

Abstract
Sphingadienes (SDs) derived from soy and other natural sphingolipids are cytotoxic to colon cancer cells via an Akt-dependent mechanism and reduce adenoma formation in Apc Min/+ mice. Wnt signaling is fundamental to colon carcinogenesis and is the basis for spontaneous tumorigenesis in Apc Min/+ mice and patients with familial adenomatous polyposis. In the present study, we investigated the impact of SDs on Wnt signaling. Oral SD administration reduced levels of active β-catenin and Wnt targets c-Myc and cyclin D1 in Apc Min/+ mouse intestinal tissues. Colon cancer cells treated with SDs exhibited reduced Wnt transcriptional activity, as well as reduced nuclear β-catenin localization and subsequent reduction in active-β-catenin levels. Further, we observed a decrease in phosphorylated (inactive) GSK3β in SD-treated mice and colon cancer cells. Expression of constitutively active myristoylated-Akt or inactivation of GSK3β using LiCl attenuated SD-mediated inhibition of Wnt transcriptional activity and active-β-catenin levels. SDs exhibited additive effects with inhibitors of the phosphatidylinositol-3-kinase/Akt/mTOR pathway to induce cytotoxicity. Further, a combination regime of SDs and low-dose rapamycin decreased visible polyps in Apc Min/+ mice and reduced the levels of Wnt target gene expression and mTOR target activation. SD-mediated inhibition of Akt and Wnt pathways and cytotoxicity in colon cancer cells was dependent upon the activity of protein phosphatase 2A, as shown by reversal of these effects by pretreatment with okadaic acid or calyculin A. Our cumulative findings indicate that SDs inhibit Wnt signaling through a protein phosphatase 2A/Akt/GSK3β-dependent mechanism that may contribute to their chemopreventive effects in intestinal tumorigenesis.
Abbreviations:
- 4EBP-1
eIF4E-binding protein1
- APC
adenomatous polyposis coli
- GSK3β
Glycogen synthase Kinase 3β
- LCM
laser capture microscopy
- LEF
lymphoid enhancer factor
- mTOR
mammalian target of Rapamycin
- PI3K
Phosphatidylinositol-3-kinase
- PKC
protein kinase C
- PP2A
protein phosphatase 2A
- TCF
T-cell factor
- SD
Sphingadiene
Introduction
Sphingadienes (SDs) are growth-inhibitory sphingoid bases found in insects, marine organisms, and legumes including soybeans and soy products (1–3). Enzymes at the brush border of the gut liberate the free sphingoid base form of SDs from complex sphingolipids, thereby allowing SDs to be taken up into intestinal epithelial cells (4). We recently demonstrated that SDs exert chemopreventive action in the Apc Min/+ mouse model of intestinal tumorigenesis (5). SDs are cytotoxic to colon cancer cells, promoting apoptosis and autophagy by inhibiting Akt activation, thereby derepressing conserved cell death pathways (5). Orally delivered SDs are poorly absorbed into the bloodstream, exhibit a low toxicity profile, and, unlike sphingolipids derived from mammalian sources, are poorly converted to the oncogenic lipid sphingosine 1-phosphate (S1P). These features make SDs attractive for their potential utility in colon cancer chemoprevention strategies.
The Wnt-β-catenin pathway plays an essential role in embryonic development and contributes to tissue homeostasis and tumorigenesis. Activating mutations in the Wnt pathway are considered an early, critical step in human colon carcinogenesis and are responsible for intestinal tumor formation in patients with familial polyposis and in Apc Min/+ mice that undergo spontaneous intestinal adenoma formation (6,7). The Apc Min/+ mouse is heterozygous for a truncating mutation in the Adenomatous Polyposis Coli (APC) gene, a negative regulator of the Wnt signaling pathway. The oncogenic potential of the Wnt pathway derives from β-catenin protein stabilization and relocation from the cell membrane to the nucleus, where it is recruited into T-cell factor/lymphoid enhancer factor 1 (TCF/LEF) transcriptional regulatory complexes (8). TCF/LEF complexes bind to enhancer regions of target genes involved in proliferation, invasion, and inhibition of apoptosis, including c-Myc and cyclin D1. These effects contribute directly to colon cancer development.
Akt signaling has been shown to upregulate the Wnt signaling pathway through inhibitory phosphorylation of glycogen synthase kinase β (GSK3β, thereby stabilizing β-catenin and promoting its recruitment to TCF/LEF complexes (9–11). Considering that SDs inhibit the membrane translocation and activation of Akt, we hypothesized that SDs might also affect downstream Wnt signaling. We found that SDs regulate non-phosphorylated β-catenin protein levels, nuclear accumulation of β-catenin, TCF/LEF transcriptional activity, and Wnt target gene expression. These effects on Wnt signaling appear to occur via SD-mediated inhibition of GSK3β phosphorylation through the Akt pathway. SDs showed an additive effect with the inhibitors of the PI3K/Akt/mTOR pathway to suppress cell growth and inhibit intestinal tumorigenesis in Apc Min/+ mice. Further, the effects of SDs on Akt and Wnt signaling as well as their cytotoxicity in colon cancer cells required the activity of protein phosphatase 2A (PP2A), thus identifying PP2A as an upstream target of SDs.
Materials and methods
Antibodies and reagents
[(2S,3R,4E,6E)-2-amino-tetradecadiene-1,3-diol], hereafter referred to as SDs, were synthesized as described earlier (5). LY294002, myristolated PKCζ inhibitor and wortmannin are from EMD Biosciences (San Diego, CA). Rapamycin is from Tocris Bioscience (Ellisville, MO). Ac-DEVD-pNA is from Enzo Life Sciences (Farmingdale, NY). Calyculin A and antibodies against phospho-Akt1/2/3 (Ser473), Akt, phospho-GSK3β (Ser9), GSK3β, non-phosphorylated-β-catenin (non-P-β-catenin) (Ser33/37/Thr41), phospho-p70-S6 kinase (Thr389), and phospho-4EBP1 (Thr37/46) are from Cell Signaling Technology (Danvers, MA). Antibodies against β-catenin, c-Myc, and GAPDH are from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-cyclin D1 is from Millipore (Billerica, MA). Lithium chloride, okadaic acid and anti-actin antibody are from Sigma-Aldrich (St. Louis, MO). Horseradish peroxidase-anti-rabbit IgG, Cy3-anti-rabbit IgG, and FITC-anti-mouse IgG secondary antibodies are from Jackson ImmunoResearch Laboratories (West Grove, PA). Dulbecco’s modified Eagle’s medium (DMEM) with high glucose (DME H-21), fetal bovine serum (FBS), and antibiotics are from University of California–San Francisco Tissue Culture Facility (San Francisco, CA).
Immunoblotting
Immunoblotting was performed as described earlier (12). Signals were visualized using the SuperSignal West-Pico kit (Pierce, Rockford, IL) and quantified by densitometry using ImageJ software (NIH).
Laser capture microscopy
Apc Min/+ mouse intestinal tissues lacking microscopic adenomas were isolated by laser capture microscopy (LCM) using the Arcturus PixCell II Laser Capture Microdissection workstation (Applied Biosystems, Foster City, CA). Tissues were harvested immediately after euthanasia and frozen in optimal cutting temperature compound. Serial cut 10 µm frozen sections were washed in phosphate-buffered saline to reomove optimal cutting temperature compound. Uninvolved tissues from SD-treated and control animals were scraped into eppendorf tubes containing tissue homogenization buffer and processed for immunoblotting.
Cell culture and transfections
HT29, SW480, DLD1, and HCT116 cell lines were from ATCC (Rockville, MD) and were grown in DME H-21 containing 10% FBS, 100 units/ml penicillin, and 100 µg/ml streptomycin. SDs were dissolved in ethanol. SD treatment was performed in DME-H21 media containing 2% FBS. DLD1 cells were transiently transfected with either Wnt reporter plasmids as described below, or with plasmid complementary DNA encoding enhanced green fluorescent protein or recombinant human-Akt1 containing the Src myristoylation (myr) sequence cloned into pcDNA3.0 (myr-Akt1) (13) from Addgene (Cambridge, MA) using the calcium phosphate method (14).
Luciferase reporter assay
Colon cancer cell lines were transiently transfected with the firefly luciferase reporter plasmid M50 super 8× TOPflash or its control counterpart M51 super 8× FOPflash. DLD1 and SW480 cells were transfected using the calcium phosphate method and HT29 cells were transfected with X-tremeGene HP DNA transfection reagent (Roche Applied Sciences, Indianapolis, IN). SuperTop/FOPflash plasmids were from Dr. Randall Moon (University of Washington, Seattle, WA). SuperTOPflash contains seven consensus TCF/LEF-binding sites upstream of a minimal thymidine kinase promoter driving luciferase expression. In SuperFOPflash, TCF/LEF sites are mutated (15). Cells were cotransfected with sea pansy Renilla pRL-SV40 plasmid obtained from Addgene, wherein luciferase expression is driven by the SV40 promoter (16). Forty-eight hours after transfection, cells were treated with vehicle, SDs or LiCl, and luciferase reporter activity was assayed using the Dual-Glo Luciferase Assay System (Promega, Madison, WI). Firefly luciferase activity was normalized to the Renilla luciferase activity value of each sample.
Immunofluorescence
Cells were grown on eight-well chamber slides to 60% confluence and then treated with vehicle or SDs for 2–3h. Cells were washed with cold phosphate-buffered saline, fixed with 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100. After blocking, cells were incubated with mouse monoclonal non-phosphorylated-β-catenin antibody (8E7) or total β-catenin antibody at 4°C overnight. After washing, cells were incubated with FITC-anti-mouse or Cy3-anti-rabbit IgG for 1h. After washing again, cells were stained with 4′,6-diamidino-2-phenylindole and mounted in Vectashield. Images were captured using a Zeiss Axioskop fluorescence microscope (Carl Zeiss, Thornwood, NY). Images were processed using Adobe Photoshop CS5 (Adobe, San Jose, CA).
Cell growth and caspase-3 activity
Cell growth and caspase-3 activity were measured as described previously (5).
Animals
Twelve-week-old male C57BL/6J-APCMin+/– (Apc Min/+) mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Mice were administered by gavage either vehicle (0.5% methylcellulose), 25mg/kg SDs in 0.5% methylcellulose, 2.5mg/kg rapamycin (in 10% polyethyleneglycol, 8% ethanol, 10% Tween 20) i.p. or both 25mg/kg SDs and 2.5mg/kg rapamycin daily for 10 days. Subjects were killed by CO2 asphyxiation. Visible polyps were counted and uninvolved intestinal tissues harvested, flash frozen, and used for immunoblotting. All murine experiments were performed in accordance with CHORI Institutional Animal Care and Use Committee approved protocols.
Statistical analysis
Statistical analysis was performed using Student’s t-test. If more than two groups were present, statistical analysis was performed by one-way ANOVA followed by Tukey’s post-hoc test. P value ≤0.05 was considered significant.
Results
SDs reduce Wnt signaling in the intestinal mucosa of ApcMin/+ mice
Oral administration of 25mg/kg/day SDs for 10 days to Apc Min/+ mice results in a 35% reduction in grossly visible intestinal polyps (5). To determine whether this chemopreventive regimen is associated with changes in Wnt signaling in the intestinal mucosa, Apc Min/+ mice treated with this regimen of SDs or vehicle control were killed 24h after the last dose, and polyp-free intestinal tissues were harvested and flash frozen. Activation of the Wnt pathway was evaluated in tissue extracts from treated and control groups by using immunoblotting to compare the protein levels of total and activated β-catenin. The latter is specifically non-phosphorylated at residues S37 and T41 and has increased capacity for nuclear accumulation as compared with the total pool of β-catenin (17). As shown in Figure 1A and 1B, the levels of activated β-catenin were reduced in polyp-free small intestinal tissues of SD-treated mice as compared with controls. In contrast, total β-catenin levels were not changed significantly. To confirm that Wnt signaling is reduced by SD treatment, the protein levels of Wnt downstream targets c-Myc and cyclin D1 were measured by immunoblotting in the same tissue extracts. As shown in Figure 1A and 1B, SD treatment was associated with reduced levels of both c-Myc and cyclin D1. Although the intestinal tissues examined in these experiments did not contain visible polyps, they could potentially contain microscopic adenomatous tissue. Therefore, to confirm the effects of SD treatment on Wnt signaling in uninvolved tissues, we harvested intestinal tissues that were free of microscopic adenomas using LCM. Using immunoblotting to compare non-phosphorylated β-catenin, c-Myc and cyclin D1 protein levels in microscopically uninvolved tissues of SD-treated and control mice, we confirmed that SD treatment reduced the levels of each of these proteins (Figure 1C and 1D). These findings indicate that SD treatment reduces Wnt signaling in the small intestinal mucosa of Apc Min/+ mice.
Fig. 1.
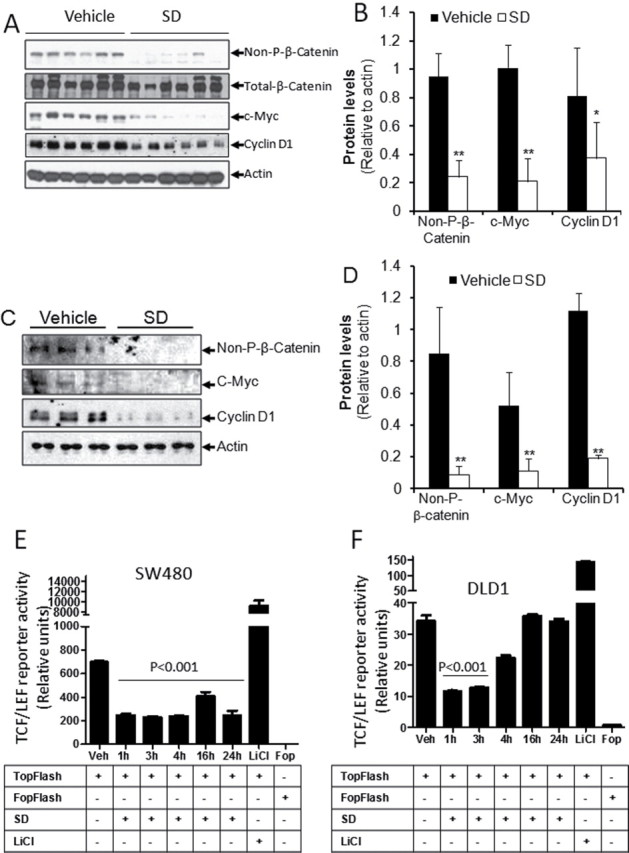
SDs reduces Wnt signaling in Apc Min/+ intestines and downregulate Wnt transcriptional activity in colon cancer cells. (A–D) Apc Min/+ mice were administered 25mg/kg body weight/day SDs or vehicle (n = six/group). Mice were killed and intestinal tissues harvested on day 10. (A) Intestinal levels of activated β-catenin (Non-P-β-catenin), total β-catenin, c-Myc and cyclin D1 were measured in extracts prepared from non-tumor tissue by immunoblotting. (C) Microadenomas were removed from frozen sections of intestine by laser capture microdissection and polyp free sections were lysed in tissue homogenization buffer and subjected to immunoblotting as in panel A. (B and D) Quantification of blots were done by ImageJ software. Densitometric analysis for non-P-β-catenin, c-Myc, and cyclin D1 is shown as ratio to actin levels. Values are the average ± standard deviation. *P < 0.05 control versus SDs; **P < 0.01 control versus SDs. (E) SW480 and (F) DLD1 colon cancer cells were transfected with the TOPflash or FOPflash TCF/LEF reporter plus SV40 Renilla plasmid. Transfected cells were incubated with 20 µM SDs for 0–24h. Cells were then harvested and TCF/LEF reporter activity was measured. Data are presented as ratio of firefly luciferase over Renilla luciferase. Results are shown as average values ± SDEV of at least three independent experiments. P < 0.001 vehicle versus indicated time points.
SDs downregulate TCF/LEF transcriptional activity in colon cancer cells
It became important to test the relevance of these findings to human colonic neoplasia and to establish an in vitro system to explore the cellular mechanism(s) by which SDs impact Wnt signaling. To determine whether SD treatment influences the transcriptional activity of the TCF/LEF-β-catenin transcriptional complex, 20 µM SDs were delivered over a time course varying from 0 to 24h to colon cancer cell lines harboring either the TOPflash reporter, which contains seven copies of the wild type TCF/LEF binding site upstream of luciferase open reading frame, or the FOPflash control reporter in which the TCF/LEF binding sites are mutated. As shown in Figure 1E and 1F, SD treatment suppressed TCF/LEF transcriptional activity, as demonstrated by a significant reduction in TOPflash reporter activity in SW480 and DLD1 cells. In SW480 cells, TCF/LEF activity was reduced by more than 70% within 1h and remained suppressed for 24h. In DLD1 cells, TCF/LEF activity was also suppressed rapidly, but the effect began to subside within 4h and returned to baseline levels by 16h after treatment. SW480 cells exhibited at least 20-fold higher levels of TCF/LEF reporter activity in comparison to DLD1 cells. Suppression of TCF/LEF activity was also observed in HCT116 cells (Supplementary Figure 1 is available at Carcinogenesis Online). These findings show that SDs suppress the transcriptional activity of the TCF/LEF-β-catenin complex in multiple colon cancer cell lines.
SDs reduce active β-catenin and downstream targets of Wnt signaling in colon cancer cells
To confirm that SD-mediated downregulation of TCF/LEF transcriptional activity correlates with reduced Wnt signaling, non-phosphorylated and total β-catenin, c-Myc and cyclin D1 protein levels were measured by immunoblotting of whole-cell extracts of colon cancer cells treated with SDs or vehicle control. Considering the rapid downregulation of TCF/LEF activity observed in response to SD treatment in vitro, effects on protein expression were examined within a 0–3h window after treatment. As shown in Figure 2A–D, cells treated with SDs exhibited a dose- and time-dependent reduction in non-phosphorylated-β-catenin and its downstream targets c-Myc and cyclin D1. In contrast, total β-catenin levels decreased after treatment with higher doses of SDs in HT29 cells, and its levels remained relatively constant in SW480 cells (Figures 2A–D). SDs also decreased non-phosphorylated-β-catenin levels in HCT116 cells (Supplementary Figure 2 is available at Carcinogenesis Online).
Fig. 2.

SDs reduce active β-catenin and downstream targets of Wnt signaling in colon cancer cells. (A) HT29 and (B) SW480 cells were incubated with vehicle or 5–30 μM SDs for 3h. HT29 (C) and SW480 (D) cells were treated with vehicle or 20 µM SDs for the indicated times. Cells were harvested, and protein levels of non-P and total β-catenin, c-Myc, and cyclin D1 were evaluated by immunoblotting. Actin and GAPDH are loading controls.
SDs reduce nuclear localization of β-catenin in SW480 cells
TCF/LEF transcriptional activity is principally regulated through alteration of β-catenin levels in the nucleus, which are themselves regulated by phosphorylation and other post-translational modifications (18). The β-catenin protein can be found in the cytosol or at membrane locations, where it binds to E-cadherin and thereby contributes to adherens junctional complexes. Accumulation and dephosphorylation of β-catenin in response to Wnt interactions with the frizzled receptor facilitate nuclear translocation and activation of β-catenin. As shown in Figure 3A and 3B, in SW480 cells we found that both non-phosphorylated and total β-catenin were predominantly localized in the nucleus. Treatment with 20 μM SDs for 2h resulted in a reduction in nuclear β-catenin, and a parallel increase in its localization within the cytosol. However, incubation of SW480 cells with SDs for 3h resulted in an overall decrease in non-phosphorylated-β-catenin levels. These results suggest that SDs affect TCF/LEF transcriptional activity by reducing nuclear β-catenin translocation and thereafter a decrease in active β-catenin levels.
Fig. 3.
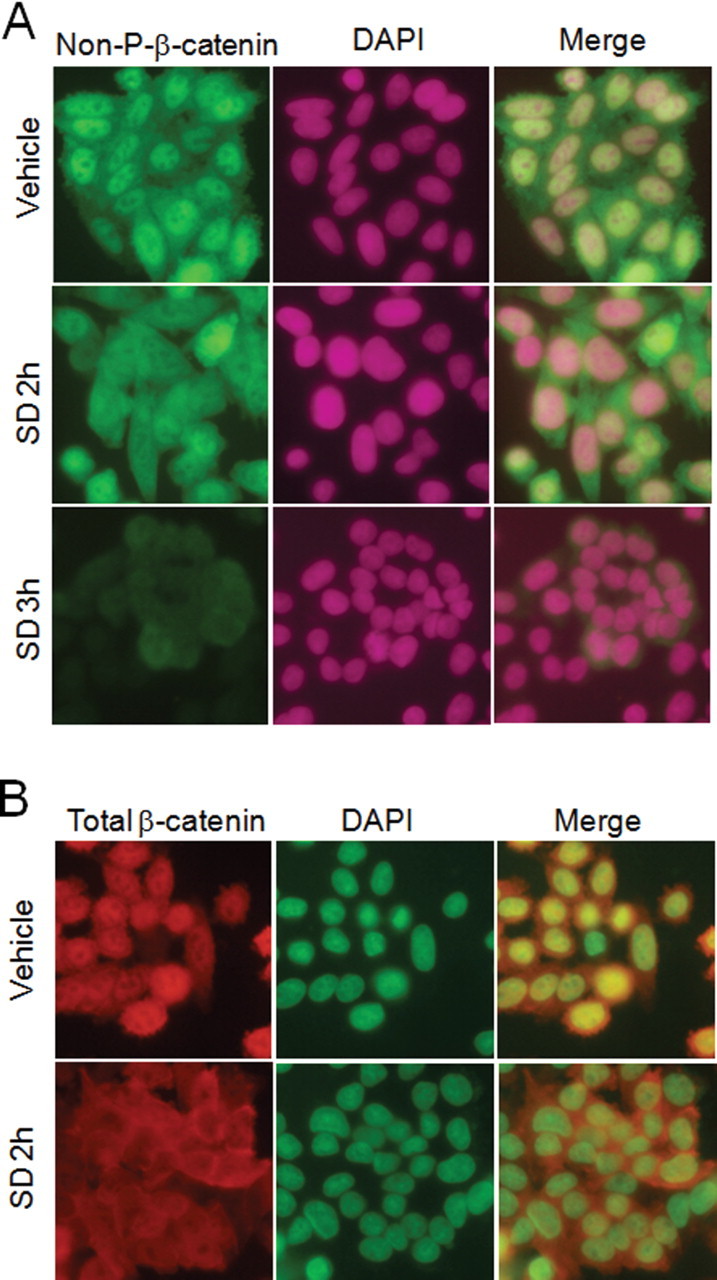
SDs reduce nuclear localization of β-catenin in colon cancer cells. SW480 cells were grown in eight-chamber slides for 48h, and then were treated with vehicle or SDs for indicated times. Cells were fixed, permeablized, and stained with antibodies against non-P-β-catenin (A) or total β-catenin (B). Cells were counterstained with 4′,6-diamidino-2-phenylindole and images acquired using a fluorescence microscope with a 40× objective lens.
Suppression of Wnt signaling by SDs is mediated in an Akt/GSK3β-dependent manner
Nuclear β-catenin levels are predominantly regulated by a multiprotein complex that targets GSK3β and casein kinase I to mediate amino-terminal serine-threonine phosphorylation and, thereby, promote proteasomal degradation of β-catenin. GSK3β activation is regulated by Akt, which phosphorylates GSK3β at Ser9 and inactivates it (19). As shown in Figure 4A and 4B, the levels of phospho-GSK3β were reduced in the intestinal mucosa of SD-treated Apc Min/+ mice. Similarly, SD treatment decreased phospho-GSK3β levels in colon cancer cells in a dose-dependent manner (Figure 4C). As reported earlier, we also observed a concomitant decrease in phospho-Akt levels in SD-treated colon cancer cells (Figure 4C). These findings suggest that SDs activate GSK3β by removing Akt-mediated repression, which would be expected to increase proteolytic degradation of β-catenin via the actions of the axin/GSK3β/APC complex. Therefore, we hypothesize that SDs reduce Wnt signaling through a mechanism involving Akt inhibition and consequent GSK3β activation. To test this possibility, we investigated the effects of constitutive Akt activation and GSK3β inhibition on TCF/LEF reporter activity and total and activated β-catenin levels in colon cancer cells treated with SD or vehicle control. As shown in Figure 4D, overexpression of constitutive active Akt (myr-Akt1) in DLD1 cells partially rescued the SD-mediated decrease in TCF/LEF reporter activity. Further, inhibition of GSK3β by LiCl attenuated the SD-induced reduction in active β-catenin (Figure 4E). These findings indicate that SDs suppress the Wnt signaling pathway through an Akt/GSK3β-mediated mechanism that involves phosphorylation of β-catenin, leading to a reduction in the nuclear and cellular levels of active, non-phosphorylated-β-catenin.
Fig. 4.
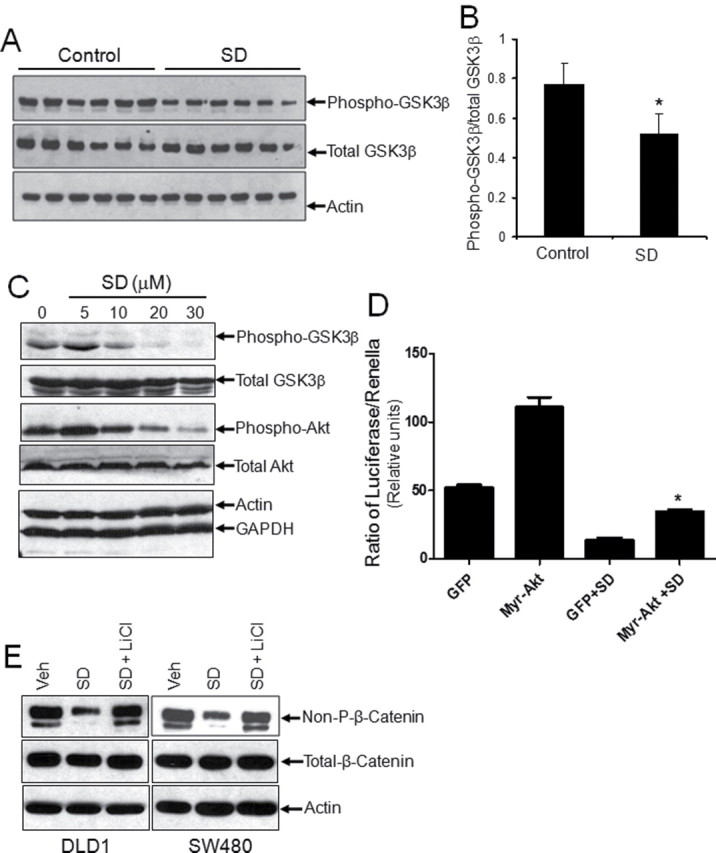
Suppression of Wnt signaling by SDs is mediated in an Akt- and GSK3β-dependent manner. Apc Min/+ mice were administered 25mg/kg body weight/day SDs or vehicle (n = six/group). Mice were killed and intestinal tissues harvested on day 10. Intestinal levels of phospho-GSK3β, total GSK3β, and actin were measured in extracts prepared from non-tumor tissue by immunoblotting (A) and quantification (B) by ImageJ software. Phospho-GSK3β levels are shown as a ratio to total GSK3β levels. *P < 0.05 control versus SDs. (C) HT29 cells were incubated with vehicle or 5–30 μM SDs for 3h. Cells were harvested, and protein levels of phospho-GSK3β, total GSK3β, phospho-Akt, and total Akt were evaluated by immunoblotting. Actin and GAPDH are loading controls. (D) DLD1 cells were transfected with green fluorescent protein or myristoylated-Akt (Myr-Akt) along with TOPflash TCF/LEF and Renilla luciferase reporter plasmids. After 48h, cells were treated with 20 μM SDs for 3h or left untreated. TCF/LEF and Renilla reporter activities were measured and shown as average of firefly luciferase/ Renilla luciferase activity ± SDEV from three independent experiments *P < 0.05 compared with green fluorescent protein+SD. (E) DLD1 and SW480 cells were treated with vehicle or 20 μM SDs in the presence or absence of LiCl (20mM) for 3h. Cells were harvested and protein levels of non-P-β-catenin, total β-catenin, and actin were determined by immunoblotting.
SDs exhibit additive effects with the inhibitors of PI3K/Akt/mTOR pathway
PI3K/Akt signaling pathway regulates cell growth metabolism, translation, and proliferation. Akt-mediated activation of mTOR is important in stimulating cell proliferation (20). We recently demonstrated that SDs inhibit Akt/mTOR signaling by preventing the translocation of Akt from the cytosol to the plasma membrane (5). Considering that the Akt, mTOR, and Wnt pathways have both shared and distinct targets of action, we considered that SDs might act in an additive manner with the inhibitors of the PI3K/Akt/mTOR pathway to inhibit cell proliferation and induce cell death. To test this possibility, we treated colon cancer cells with SDs alone or in the presence of PI3K inhibitors, wortmannin and LY294002, or the mTOR inhibitor rapamycin for 24–48h. Treatment of colon cancer cells with 10 μM SDs in the presence of wortmannin or rapamycin resulted in enhanced cytotoxicity as compared with the individual effect of SDs or PI3K/mTOR inhibitors, as shown in Supplementary Figure 3A and B (available at Carcinogenesis Online). Co-treatment of HT29 cells with SDs plus LY294002, rapamycin, or both resulted in a four to eight-fold increase in apoptosis, as assessed by caspase-3 activity (Supplementary Figure 3C is available at Carcinogenesis Online). Further, we tested whether SDs and mTOR inhibitor cooperate to suppress the Wnt signaling pathway. Thus, HT29 cells were treated with SDs (10 μM) and rapamycin (10 μM) alone or in combination for 3h, and the protein levels of phospho- and total Akt, active and total β-catenin, and c-Myc were determined by immunoblotting. As shown in Supplementary Figure 3D and E (available at Carcinogenesis Online), as compared with individual treatments, co-treatment of HT29 cells with SDs and rapamycin enhanced the reduction in active β-catenin and c-Myc levels.
SDs exhibit additive effects with rapamycin to inhibit intestinal tumorigenesis
Previously, we demonstrated that oral administration of SDs to the Apc Min/+ mice results in 35% reduction in visible polyps (5). During the early stages of carcinogenesis, Apc Min/+ mice show enhanced Akt/mTOR signaling (21). The mTOR inhibitor rapamycin shows promise as a chemotherapeutic agent (22,23). However, chronic immunosuppression and other toxicities may preclude its long-term usage as a chemopreventive agent. To test whether a combination of SDs and low-dose rapamycin would enhance the chemopreventive potential of each agent, Apc Min/+ mice were administered vehicle, SDs, rapamycin or both daily for 10 days, and visible polyps were counted in the small intestine. As shown in Figure 5A, administration of SDs or rapamycin alone resulted in a 38% reduction of visible polyps in small intestine as compared with vehicle-treated mice, whereas the combination of SDs and low-dose rapamycin led to a 67% decrease in visible polyps in the small intestine. We then compared the impact of a combination regimen of SDs and low-dose rapamycin to the impact of each agent alone on β-catenin and Akt/mTOR signaling in the intestines of Apc Min/+ mice. As shown in Figures 5B and 5C, as compared with individual treatment of SD or low-dose rapamycin alone, a combination regimen of SDs and low-dose rapamycin significantly reduced the protein levels of active-β-catenin and its downstream targets c-Myc and cyclin D1. Protein levels of mTOR downstream targets phospho-p70 S6 kinase and phospho-4EBP1 were also reduced in the small intestine of mice treated with the combined regimen (Figures 5D and 5E).
Fig. 5.
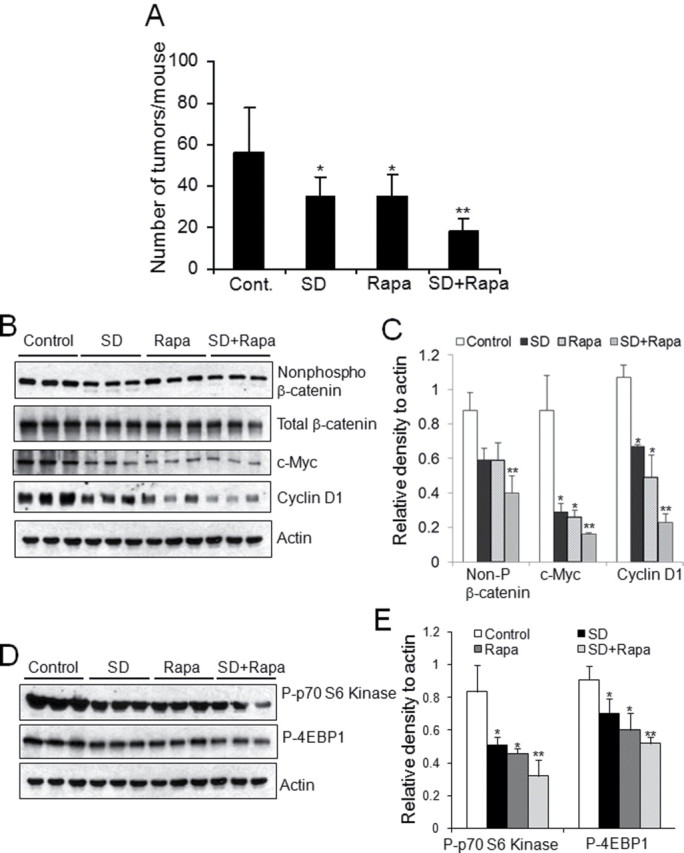
SDs exhibit additive effect with inhibitors of PI3K/mTOR pathway to inhibit intestinal tumorigenesis. Apc Min/+ mice were administered vehicle, 25mg/kg body weight/day SDs, 2.5mg/kg body weight/day rapamycin, or both (SDs + rapamycin; n = five/group). Mice were killed and intestinal tissues harvested on day 10. (A) Gross polyps were counted in the small intestines of Apc Min/+ mice. *P < 0.05 control versus SDs or control versus rapamycin; **P < 0.001 SDs + rapamycin versus control. (B–E) Intestinal levels of activated β-catenin (non-P-β-catenin), total β-catenin, c-Myc, and cyclin D1, phospho-p70 S6 kinase, and phospho-4EBP1 were measured in extracts prepared from non-tumor tissue by immunoblotting. (C and E) ratios of non-P-β-catenin, c-Myc, cyclin D1, phospho-p70 S6 kinase, and phospho-4EBP1 to actin were quantitated by densitometric analysis (n = 3) *P < 0.05 as compared with control; **P < 0.01 as compared with control.
SDs inhibit growth-activating signaling in a PP2A-dependent manner
Having established that SDs induce cytotoxicity at least in part by suppressing the activity of Akt and Wnt pathways interconnected through GSK3β, it became important to identify an upstream target of SDs that might be responsible for mediating these effect of SDs. PP2A is a signaling phosphatase implicated in the regulation of Akt, GSK3β and β-catenin activities (24–27). Further, PP2A has been shown to be activated by ceramide and, thus, represents a sphingolipid-regulated enzyme (28,29). Others have provided evidence that ceramide inactivates Akt through its effects on PKCζ (30). To explore the potential roles of PP2A and PKCζ in mediating the effects of SDs on Akt, we measured Akt phosphorylation by immunoblotting in extracts of cells treated with the Akt activator IGF1 plus SDs alone and in combination with inhibitors of PP2A and PKCζ. As shown in Figures 6A and 6B, IGF1 treatment resulted in robust Akt activation, whereas cells treated with Akt plus SDs exhibited approximately half maximal activation. The addition of the PKCζ inhibitor neither appreciably changed Akt activation by IGF1 nor suppressed the activation of Akt by SDs as compared with controls. In contrast, the PP2A inhibitor okadaic acid completely reversed the inhibitory effect of SDs on Akt. Okadaic acid pretreatment also reversed the ability of SDs to inhibit TCF/LEF signaling, measured by TOPflash reporter assay (Figure 6C). Lastly, we examined the cytotoxic effect of SDs on colon cancer cells in the presence and absence of two different PP2A inhibitors, okadaic acid and calyculin A. As shown in Figure 6D, inhibition of PP2A by either method prevented the ability of SDs to inhibit colon cancer cell growth. These results indicate that PP2A is an upstream target of SDs responsible for mediating its effects on cell growth-activating signaling pathways involved in colon carcinogenesis.
Fig. 6.
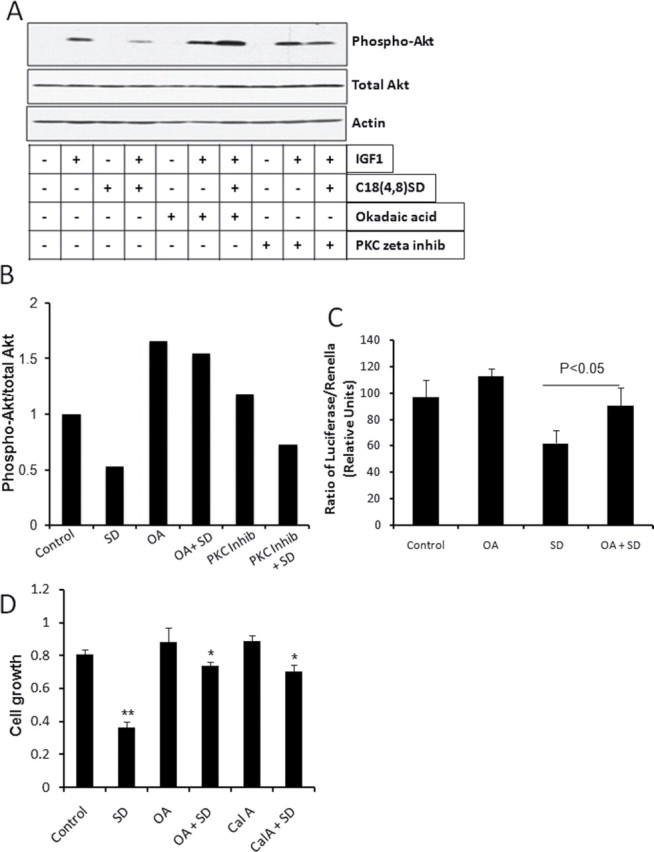
SDs inhibit Akt and Wnt signaling in a PP2A-dependent manner. (A, B) SW480 cells were grown to 70% confluency, and then cells were serum starved for 20h. Cells were treated with a PP2A inhibitor, 100nM okadaic acid (OA) or 5 µM myrisolated PKCζ peptide inhibitor and 20 μM C18 (4,8) SDs alone or in combination. Then, cells were treated with 10nM IGF1 for 30min. Cell lysates were immunoblotted (A) with indicated antibodies and densitometric analysis (B) was shown for lanes treated with IGF1. (C) HT29 cells were transfected with the TOPflash TCF/LEF reporter plus SV40 Renilla plasmid. Transfected cells were preincubated with 50nM Okadaic acid (OA) in DMEM containing 2% FBS for 3h and treated 20 µM SDs for 3h. Cells were then harvested and TCF/LEF reporter activity was measured. Data are presented as ratio of firefly luciferase over Renilla luciferase. Results are shown as average values ± SDEV of at least three independent experiments. *P < 0.05 SD versus OA+SD. (D) DLD1 cells were grown in complete media to 70% confluency, then cells switched to DMEM media containing 2% FBS and pretreated with 10nM Okadaic acid (OA) and 1nM Calyculin A (Cal A) and cells were treated with 20 µM SD for 24h. Cell growth was determined by MTT assay. **P < 0.01 control versus SD, *P < 0.05 SD versus SD + OA or SD versus Cal A + SD.
Discussion
Sphingolipids are found in the cell membranes of most eukaryotic species and are present in plant and animal constituents of the human diet (2,4,31). Normal consumption of sphingolipids is estimated to be 0.3–0.4g/day. Complex sphingolipids such as sphingomyelin (SM) and plant and animal glucosylceramides are digested in the gut lumen by a series of ecto-enzymes expressed on the luminal side of the intestinal epithelium. Dietary and intracellular SM is metabolized by sphingomyelinases to yield ceramide, which in turn is deacylated to sphingosine by the enzyme ceramidase. SM and other complex sphingolipids are not appreciably imported into cells of the intestinal epithelium, while ceramide uptake is inefficient. In contrast, sphingoid bases including sphingosine and SDs derived from dietary sphingolipids are readily taken up by intestinal cells (32).
Both sphingosine and SDs exert growth-inhibitory effects on intestinal cells (33,34). Importantly, however, sphingosine can be phosphorylated by the oncogenic enzyme sphingosine kinase 1 (SphK1), giving rise to the bioactive signaling molecule, S1P. S1P signaling through G-protein coupled receptors promotes angiogenesis, STAT3-dependent pro-tumorigenic inflammatory signaling, Akt activation, and oncogenic Ras-mediated transformation (35–37). Further, SphK1 is overexpressed in colon cancer and may contribute to colon carcinogenesis (38–40). Thus, there is concern that the growth-inhibitory actions of sphingosine derived from mammalian sphingolipids may be self-limited or even tumor promoting, especially when given over long time periods and/or in the presence of neoplastic intestinal tissues that can readily convert it to S1P.
In contrast to sphingosine, SDs are poorly metabolized by colon cancer cells and are retained as the unphosphorylated long-chain base for prolonged time periods (5,41). SDs are minimally absorbed into the bloodstream, reducing the risk of toxicities. SDs induce apoptosis and autophagy by releasing conserved cell death pathways from Akt-mediated repression, whereas the parent glycosphingolipids from which they are derived do not exert appreciable effects on Akt (5). SDs are more potent than sphingosine or ceramide in cytotoxicity assays against colon cancer cells, likely due the presence of the second double bond in SDs (42).
Although SDs were shown to influence Akt activation, their potential influence on the downstream Wnt signaling pathway central to colon carcinogenesis has not been examined until now. In this study, we have shown that SDs are inhibitors of Wnt signaling, TCF/LEF transcriptional activity, and Wnt target gene activation. SD treatment inhibited Wnt signaling in a variety of human colon cancer cell lines and a well-established murine model of intestinal carcinogenesis, the Apc Min/+ mouse. Addition of sphingolipids to rodent diets has been associated with changes in cellular gene expression and protein localization, including modulation of intracellular β-catenin localization and other factors, which may contribute to the ability of these lipids to influence tumorigenesis (43,44). Sphingolipid supplementation caused β-catenin to relocalize to membranes and away from the nucleus in intestinal tumors of Apc Min/+ mice. Further, treatment of colon cancer cell lines with sphingolipids induced β-catenin membrane localization, cell growth inhibition, and death. Further, S1P and SphK1 interact with the catenin family members (45–49). Thus, our findings are consistent with previous reports linking sphingolipids to Wnt signaling and further extend this link to include a novel family of sphingolipids with significant advantages as chemopreventive agents.
We observed that GSK3β inhibitory phosphorylation was reduced in SD-treated mice and colon cancer cell lines, consistent with the inhibitory effect of SDs on Akt signaling. Using a combination of constitutive Akt activation and inhibition of GSK3β by LiCl treatment, we provide evidence that the inhibitory effects of SDs on Wnt signaling are at least partly due to their ability to mediate Akt inhibition and thereby prevent phosphorylation of GSK3β. Although it has been reported that Akt and GSK3β/β-catenin are segregated pathways in colon cancer (50), we find a direct interaction between Akt and Wnt signaling that allows SDs to repress Wnt signaling through GSK3β in our model systems.
A specific molecular target with which SDs may directly interact to mediate their effects on cell signaling has not yet been identified. In this study, we have identified an upstream target of SDs, namely the protein phosphatase PP2A. This enzyme is implicated in the regulation of Akt, GSK3β and Wnt activation and has additionally been shown to be regulated by another sphingolipid metabolite, ceramide. Our current findings suggest that SDs mediate their effects on key growth-regulatory signaling pathways via upstream activation of PP2A. Whether PP2A is a direct target of SDs will require further study.
PI3K inhibitors are an important and novel class of pharmacological agents that may prove useful in colon cancer treatment. Chemoprevention using the mTOR inhibitor rapamycin is also being explored for patients at risk of developing the disease. However, rapamycin has been shown to exert effects on multiple targets, and its long-term usage as a chemopreventive agent may prove problematic due to toxicities associated with chronic immunosuppression, alterations in lipid metabolism and other “off-target” effects of the drug. In our study, we provide evidence that a low-dose rapamycin and SDs combination is tumor-preventive and affects targets of each agent in additive fashion. This suggests that combination chemopreventive strategies may allow dose reductions that could potentially reduce toxicities without compromising efficacy in a manner similar to strategies used for cancer treatment. It should be noted that the SD and rapamycin combination treatment will require dosage optimization in preclinical models to establish the maximum chemopreventive benefit that can be achieved without intolerable toxicities prior to testing the combination in FAP patient clinical trials. Considering the poor bioavailability of SDs, we predict that it will be possible to administer them at high doses without incurring significant toxicities. However, this remains to be tested in vivo.
In summary, our current and previous studies show that SDs have chemopreventive action when administered to rodent models of colon cancer. Future studies testing the safety and efficacy of orally delivered SDs in humans are expected to establish the relevance of our findings to chemoprevention strategies for colon cancer. Our finding that SDs reduce colon cancer cell viability at least in part through inhibition of Wnt signaling suggests that SDs could potentially also have utility in the treatment of malignancies in which the Wnt pathway is implicated. The list of such malignancies includes desmoid tumors, hepatocellular carcinoma, gastric cancer, thyroid cancer, endometrial ovarian cancer, breast cancer, prostate cancer, melanoma and pediatric malignancies including medulloblastoma, hepatoblastoma and Wilms’ tumor (51). However, it should be noted that in colon cancer chemoprevention the poor absorption of orally delivered SDs serves as an advantage that maximizes local effects while reducing the risk of systemic toxicities. In contrast, the treatment of non-gastrointestinal tumors with SDs will require development of effective methods for parenteral delivery to distant tumor sites and analysis of the toxicity profile of systemic SD therapy. Toward that end, we are currently investigating methods that may allow us to expand the investigation of SDs from the realm of chemoprevention to that of chemotherapy.
Supplementary material
Supplementary Figures 1–3 can be found at http://carcin.oxfordjournals.org/
Acknowledgments
Funding
This work was supported by National Institutes of Health [RAT005336A and CA129438 to JDS, HL-083187 to RB, 5T32CA009041 to FL]; American Institute for Cancer Research Grant [09A041 to JDS]; the Swim Across America Foundation (JDS).
Conflict of Interest: No potential conflicts of interest were disclosed.
References
- 1. Row L.C., et al. (2007). Cerebrosides and tocopherol trimers from the seeds of Euryale ferox. J. Nat. Prod., 70, 1214–1217 [DOI] [PubMed] [Google Scholar]
- 2. Sullards M.C., et al. (2000). Structure determination of soybean and wheat glucosylceramides by tandem mass spectrometry. J. Mass Spectrom., 35, 347–353 [DOI] [PubMed] [Google Scholar]
- 3. Fyrst H., et al. (2008). Identification and characterization by electrospray mass spectrometry of endogenous Drosophila sphingadienes. J. Lipid Res., 49, 597–606 [DOI] [PubMed] [Google Scholar]
- 4.Nilsson A et al. (2006). Absorption and lipoprotein transport of sphingomyelin. J. Lipid Res., 47, 154–171 [DOI] [PubMed] [Google Scholar]
- 5.Fyrst H., et al. 2009). Natural sphingadienes inhibit Akt-dependent signaling and prevent intestinal tumorigenesis. Cancer Res., 69, 9457–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moser A., et al. (1990). A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science, 247, 322–324 [DOI] [PubMed] [Google Scholar]
- 7. de Lau W., et al. (2007). WNT signaling in the normal intestine and colorectal cancer. Front. Biosci., 12, 471–491 [DOI] [PubMed] [Google Scholar]
- 8. Fuchs S.Y et al. 2005). Oncogenic beta-catenin signaling networks in colorectal cancer. Cell Cycle, 4, 1522–1539 [DOI] [PubMed] [Google Scholar]
- 9. Satyamoorthy K., et al. (2001). Insulin-like growth factor-1 induces survival and growth of biologically early melanoma cells through both the mitogen-activated protein kinase and beta-catenin pathways. Cancer Res., 61, 7318–7324 [PubMed] [Google Scholar]
- 10.Lovatt M., et al. Stabilisation of beta-catenin downstream of T cell receptor signalling. PLoS One, (2010);5,:e12794. doi: 10.1371/journal.pone.0012794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McManus E.J., et al. (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J., 24, 1571–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oskouian B., et al. (2006). Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is downregulated in colon cancer. Proc. Natl. Acad. Sci. U. S. A., 103, 17384–17389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramaswamy S., et al. (1999). Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. U. S. A., 96, 2110–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen C.A., et al. (1988). Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques, 6, 632–638 [PubMed] [Google Scholar]
- 15. Veeman M.T., et al. (2003). Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol., 13, 680–685 [DOI] [PubMed] [Google Scholar]
- 16. Chen X., et al. (1999). Serum-induced expression of the cdc25A gene by relief of E2F-mediated repression. Mol. Cell. Biol., 19, 4695–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Staal F.J., et al. (2002). Wnt signals are transmitted through N-terminally dephosphorylated beta-catenin. EMBO Rep, 3, 63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kikuchi A., et al. (2006). Regulation of Wnt signaling by protein-protein interaction and post-translational modifications. Exp. Mol. Med., 38, 1–10 [DOI] [PubMed] [Google Scholar]
- 19. Cross D.A., et al. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature, 378, 785–789 [DOI] [PubMed] [Google Scholar]
- 20. Hennessy B.T., et al. (2005). Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov, 4, 988–1004 [DOI] [PubMed] [Google Scholar]
- 21. Moran A., et al. (2004). Apc deficiency is associated with increased Egfr activity in the intestinal enterocytes and adenomas of C57BL/6J-Min/+ mice. J. Biol. Chem., 279, 43261–43272 [DOI] [PubMed] [Google Scholar]
- 22. Zoncu R., et al. (2011). mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol, 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Don A.S., et al. (2011). Recent clinical trials of mTOR-targeted cancer therapies. Rev Recent Clin Trials, 6, 24–35 [DOI] [PubMed] [Google Scholar]
- 24. Zolnierowicz S. (2000). Type 2A protein phosphatase, the complex regulator of numerous signaling pathways. Biochem. Pharmacol., 60, 1225–1235 [DOI] [PubMed] [Google Scholar]
- 25. Yang J., et al. (2010). Functions of B56-containing PP2As in major developmental and cancer signaling pathways. Life Sci., 87, 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jeon K.I., et al. (2010). Ca2+/calmodulin-stimulated PDE1 regulates the beta-catenin/TCF signaling through PP2A B56 gamma subunit in proliferating vascular smooth muscle cells. FEBS J, 277, 5026–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang W., et al. (2009). PR55 alpha, a regulatory subunit of PP2A, specifically regulates PP2A-mediated beta-catenin dephosphorylation. J. Biol. Chem., 284, 22649–22656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dobrowsky R.T., et al. (1993). Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem., 268, 15523–15530 [PubMed] [Google Scholar]
- 29. Ivaska J., et al. (2002). Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol. Cell. Biol., 22, 1352–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Powell D.J., et al. (2003). Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol. Cell. Biol., 23, 7794–7808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abnet C., et al. (2001). A cross-sectional study of human serum sphingolipids, diet and physiological parameters. J. Nutr., 131, 2748–2752 [DOI] [PubMed] [Google Scholar]
- 32. Schmelz E.M., et al. (1996). Sphingomyelin consumption suppresses aberrant colonic crypt foci and increases the proportion of adenomas versus adenocarcinomas in CF1 mice treated with 1,2-dimethylhydrazine: implications for dietary sphingolipids and colon carcinogenesis. Cancer Res., 56, 4936–4941 [PubMed] [Google Scholar]
- 33. Ahn E.H., et al. (2002). Sphingoid bases and ceramide induce apoptosis in HT-29 and HCT-116 human colon cancer cells. Exp Biol Med (Maywood), 227, 345–353 [DOI] [PubMed] [Google Scholar]
- 34. Sweeney E.A., et al. (1996). Sphingosine and its methylated derivative N,N-dimethylsphingosine (DMS) induce apoptosis in a variety of human cancer cell lines. Int. J. Cancer, 66, 358–366 [DOI] [PubMed] [Google Scholar]
- 35. Lee M.J., et al. (1999). Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell, 99, 301–312 [DOI] [PubMed] [Google Scholar]
- 36. Lee H., et al. (2010). STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med., 16, 1421–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xia P., et al. (2000). An oncogenic role of sphingosine kinase. Curr. Biol., 10, 1527–1530 [DOI] [PubMed] [Google Scholar]
- 38. Kawamori T., et al. (2006). Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J., 20, 386–388 [DOI] [PubMed] [Google Scholar]
- 39. Kawamori T., et al. (2009). Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J., 23, 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chumanevich A.A., et al. (2010). Suppression of colitis-driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis, 31, 1787–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sugawara T., et al. (2010). Intestinal absorption of dietary maize glucosylceramide in lymphatic duct cannulated rats. J. Lipid Res., 51, 1761–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Struckhoff A.P., et al. (2004). Novel ceramide analogs as potential chemotherapeutic agents in breast cancer. J. Pharmacol. Exp. Ther., 309, 523–532 [DOI] [PubMed] [Google Scholar]
- 43. Schmelz E.M., et al. (2001). Modulation of intracellular beta-catenin localization and intestinal tumorigenesis in vivo and in vitro by sphingolipids. Cancer Res., 61, 6723–6729 [PubMed] [Google Scholar]
- 44. Symolon H., et al. (2004). Dietary soy sphingolipids suppress tumorigenesis and gene expression in 1,2-dimethylhydrazine-treated CF1 mice and ApcMin/+ mice. J. Nutr., 134, 1157–1161 [DOI] [PubMed] [Google Scholar]
- 45. Lee J.F., et al. (2006). Dual roles of tight junction-associated protein, zonula occludens-1, in sphingosine 1-phosphate-mediated endothelial chemotaxis and barrier integrity. J. Biol. Chem., 281, 29190–29200 [DOI] [PubMed] [Google Scholar]
- 46. Paik J.H., et al. (2004). Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev., 18, 2392–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Singer II, et al. (2005). Sphingosine-1-phosphate agonists increase macrophage homing, lymphocyte contacts, and endothelial junctional complex formation in murine lymph nodes. J. Immunol., 175, 7151–7161 [DOI] [PubMed] [Google Scholar]
- 48. Matsubara S., et al. (2001). Expression of alpha-catenin in alpha-catenin-deficient cells increases resistance to sphingosine-induced apoptosis. J. Cell Biol., 154, 573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fujita T., et al. (2004). Delta-catenin/NPRAP (neural plakophilin-related armadillo repeat protein) interacts with and activates sphingosine kinase 1. Biochem. J., 382, 717–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ng S.S., et al. (2009). Phosphatidylinositol 3-kinase signaling does not activate the wnt cascade. J. Biol. Chem., 284, 35308–35313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Polakis P. ( 2000). Wnt signaling and cancer. Genes Dev., 14, 1837–1851 [PubMed] [Google Scholar]