR is critical in breast cancer and that several α9nAChR-specific antagonists (such as α-ImI, α-ImI, Vc1.1, RgIA, and It14a) produce an analgesic effect in many in vivo studies. Also, for cancer therapy, natural compounds such as garcinol have been found to inhibit the α9-nAChR signaling pathway, thereby blocking the nicotine and estrogen-induced breast cancer cell proliferation (117). Interestingly, it has also been shown in PC that nicotine-mediated upregulation of α7nAChRs and desensitization of α4β2nAChR in smokers shifts the balance in favor of α7nAChR signaling with strong stimulatory effects on cancer cells. On the other hand, γ-aminobutyric acid (GABA) balances these effects by inhibiting adenylyl cyclase downstream of the Gαi-coupled GABA receptor (GABAR) through a α4β2nAChR-dependent mechanism (118,119).
Recently, studies have reported that, similar to nicotine, NNK may also modulate the nAChRs, α7nAchR sensitization, and desensitization of the α4nAChR (120), causing increased MAPK/CREB signaling and a concomitant increase in stress neurotransmitters and decrease in inhibitory neurotransmitters (GABA) (118). Both nicotine and NNK are selective agonists for α7nAChR, which upon binding causes the cell depolarization and leads to an enhanced influx of cations including calcium ions through voltage-dependent channels. Enhanced calcium influx triggers the release and synthesis of excitatory neurotransmitters, which activates the adenylyl cyclase downstream of Gαs-coupled receptors. These findings suggest that nicotine and NNK-induced alterations in regulatory nAChRs may contribute to the development of smoking-associated PC by disturbing the balance between cancer stimulating and inhibiting neurotransmitters (118,121) (Figure 1).
Other Intracellular Pathways
Nicotine induces cell proliferation, invasion, and epithelial–mesenchymal transition (EMT) in a variety of human cancer cell lines (110). Studies by Chipitsyna et al. showed that nicotine significantly upregulates the OPN messenger RNA (mRNA) and protein secretion in PC and further activates MAPK signaling pathways that induce cell survival, proliferation, invasion, and metastasis (122). A recent study has shown that a pro-metastatic splice variant of OPN, OPNc, confers a migratory phenotype in PC (104). Later, the same group showed that matrix metallopeptidase-9 (MMP-9) mRNA levels were significantly higher in smokers compared with non-smokers in invasive PC lesions. Also, the vascular endothelial growth factor (VEGF) protein co-localized with MMP-9 and OPN in the malignant ducts correlated well with their higher levels in invasive PC lesions in smokers. Hence, this study proved that cigarette-smoking and nicotine-induced OPN plays a central role in mediating PC metastasis through the induction of MMP-9 and VEGF (123). Nicotine, through EGFR/AKT/NFκB-mediated pathways, can also induce changes in gene expression consistent with EMT, characterized by a reduction of epithelial markers like E-cadherin expression and concomitant increase in levels of mesenchymal proteins like vimentin and fibronectin (110). Nicotine increases the cellular proliferation of the AR42J PC cells by activating pERK-1/2 signaling. This process is also known to occur through an independent pathway, that is, the stimulation of the G-protein coupled receptor (GPCR)-mediated secretory response of nicotine (124). Also, studies by Dasgupta et al. showed that nicotine induces cell proliferation by activation of the Src and Rb-Raf-1 pathways upon stimulation of GPCRs. They illustrated that Src forms a complex with the scaffolding protein β-arrestin-1 and gets recruited to the nAChRs. The mitogenic effects of nicotine were mediated via the α7nAChR subunit and resulted in enhanced recruitment of E2F1 and Raf-1 on proliferative promoters (cdc25A and cdc6) and dissociation of Rb from these promoters, in NSCLC cell lines and human lung tumors, correlating with transcriptional activation and S-phase entry (125).
Studies have shown that NNK is also a selective agonist for β-adrenergic receptor and that it stimulates the MAP kinase pathway by activation of Src tyrosine kinase (126). Binding of NNK to the β-adrenergic receptor may also regulate the release and synthesis of endothelial growth factor (EGF), VEGF, or arachidonic acid that acts as a second messenger, leading to the formation of mitogenic metabolites that stimulate DNA synthesis and cell proliferation (126). Moreover, NNK and nicotine indirectly lead to the stimulation of the cell proliferation, migration, and angiogenesis through protein kinase A (PKA)-mediated transactivation of EGFR. The NNK-β-adrenergic receptor-mediated transactivation of the EGFR and phosphorylation of Erk1/2 in immortalized human pancreatic ductal epithelial cells contribute to the development of tobacco-mediated pancreatic carcinogenesis (127). This leads to the nicotine/NNK-mediated indirect induction of the Ras-Raf-MEK ERK pathway. MAPK-mediated JAK2/STAT3 pathway further stimulates the upregulation of the genes such as MYC, CYP, KRAS, FOS, JUN, etc., in the nucleus, inhibiting the apoptosis of the cells and causing cells to proliferate and grow (Figure 1).
The carcinogens NNK and PAH are metabolically activated to intermediates that react with DNA-forming DNA adducts. If the DNA adducts are repaired by cellular repair enzymes, DNA is returned to its normal undamaged state. Cells with damaged or mutated DNA can be removed by apoptosis, which is blocked by nicotine-mediated effects. Nicotine induces oxidative stress and activates NFκB leading to decrease in apoptosis in colon cancer cells (128). Several studies have shown that the NFκB pathway confers an antiapoptotic trait to the PC cells (129–131). Nicotine regulates EGFR leading to activation of the serine threonine kinase, AKT, and other factors such as NFκB, Bcl2, and inactivates XIAP and survivin, thereby causing decreased apoptosis. PAH also contributes to the cell metastasis by inducing the release of arachidonic acid and inhibiting the gap junctional intercellular communication. Experimental evidence suggest that upon implantation of a PAH component namely DMBA, into the area of the pancreatic head of mice, the mice quickly developed pancreatic duct alterations in a notch signaling-dependent manner (132). Thus, the individual components of CS have adverse effects on the pancreas by altering various processes such as increasing the proliferation rate, angiogenesis, migration potential, and EMT phenotype as well as decreasing the apoptosis of the pancreatic cells (Figure 1).
Future Implications and Clinical Perspectives
PC is a silent killer because it remains undiagnosed at an early stage due to lack of symptoms. Cigarette smoking is the most well-established environmental factor that has been associated with high risk of PC. To quit or avoid smoking remains the best prevention from this malignancy, but apart from that, there is an urgent need for the design of therapies that are more effective than the current regimens. For this, a deeper insight into the genetic alterations and the molecular mechanisms that contribute to the aggressive nature of PC is highly essential. This aforementioned review hints that the interplay between the susceptible polymorphisms, genotoxicity, and altered signaling pathways caused by the various constituents contained in CS contributes to the CS-induced pathogenesis. This knowledge will be helpful to target the specific key molecules and key genes that are altered in the CS-mediated regulatory pathways, hence inhibiting the cancer promoting events (Figure 2). Overall, our review has attempted for better understanding of the smoke-induced PC, which must overcome the limitations in PC therapeutics and provide a better survival benefit.
Fig. 2.

The various inhibitors/activators (shown in yellow boxes) against the key players involved in CS-mediated PC pathogenesis. (a) One of the therapies suggested is the maintenance between the release of α7nAChR-mediated stimulatory neurotransmitters (adrenalin, noradrenalin) and α4/β2nAchR-mediated inhibitory neurotransmitter (GABA). Thus, the desensitization of α7nAChR using antagonists such as α-bungarotoxin, α-conotoxin, cobratoxin, and atropine as well as sensitization of α4/β2 nAchR and stimulation of GABA-B-receptor by GABA or baclofen can attenuate the cAMP-dependent proliferative effects of nicotine/NNK. (b) Blockers of β-adrenergic receptors such as propranolol, ICI-118 551, and voltage-gated Ca2+ channels blockers might be yet another alternative for reversing the NNK/nicotine-mediated upregulation p-CREB/p-ERK1/2 pathways. (c) NNK/PAH-induced DNA adducts can be prevented using certain inhibitors such as methoxalen and some benzyl and phenylethyl isothiocyanates against both CYP2A6 and CYP2A13 enzymes. Phenyl isothiocyanate treatment also inhibits the generation of NNAL and decreases the NNK-mediated carcinogenic potential. (d) Further inhibitors against induced arachidonic acid metabolizing enzymes, such as ibuprofen, NSAIDS, and celecoxib (COX-2 inhibitors) and MK886 (5-lox inhibitor) may be used as chemopreventive agents for NNK/PAH-induced ductal pancreatic adenocarcinomas in smokers. The growing knowledge on the plausible strategies against the potential CS-mediated signaling cascades must prove to be helpful for developing advanced clinical and therapeutic strategies for a better survival benefit.
One of the evolving therapeutic strategies is the usage of α7nAChR as a molecular target for combating cancers (113,114). Previous studies involving lung adenocarcinoma cell lines have shown that the antagonists of α7nAChR, α-bungarotoxin or methyllycaconitine, can attenuate the proliferative effects of nicotine (133–135). The antagonists of α7nAChR might be used with caution because these receptors also regulate many vital cellular and organelle functions, such as inflammatory reactions and respiratory and cardiovascular functions, thereby leading to mental and psychological problems. Future studies involving the design of nAChR antagonists with improved selectivity might identify novel strategies for the treatment of tobacco-related PC.
In smokers with PC, the balance shifts in favor of the α7nAChR-mediated upregulation of stimulatory neurotransmitter, imparting strong stimulatory effects on cancer cells, with concurrent decrease in the inhibitory neurotransmitter, GABA (118). GABA or baclofen-mediated stimulation of the GABA-B-R inhibiting the isoproterenol-induced cyclic adenosine 3′,5′-monophosphate (cAMP) signaling suggests GABA-B-R as a potential target for the treatment and prevention of PC (136). Blockers of voltage-gated Ca2+ channels might be yet another alternative for desensitization of the hyperactive α7nAChR.
Experimental evidence has shown that the treatment of the adenocarcinoma cells with the β-adrenergic receptor blockers, such as propranolol, atenolol, or ICI-118551, reduces the mitogenic response to NNK. Studies by Hussein et al. have shown that propranolol prevents development of PC in hamsters by reversing the NNK-mediated upregulation of the α7nAChR, associated with significant inductions of p-CREB, p-ERK1/2, EGF, and VEGF (118). Subsequently, animal experiments demonstrated that phenyl isothiocyanate treatment, which inhibits the α-hydroxylation of NNK also inhibited the generation of NNAL and decreased its carcinogenic potential. However, this inhibitory treatment seems to be non-specific (137,138). Zhang et al. showed that β2-adrenergic and β1/2-adrenergic antagonists significantly suppressed the cell invasion and proliferation of PC cells in comparison to β1-adrenergic antagonist. Although the β1-adrenergic antagonist (metoprolol) inhibits the cAMP/PKA pathway, the β2-adrenergic antagonists (ICI-118551 and propranolol) suppressed both cAMP/PKA and Ras pathways, suggesting that they may be useful as novel preventive and therapeutic strategies for PC (139).
Antioxidants are molecules that, by definition, neutralize ROS. For example, in a study of 532 cases and 1701 age and sex-matched controls, consumption of fruits and vegetables, specifically green leafy vegetables, cruciferous vegetables, yellow vegetables, and beans/legumes, was shown to reduce PC risk by close to 50% (140). A study by von Weymarn et al. (141) reported that benzyl and phenylethyl isothiocyanates that are found in cruciferous vegetables such as broccoli and cabbage were effective competitive inhibitors of both CYP2A6 and CYP2A13 enzymes. Another inhibitor of CYP2A6, methoxalen, has been shown to be effective in increasing the bioavailability of nicotine in smokers, resulting in a decrease in the number of cigarettes they smoke per day (142). Chowdhury et al. demonstrated an enhanced proliferation and CCK-stimulated amylase release in AR42J cells by nicotine. Treatment with allopurinol, a XOD inhibitor, significantly suppressed these effects in PC. Allopurinol also ameliorates the caerulein-induced pancreatitis (143) and suppresses an important oxidant-generating system in granulocytes (144,145), scavenges hydroxyl radicals (146), and prevents irreversible loss of adenosine triphosphate (ATP) by inhibition of 5′-nucleotidase and facilitation of mitochondrial electron transport (96,147,148). Thus, treatment with XOD inhibitors might be useful in combating the ROS-mediated effects. These studies can be used as strategies to prevent or reduce the tobacco-mediated effects and may have the potential to increase the success of smoking cessation programs.
PC is the most lethal of all solid tumors partially because of its chemoresistance. Systemic chemotherapy still relies on a few drugs of which gemcitabine is now the standard drug for advanced PC (149). Unfortunately, studies till date, with gemcitabine alone, have produced unsatisfactory results with a low survival benefit (150). Many other chemotherapeutic agents have been examined, but only a few of them, such as oxaliplatin and irinotecan, have been consistently shown to have a beneficial effect, particularly as second line of chemotherapy as well as in combination with gemcitabine (151,152). Studies by Dasgupta et al. have shown that nicotine inhibits apoptosis induced by various chemotherapeutic drugs such as gemcitabine, cisplatin, and taxol by depletion of XIAP and survivin (125). The susceptible polymorphisms, along with CS-mediated genotoxicity and altered signaling pathways, aggravate the PC risk and chemotherapeutic resistance (153–157). This issue deserves intensive investigation and the future challenge of chemotherapy against PC relies on the identification of molecular and genetic markers that are predictive of response. It is to be expected that combinatorial or sequential treatment with different drugs might prove to be more promising with a better survival rate. However, the current knowledge of the side effects for the various inhibitors is limited and requires further investigation.
We foresee PC research and a therapeutic outlook toward it developing into much understandable form. This can be achieved thorough in-depth knowledge of biology of pancreatic tumor along with its hostile tumor microenvironment, association with high-impact risk factors and carcinogenesis as reflected through several genetic and signaling alterations. Keeping in mind that smoking is a highly potential but modifiable risk factor for PC, a holistic clarity and solid basis in the field of smoking-mediated PC carcinogenesis would lead to the prevention and cure of this deadly malignancy.
Funding
The authors on this work are supported, in part, by grants from the National Institutes of Health (RO1 CA133774, RO1 CA78590, UO1 CA111294, RO1 CA131944, P50 CA127297 and U54 CA163120).
Acknowledgements
We thank Ms. Kristi L.Berger for editing the manuscript.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- ALDH2
aldehyde dehydrogenase 2
- CCR5
Chemokine (C-C motif) receptor 5
- CYP2A6
cytochrome P450 2A6
- DMBA
dimethylbenzanthracene
- GABA
γ-aminobutyric acid
- HCA
heterocyclic amine
- MAPK
mitogen-activated protein kinase
- MTHFR
methylenetetrahydrofolate reductase
- nAChR
nicotinic acetylcholine receptor
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NNAL
4-(methylnitrosamino)-1-(3-pyridyl)-1- butanol
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NO
nitric oxide
- OPN
osteopontin
- PC
pancreatic cancer
Footnotes
Funding
Conflict of Interest Statement: None declared.
References
- 1. Lowenfels A.B., et al. (2005). Risk factors for pancreatic cancer. J. Cell. Biochem. 95 649–656 [DOI] [PubMed] [Google Scholar]
- 2. Lowenfels A.B., et al. (2006). Epidemiology and risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 20 197–209 [DOI] [PubMed] [Google Scholar]
- 3. Freitas D., et al. (2009). Medical management of pancreatic adenocarcinoma. Pancreatology 9 223–232 [DOI] [PubMed] [Google Scholar]
- 4. Mendieta Z.H., et al. (2009). Limitations in improving detection of pancreatic adenocarcinoma Future Oncol. 5 657–668 [DOI] [PubMed] [Google Scholar]
- 5. Qiu D., et al. JACC Study Group (2005). Overview of the epidemiology of pancreatic cancer focusing on the JACC Study. J. Epidemiol. 15 Suppl 2 S157–S167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wittel U.A., et al. (2006). Chronic pancreatic inflammation induced by environmental tobacco smoke inhalation in rats. Am. J. Gastroenterol. 101 148–159 [DOI] [PubMed] [Google Scholar]
- 7. Wittel U.A., et al. (2008). Cigarette smoke-induced pancreatic damage: experimental data. Langenbecks. Arch. Surg. 393 581–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bryant A., et al. (2007). Differences in epidemiology, histology, and survival between cigarette smokers and never-smokers who develop non-small cell lung cancer. Chest 132 185–192 [DOI] [PubMed] [Google Scholar]
- 9. Fuchs C.S., et al. (1996). A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch. Intern. Med. 156 2255–2260 [PubMed] [Google Scholar]
- 10. Mack T.M., et al. (1986). Pancreas cancer and smoking, beverage consumption, and past medical history. J. Natl. Cancer Inst. 76 49–60 [PubMed] [Google Scholar]
- 11. Silverman D.T., et al. (1994). Cigarette smoking and pancreas cancer: a case-control study based on direct interviews. J. Natl. Cancer Inst. 86 1510–1516 [DOI] [PubMed] [Google Scholar]
- 12. Iodice S., et al. (2008). Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks. Arch. Surg. 393 535–545 [DOI] [PubMed] [Google Scholar]
- 13. Vrieling A., et al. (2010). Cigarette smoking, environmental tobacco smoke exposure and pancreatic cancer risk in the European Prospective Investigation into Cancer and Nutrition. Int. J. Cancer 126 2394–2403 [DOI] [PubMed] [Google Scholar]
- 14. Talamini R., et al. (2010). Tobacco smoking, alcohol consumption and pancreatic cancer risk: a case-control study in Italy. Eur. J. Cancer 46, 370–376 [DOI] [PubMed] [Google Scholar]
- 15. Boyle P., et al. (1996). Cigarette smoking and pancreas cancer: a case control study of the search programme of the IARC. Int. J. Cancer 67, 63–71 [DOI] [PubMed] [Google Scholar]
- 16. Raimondi S., et al. (2007). Early onset pancreatic cancer: evidence of a major role for smoking and genetic factors. Cancer Epidemiol. Biomarkers Prev. 16 1894–1897 [DOI] [PubMed] [Google Scholar]
- 17. Rebours V., et al. (2008). Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am. J. Gastroenterol. 103 111–119 [DOI] [PubMed] [Google Scholar]
- 18. Stewart S.L., et al. Centers for Disease Control and Prevention (CDC) (2008). Surveillance for cancers associated with tobacco use–United States, 1999-2004. MMWR. Surveill. Summ. 57 1–33 [PubMed] [Google Scholar]
- 19. Arnold L.D., et al. (2009). Are racial disparities in pancreatic cancer explained by smoking and overweight/obesity? Cancer Epidemiol. Biomarkers Prev. 18, 2397–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cerami C., et al. (1997). Tobacco smoke is a source of toxic reactive glycation products. Proc. Natl. Acad. Sci. U.S.A. 94 13915–13920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kodama M., et al. (1997). Free radical chemistry of cigarette smoke and its implication in human cancer. Anticancer Res. 17 433–437 [PubMed] [Google Scholar]
- 22. Pryor W.A., et al. (1993). Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. N. Y. Acad. Sci. 686 12–27; discussion 27 [DOI] [PubMed] [Google Scholar]
- 23. Hoffmann D., et al. (1993). A study of tobacco carcinogenesis: effect of the fat content of the diet on the carcinogenic activity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res. 53 2758–2761 [PubMed] [Google Scholar]
- 24. Stolerman I.P., et al. (1995). The scientific case that nicotine is addictive. Psychopharmacology (Berl.) 117 2–10; discussion 14 [DOI] [PubMed] [Google Scholar]
- 25. Majumdar A.P., et al. (1985). Nicotine stimulation of protein secretion from isolated rat pancreatic acini. Am. J. Physiol. 248 G158–G163 [DOI] [PubMed] [Google Scholar]
- 26. Bersch V.P., et al. (2009). Effect of nicotine and cigarette smoke on an experimental model of intraepithelial lesions and pancreatic adenocarcinoma induced by 7,12-dimethylbenzanthracene in mice. Pancreas 38, 65–70 [DOI] [PubMed] [Google Scholar]
- 27. Jianyu H., et al. (2009). Evidence for cigarette smoke-induced oxidative stress in the rat pancreas Inhal. Toxicol. 21 1007–1012 [DOI] [PubMed] [Google Scholar]
- 28. Chowdhury P., et al. (1992). Induction of pancreatic acinar pathology via inhalation of nicotine. Proc. Soc. Exp. Biol. Med. 201, 159–164 [DOI] [PubMed] [Google Scholar]
- 29. Brown P. (1976). The influence of smoking on pancreatic function in man. Med. J. Aust. 2 290–293 [DOI] [PubMed] [Google Scholar]
- 30. Sliwinska-Mosson M., et al. (2005). [Influence of tobacco smoking on lipase activity in patients with pancreatitis]. Prz. Lek. 62 1058–1061 [PubMed] [Google Scholar]
- 31. Milnerowicz H., et al. (2004). [Effect of tobacco smoking on amylase activity in patients with pancreatitis]. Prz. Lek. 61 1071–1072 [PubMed] [Google Scholar]
- 32. Hecht S.S., et al. (1991). N-nitroso compounds and tobacco-induced cancers in man. IARC Sci. Publ. 105, 54–61 [PubMed] [Google Scholar]
- 33. Prokopczyk B., et al. (2002). Identification of tobacco-derived compounds in human pancreatic juice. Chem. Res. Toxicol. 15 677–685 [DOI] [PubMed] [Google Scholar]
- 34. Schuller H.M. (2002). Mechanisms of smoking-related lung and pancreatic adenocarcinoma development. Nat. Rev. Cancer 2 455–463 [DOI] [PubMed] [Google Scholar]
- 35. Rivenson A., et al. (1988). Induction of lung and exocrine pancreas tumors in F344 rats by tobacco-specific and Areca-derived N-nitrosamines. Cancer Res. 48 6912–6917 [PubMed] [Google Scholar]
- 36. Trushin N., et al. (2008). The tobacco carcinogen NNK is stereoselectively reduced by human pancreatic microsomes and cytosols. Langenbecks. Arch. Surg. 393 571–579 [DOI] [PubMed] [Google Scholar]
- 37. Upadhyaya P., et al. (1999). Tumorigenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers and metabolites in the A/J mouse. Carcinogenesis 20 1577–1582 [DOI] [PubMed] [Google Scholar]
- 38. Zhang S., et al. (2009). Analysis of pyridyloxobutyl and pyridylhydroxybutyl DNA adducts in extrahepatic tissues of F344 rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 22 926–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Severson R.F., et al. (1976). Gas chromatographic quantitation of polynuclear aromatic hydrocarbons in tobacco smoke. Anal. Chem. 48 1866–1872 [DOI] [PubMed] [Google Scholar]
- 40. Upham B.L., et al. (2008). Tumor promoting properties of a cigarette smoke prevalent polycyclic aromatic hydrocarbon as indicated by the inhibition of gap junctional intercellular communication via phosphatidylcholine-specific phospholipase C. Cancer Sci. 99, 696–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Black O., Jr, et al. (1981). Metabolism of a carcinogen (7,12-dimethylbenzanthracene) in the pancreas of the Long-Evans rat. Res. Commun. Chem. Pathol. Pharmacol. 33 103–118 [PubMed] [Google Scholar]
- 42. Jones S., et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321 1801–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fryzek J.P., et al. (2006). The association between selected risk factors for pancreatic cancer and the expression of p53 and K-ras codon 12 mutations. Int. J. Gastrointest. Cancer 37 139–145 [DOI] [PubMed] [Google Scholar]
- 44. Hruban R.H., et al. (1993). K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol. 143 545–554 [PMC free article] [PubMed] [Google Scholar]
- 45. Berger D.H., et al. (1999). Mutational activation of K-ras in nonneoplastic exocrine pancreatic lesions in relation to cigarette smoking status. Cancer 85 326–332 [DOI] [PubMed] [Google Scholar]
- 46. Li D., et al. (2002). DNA adducts, genetic polymorphisms, and K-ras mutation in human pancreatic cancer. Mutat. Res. 513 37–48 [DOI] [PubMed] [Google Scholar]
- 47. Georgiadis P., et al. (1991). Nitrosamine-induced cancer: selective repair and conformational differences between O6-methylguanine residues in different positions in and around codon 12 of rat H-ras. Cancer Res. 51 5843–5850 [PubMed] [Google Scholar]
- 48. Li D. (2001). Molecular epidemiology of pancreatic cancer. Cancer J. 7 259–265 [PubMed] [Google Scholar]
- 49. Tai M.H., et al. (2007). Cigarette smoke components inhibited intercellular communication and differentiation in human pancreatic ductal epithelial cells. Int. J. Cancer 120 1855–1862 [DOI] [PubMed] [Google Scholar]
- 50. Pfeifer G.P., et al. (2002). Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 21 7435–7451 [DOI] [PubMed] [Google Scholar]
- 51. Yoon J.H., et al. (2001). Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Res. 61 7110–7117 [PubMed] [Google Scholar]
- 52. Foster J.R., et al. (1993). Induction of drug-metabolizing enzymes in human pancreatic cancer and chronic pancreatitis. J. Pathol. 169 457–463 [DOI] [PubMed] [Google Scholar]
- 53. Shimada T., et al. (2006). Inhibition of human cytochrome P450 1A1-, 1A2-, and 1B1-mediated activation of procarcinogens to genotoxic metabolites by polycyclic aromatic hydrocarbons. Chem. Res. Toxicol. 19 288–294 [DOI] [PubMed] [Google Scholar]
- 54. Messina E.S., et al. (1997). A major role for CYP2A6 in nicotine C-oxidation by human liver microsomes. J. Pharmacol. Exp. Ther. 282 1608–1614 [PubMed] [Google Scholar]
- 55. Nakajima M., et al. (1996). Role of human cytochrome P4502A6 in C-oxidation of nicotine. Drug Metab. Dispos. 24 1212–1217 [PubMed] [Google Scholar]
- 56. Chowdhury P., et al. (2002). Pathophysiological effects of nicotine on the pancreas: an update. Exp. Biol. Med. (Maywood) 227 445–454 [DOI] [PubMed] [Google Scholar]
- 57. Hecht S.S. (1999). DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 424 127–142 [DOI] [PubMed] [Google Scholar]
- 58. Eaton D.L., et al. (1995). Role of cytochrome P4501A2 in chemical carcinogenesis: implications for human variability in expression and enzyme activity. Pharmacogenetics 5 259–274 [DOI] [PubMed] [Google Scholar]
- 59. Li D., et al. (2006). Polymorphisms of cytochrome P4501A2 and N-acetyltransferase genes, smoking, and risk of pancreatic cancer. Carcinogenesis 27 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mori Y., et al. (1995). Effect of cigarette smoke on the mutagenic activation of various carcinogens in hamster. Mutat. Res. 346 1–8 [DOI] [PubMed] [Google Scholar]
- 61. Blackford A., et al. (2009). Genetic mutations associated with cigarette smoking in pancreatic cancer. Cancer Res. 69 3681–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kaino M., et al. (1999). Detection of K-ras and p53 gene mutations in pancreatic juice for the diagnosis of intraductal papillary mucinous tumors. Pancreas 18 294–299 [DOI] [PubMed] [Google Scholar]
- 63. Brennan J.A., et al. (1995). Association between cigarette smoking and mutation of the p53 gene in squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 332 712–717 [DOI] [PubMed] [Google Scholar]
- 64. Conway K., et al. (2002). Prevalence and spectrum of p53 mutations associated with smoking in breast cancer. Cancer Res. 62 1987–1995 [PubMed] [Google Scholar]
- 65. Nakayama T., et al. (1985). Cigarette smoke induces DNA single-strand breaks in human cells. Nature 314 462–464 [DOI] [PubMed] [Google Scholar]
- 66. Seree E.M., et al. (1996). High inducibility of mouse renal CYP2E1 gene by tobacco smoke and its possible effect on DNA single strand breaks. Biochem. Biophys. Res. Commun. 219 429–434 [DOI] [PubMed] [Google Scholar]
- 67. Karube T., et al. (1989). Analyses of transplacentally induced sister chromatid exchanges and micronuclei in mouse fetal liver cells following maternal exposure to cigarette smoke. Cancer Res. 49 3550–3552 [PubMed] [Google Scholar]
- 68. DeMarini D.M. (2004). Genotoxicity of tobacco smoke and tobacco smoke condensate: a review. Mutat. Res. 567 447–474 [DOI] [PubMed] [Google Scholar]
- 69. Ando M., et al. (2003). Association of CYP2A6 gene deletion with cigarette smoking status in Japanese adults. J. Epidemiol. 13 176–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schoedel K.A., et al. (2004). Ethnic variation in CYP2A6 and association of genetically slow nicotine metabolism and smoking in adult Caucasians. Pharmacogenetics 14, 615–626 [DOI] [PubMed] [Google Scholar]
- 71. Chen J., et al. (2010). Genetic variants of p21 and p27 and pancreatic cancer risk in non-Hispanic Whites: a case-control study. Pancreas 39, 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hahn S.A., et al. (2003). BRCA2 germline mutations in familial pancreatic carcinoma. J. Natl. Cancer Inst. 95 214–221 [DOI] [PubMed] [Google Scholar]
- 73. Li D., et al. (2011). Association between somatostatin receptor 5 gene polymorphisms and pancreatic cancer risk and survival. Cancer 117 2863–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wolpin B.M., et al. (2007). Circulating insulin-like growth factor binding protein-1 and the risk of pancreatic cancer. Cancer Res. 67, 7923–7928 [DOI] [PubMed] [Google Scholar]
- 75. Suzuki H., et al. (2008). Effect of insulin-like growth factor gene polymorphisms alone or in interaction with diabetes on the risk of pancreatic cancer. Cancer Epidemiol. Biomarkers Prev. 17 3467–3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wheatley-Price P., et al. (2008). Myeloperoxidase and superoxide dismutase polymorphisms are associated with an increased risk of developing pancreatic adenocarcinoma. Cancer 112, 1037–1042 [DOI] [PubMed] [Google Scholar]
- 77. Kaklamani V.G., et al. (2003). TGFBR1*6A and cancer risk: a meta-analysis of seven case-control studies. J. Clin. Oncol. 21 3236–3243 [DOI] [PubMed] [Google Scholar]
- 78. Duell E.J., et al. (2002). A population-based study of the Arg399Gln polymorphism in X-ray repair cross- complementing group 1 (XRCC1) and risk of pancreatic adenocarcinoma. Cancer Res. 62 4630–4636 [PubMed] [Google Scholar]
- 79. Jiao L., et al. (2006). Selected polymorphisms of DNA repair genes and risk of pancreatic cancer. Cancer Detect. Prev. 30 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Duell E.J., et al. (2008). Detecting pathway-based gene-gene and gene-environment interactions in pancreatic cancer. Cancer Epidemiol. Biomarkers Prev. 17 1470–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jiao L., et al. (2007). The XPD Asp312Asn and Lys751Gln polymorphisms, corresponding haplotype, and pancreatic cancer risk. Cancer Lett. 245 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Suzuki H., et al. (2008). Interaction of the cytochrome P4501A2, SULT1A1 and NAT gene polymorphisms with smoking and dietary mutagen intake in modification of the risk of pancreatic cancer. Carcinogenesis 29 1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sonoyama T., et al. (2011). TP53 codon 72 polymorphism is associated with pancreatic cancer risk in males, smokers and drinkers. Mol. Med. Report. 4 489–495 [DOI] [PubMed] [Google Scholar]
- 84. Miyasaka K., et al. (2005). Inactive aldehyde dehydrogenase-2 increased the risk of pancreatic cancer among smokers in a Japanese male population. Pancreas 30 95–98 [DOI] [PubMed] [Google Scholar]
- 85. Hanahan D., et al. (2011). Hallmarks of cancer: the next generation. Cell 144 646–674 [DOI] [PubMed] [Google Scholar]
- 86. Malfertheiner P., et al. (2006). Smoking–a trigger for chronic inflammation and cancer development in the pancreas. Am. J. Gastroenterol. 101 160–162 [DOI] [PubMed] [Google Scholar]
- 87. Macarthur M., et al. (2004). Inflammation and Cancer II. Role of chronic inflammation and cytokine gene polymorphisms in the pathogenesis of gastrointestinal malignancy. Am. J. Physiol. Gastrointest. Liver Physiol. 286 G515–G520 [DOI] [PubMed] [Google Scholar]
- 88. Whitcomb D.C. (2004). Inflammation and Cancer V. Chronic pancreatitis and pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 287 G315–G319 [DOI] [PubMed] [Google Scholar]
- 89. Coussens L.M., et al. (2002). Inflammation and cancer. Nature 420 860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hussain S.P., et al. (2003). Radical causes of cancer. Nat. Rev. Cancer 3, 276–285 [DOI] [PubMed] [Google Scholar]
- 91. Goecke H., et al. (2000). Macrophages infiltrating the tissue in chronic pancreatitis express the chemokine receptor CCR5. Surgery 128 806–814 [DOI] [PubMed] [Google Scholar]
- 92. Mulé J.J., et al. (1996). RANTES secretion by gene-modified tumor cells results in loss of tumorigenicity in vivo: role of immune cell subpopulations. Hum. Gene Ther. 7 1545–1553 [DOI] [PubMed] [Google Scholar]
- 93. Duell E.J., et al. (2006). Inflammation, genetic polymorphisms in proinflammatory genes TNF-A, RANTES, and CCR5, and risk of pancreatic adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 15 726–731 [DOI] [PubMed] [Google Scholar]
- 94. Hartwig W., et al. (2000). Cigarette smoke enhances ethanol-induced pancreatic injury. Pancreas 21, 272–278 [DOI] [PubMed] [Google Scholar]
- 95. Song Z., et al. (2010). Potential carcinogenic effects of cigarette smoke and Swedish moist snuff on pancreas: a study using a transgenic mouse model of chronic pancreatitis. Lab. Invest. 90 426–435 [DOI] [PubMed] [Google Scholar]
- 96. Dong M., et al. (2006). Development of enzymatic probes of oxidative and nitrosative DNA damage caused by reactive nitrogen species. Mutat. Res. 594 120–134 [DOI] [PubMed] [Google Scholar]
- 97. Franco L., et al. (2004). Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in pancreatic cancer. Prostaglandins Other Lipid Mediat. 73 51–58 [DOI] [PubMed] [Google Scholar]
- 98. Vickers S.M., et al. (1999). Association of increased immunostaining for inducible nitric oxide synthase and nitrotyrosine with fibroblast growth factor transformation in pancreatic cancer. Arch. Surg. 134 245–251 [DOI] [PubMed] [Google Scholar]
- 99. Chen G.G., et al. (2008). Increased inducible nitric oxide synthase in lung carcinoma of smokers. Cancer 112 372–381 [DOI] [PubMed] [Google Scholar]
- 100. Shen J., et al. (2004). A novel genetic polymorphism of inducible nitric oxide synthase is associated with an increased risk of gastric cancer. World J. Gastroenterol. 10 3278–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schlosser W., et al. (2002). Cyclooxygenase-2 is overexpressed in chronic pancreatitis. Pancreas 25 26–30 [DOI] [PubMed] [Google Scholar]
- 102. Lazar M., et al. (2010). Induction of monocyte chemoattractant protein-1 by nicotine in pancreatic ductal adenocarcinoma cells: role of osteopontin. Surgery 148, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wessler I., et al. (2008). Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br. J. Pharmacol. 154 1558–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sullivan J., et al. (2009). Expression of a prometastatic splice variant of osteopontin, OPNC, in human pancreatic ductal adenocarcinoma. Surgery 146, 232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schwartz R.D., et al. (1983). Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science 220 214–216 [DOI] [PubMed] [Google Scholar]
- 106. Wonnacott S. (1990). The paradox of nicotinic acetylcholine receptor upregulation by nicotine. Trends Pharmacol. Sci. 11 216–219 [DOI] [PubMed] [Google Scholar]
- 107. Marks M.J., et al. (1983). Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J. Pharmacol. Exp. Ther. 226 817–825 [PubMed] [Google Scholar]
- 108. Marks M.J., et al. (1985). Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J. Pharmacol. Exp. Ther. 235 619–628 [PubMed] [Google Scholar]
- 109. Chowdhury P., et al. (2008). A cell-based approach to study changes in the pancreas following nicotine exposure in an animal model of injury. Langenbecks. Arch. Surg. 393 547–555 [DOI] [PubMed] [Google Scholar]
- 110. Dasgupta P., et al. (2009). Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 124 36–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Karlin A., et al. (1995). Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron 15 1231–1244 [DOI] [PubMed] [Google Scholar]
- 112. Chen R.J., et al. (2008). Rapid activation of Stat3 and ERK1/2 by nicotine modulates cell proliferation in human bladder cancer cells. Toxicol. Sci. 104 283–293 [DOI] [PubMed] [Google Scholar]
- 113. Egleton R.D., et al. (2008). Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol. Sci. 29 151–158 [DOI] [PubMed] [Google Scholar]
- 114. Paleari L., et al. (2009). Inhibition of nonneuronal alpha7-nicotinic receptor for lung cancer treatment. Am. J. Respir. Crit. Care Med. 179 1141–1150 [DOI] [PubMed] [Google Scholar]
- 115. Improgo M.R., et al. (2010). From smoking to lung cancer: the CHRNA5/A3/B4 connection. Oncogene 29 4874–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dasgupta P., et al. (2006). Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc. Natl. Acad. Sci. U.S.A. 103 6332–6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wu C.H., et al. (2011). Nicotinic acetylcholine receptor-based blockade: applications of molecular targets for cancer therapy. Clin. Cancer Res. 17 3533–3541 [DOI] [PubMed] [Google Scholar]
- 118. Al-Wadei H.A., et al. (2009). Nicotine stimulates pancreatic cancer xenografts by systemic increase in stress neurotransmitters and suppression of the inhibitory neurotransmitter gamma-aminobutyric acid. Carcinogenesis 30 506–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Schuller H.M., et al. (2010). Neurotransmitter receptors as central regulators of pancreatic cancer. Future Oncol. 6 221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Al-Wadei H.A., et al. (2010). Chronic exposure to estrogen and the tobacco carcinogen NNK cooperatively modulates nicotinic receptors in small airway epithelial cells. Lung Cancer 69 33–39 [DOI] [PubMed] [Google Scholar]
- 121. Schuller H.M. (2009). Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 9 195–205 [DOI] [PubMed] [Google Scholar]
- 122. Chipitsyna G., et al. (2009). Induction of osteopontin expression by nicotine and cigarette smoke in the pancreas and pancreatic ductal adenocarcinoma cells. Int. J. Cancer 125 276–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lazar M., et al. (2010). Involvement of osteopontin in the matrix-degrading and proangiogenic changes mediated by nicotine in pancreatic cancer cells. J. Gastrointest. Surg. 14 1566–1577 [DOI] [PubMed] [Google Scholar]
- 124. Bose C., et al. (2005). Activation of p-ERK1/2 by nicotine in pancreatic tumor cell line AR42J: effects on proliferation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 289 G926–G934 [DOI] [PubMed] [Google Scholar]
- 125. Dasgupta P., et al. (2006). Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Invest. 116 2208–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Weddle D.L., et al. (2001). Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis 22 473–479 [DOI] [PubMed] [Google Scholar]
- 127. Askari M.D., et al. (2005). The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J. Cancer Res. Clin. Oncol. 131 639–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Crowley-Weber C.L., et al. (2003). Nicotine increases oxidative stress, activates NF-kappaB and GRP78, induces apoptosis and sensitizes cells to genotoxic/xenobiotic stresses by a multiple stress inducer, deoxycholate: relevance to colon carcinogenesis. Chem. Biol. Interact. 145 53–66 [DOI] [PubMed] [Google Scholar]
- 129. De P.D., et al. (2007). Effect of iNOS and NF-kappaB gene silencing on beta-cell survival and function J. Drug Target 15 358–369 [DOI] [PubMed] [Google Scholar]
- 130. Rengifo-Cam W., et al. (2007). Antiapoptotic effects of progastrin on pancreatic cancer cells are mediated by sustained activation of nuclear factor-{kappa}B. Cancer Res. 67 7266–7274 [DOI] [PubMed] [Google Scholar]
- 131. Zhang Z., et al. (2006). NF-kappaB, inflammation and pancreatic carcinogenesis: NF-kappaB as a chemoprevention target (review). Int. J. Oncol. 29 185–192 [PubMed] [Google Scholar]
- 132. Kimura K., et al. (2007). Activation of Notch signaling in tumorigenesis of experimental pancreatic cancer induced by dimethylbenzanthracene in mice. Cancer Sci. 98 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Conroy W.G., et al. (1998). Nicotinic receptor subtypes in the developing chick brain: appearance of a species containing the alpha4, beta2, and alpha5 gene products. Mol. Pharmacol. 53 392–401 [DOI] [PubMed] [Google Scholar]
- 134. Cui C., et al. (2003). The beta3 nicotinic receptor subunit: a component of alpha-conotoxin MII-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. J. Neurosci. 23 11045–11053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Klink R., et al. (2001). Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J. Neurosci. 21 1452–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Schuller H.M., et al. (2008). GABA B receptor is a novel drug target for pancreatic cancer. Cancer 112 767–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Morse M.A., et al. (1989). Effects of aromatic isothiocyanates on tumorigenicity, O6-methylguanine formation, and metabolism of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in A/J mouse lung. Cancer Res. 49 2894–2897 [PubMed] [Google Scholar]
- 138. Staretz M.E., et al. (1995). Effects of phenethyl isothiocyanate on the tissue distribution of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and metabolites in F344 rats. Cancer Res. 55 5580–5588 [PubMed] [Google Scholar]
- 139. Zhang D., et al. (2010). ß2-adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NF?B and AP-1. Cancer Biol. Ther. 10 19–29 [DOI] [PubMed] [Google Scholar]
- 140. Chan J.M., et al. (2005). Vegetable and fruit intake and pancreatic cancer in a population-based case-control study in the San Francisco bay area. Cancer Epidemiol. Biomarkers Prev. 14 2093–2097 [DOI] [PubMed] [Google Scholar]
- 141. von Weymarn L.B., et al. (2006). Effects of benzyl and phenethyl isothiocyanate on P450s 2A6 and 2A13: potential for chemoprevention in smokers. Carcinogenesis 27 782–790 [DOI] [PubMed] [Google Scholar]
- 142. Sellers E.M., et al. (2000). Inhibition of cytochrome P450 2A6 increases nicotine’s oral bioavailability and decreases smoking. Clin. Pharmacol. Ther. 68, 35–43 [DOI] [PubMed] [Google Scholar]
- 143. Wisner J., et al. (1988). Evidence for a role of oxygen derived free radicals in the pathogenesis of caerulein induced acute pancreatitis in rats. Gut 29 1516–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Das D.K., et al. (1987). Role of xanthine oxidase inhibitor as free radical scavenger: a novel mechanism of action of allopurinol and oxypurinol in myocardial salvage. Biochem. Biophys. Res. Commun. 148 314–319 [DOI] [PubMed] [Google Scholar]
- 145. Grisham M.B., et al. (1986). Xanthine oxidase and neutrophil infiltration in intestinal ischemia. Am. J. Physiol. 251, G567–G574 [DOI] [PubMed] [Google Scholar]
- 146. Moorhouse P.C., et al. (1987). Allopurinol and oxypurinol are hydroxyl radical scavengers. FEBS Lett. 213 23–28 [DOI] [PubMed] [Google Scholar]
- 147. Fox I.H. (1976). Inborn errors of purine and pyrimidine metabolism. Clin. Perinatol. 3 133–140 [PubMed] [Google Scholar]
- 148. Peterson D.A., et al. (1986). Allopurinol can act as an electron transfer agent. Is this relevant during reperfusion injury? Biochem. Biophys. Res. Commun. 137 76–79 [DOI] [PubMed] [Google Scholar]
- 149. Abbruzzese J.L. (2002). New applications of gemcitabine and future directions in the management of pancreatic cancer. Cancer 95 941–945 [DOI] [PubMed] [Google Scholar]
- 150. Li D., et al. (2004). Pancreatic cancer. Lancet 363 1049–1057 [DOI] [PubMed] [Google Scholar]
- 151. Louvet C., et al. GERCOR; GISCAD (2005). Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J. Clin. Oncol. 23 3509–3516 [DOI] [PubMed] [Google Scholar]
- 152. Rocha Lima C.M., et al. (2004). Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. J. Clin. Oncol. 22 3776–3783 [DOI] [PubMed] [Google Scholar]
- 153. Chefrour M., et al. (2010). Erlotinib in combination with capecitabine (5’dFUR) in resistant pancreatic cancer cell lines. J. Chemother. 22 129–133 [DOI] [PubMed] [Google Scholar]
- 154. Gautschi O., et al. (2006). Cyclin D1 (CCND1) A870G gene polymorphism modulates smoking-induced lung cancer risk and response to platinum-based chemotherapy in non-small cell lung cancer (NSCLC) patients. Lung Cancer 51, 303–311 [DOI] [PubMed] [Google Scholar]
- 155. Gong X.G., et al. (2010). Gemcitabine resistance induced by interaction between alternatively spliced segment of tenascin-C and annexin A2 in pancreatic cancer cells. Biol. Pharm. Bull. 33 1261–1267 [DOI] [PubMed] [Google Scholar]
- 156. Rosell R., et al. (2003). Nucleotide excision repair pathways involved in Cisplatin resistance in non-small-cell lung cancer. Cancer Control 10 297–305 [DOI] [PubMed] [Google Scholar]
- 157. Rosell R., et al. (2003). Influence of genetic markers on survival in non-small cell lung cancer. Drugs Today 39 775–786 [DOI] [PubMed] [Google Scholar]
- 158. Mohelnikova-Duchonova B., et al. (2010). CYP2A13, ADH1B, and ADH1C gene polymorphisms and pancreatic cancer risk. Pancreas 39 144–148 [DOI] [PubMed] [Google Scholar]
- 159. Jiao L., et al. (2007). Haplotype of N-acetyltransferase 1 and 2 and risk of pancreatic cancer. Cancer Epidemiol. Biomarkers Prev. 16 2379–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Duell E.J., et al. (2002). A population-based, case-control study of polymorphisms in carcinogen-metabolizing genes, smoking, and pancreatic adenocarcinoma risk. J. Natl. Cancer Inst. 94 297–306 [DOI] [PubMed] [Google Scholar]
- 161. Li D., et al. (2005). 5,10-Methylenetetrahydrofolate reductase polymorphisms and the risk of pancreatic cancer. Cancer Epidemiol. Biomarkers Prev. 14 1470–1476 [DOI] [PubMed] [Google Scholar]
- 162. Wang L., et al. (2005). Genetic polymorphisms in methylenetetrahydrofolate reductase and thymidylate synthase and risk of pancreatic cancer. Clin. Gastroenterol. Hepatol. 3 743–751 [DOI] [PubMed] [Google Scholar]
- 163. Fesinmeyer M.D., et al. (2009). Association between the peroxisome proliferator-activated receptor gamma Pro12Ala variant and haplotype and pancreatic cancer in a high-risk cohort of smokers: a pilot study. Pancreas 38 631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Zhao D., et al. (2009). Interaction of cyclooxygenase-2 variants and smoking in pancreatic cancer: a possible role of nucleophosmin. Gastroenterology 136 1659–1668 [DOI] [PubMed] [Google Scholar]
- 165. Jiao L., et al. (2008). XRCC2 and XRCC3 gene polymorphism and risk of pancreatic cancer. Am. J. Gastroenterol. 103 360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. McWilliams R.R., et al. (2008). Polymorphisms in DNA repair genes, smoking, and pancreatic adenocarcinoma risk. Cancer Res. 68 4928–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Klein A.P. (2012). Genetic susceptibility to pancreatic cancer. Mol. Carcinog. 51 14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Wolpin B.M., et al. (2010). Variant ABO blood group alleles, secretor status, and risk of pancreatic cancer: results from the pancreatic cancer cohort consortium. Cancer Epidemiol. Biomarkers Prev. 19, 3140–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]