

Abstract
Lung cancer has become the top killer among malignant tumors in China and is significantly associated with somatic genetic alterations. We performed exome sequencing of 14 non–small cell lung carcinomas (NSCLCs) with matched adjacent normal lung tissues extracted from Chinese patients. In addition to the lung cancer–related genes (TP53, EGFR, KRAS, PIK3CA, and ROS1), this study revealed “novel” genes not previously implicated in NSCLC. Especially, matrix-remodeling associated 5 was the second most frequently mutated gene in NSCLC (first is TP53). Subsequent Sanger sequencing of matrix-remodeling associated 5 in an additional sample set consisting of 52 paired tumor-normal DNA samples revealed that 15% of Chinese NSCLCs contained somatic mutations in matrix-remodeling associated 5. These findings, together with the results from pathway analysis, strongly indicate that altered extracellular matrix-remodeling may be involved in the etiology of NSCLC.
Introduction
Lung cancer has become the most lethal malignant tumor in China and around the world. In China, approximately 300 000 new patients with lung cancer and more than 250 000 deaths from the disease are predicted each year (1). Lung cancer consists of two major types, non–small cell lung carcinoma (NSCLC) and small-cell lung carcinoma (SCLC). NSCLC is the most common type and accounts for approximately 85% of lung cancer. NSCLCs usually grow and spread more slowly than SCLCs but are relatively insensitive to chemotherapy. There are three common forms of NSCLCs: (i) adenocarcinomas (ADCs) that are often found in an outer area of the lung, (ii) squamous cell carcinomas (SCCs) that are usually found in the center of the lung next to an air tube (bronchus), and (iii) large cell lung carcinomas that can occur in any part of the lung. In addition, bronchioloalveolar carcinoma (BAC) is one major specific histological subtype of lung ADC arising in the distal bronchioles or alveoli that initially exhibit a specific non-invasive growth pattern. In addition to environmental factors such as smoking, genetic alterations, especially somatic mutations, play a crucial role in the etiology of the disease. To date, large-scale genome-wide sequencing projects have identified several important lung cancer–related genes such as TP53, PTEN, EGFR, KRAS, NF1, ATM, APC, ERBB4, KDR, and FGFR4 (2–4). However, our understandings of the genetic architecture of NSCLCs are far from satisfactory, especially in non-Caucasian populations such as Chinese.
To enhance our knowledge of the genetic causes of NSCLC, we performed an exome sequencing project in a cohort of 14 Chinese patients with NSCLC, including 3 ADCs, 5 SCCs, and 6 BACs. This study identified novel frequently mutated genes in these types of lung tumors in Chinese. We also screened one most frequently mutated novel gene, matrix-remodeling associated 5 (MXRA5), in an additional larger Chinese sample set of 52 patients with NSCLC and confirmed its high somatic mutation frequency. The data provide new insights into the genetic etiology of lung cancer and suggest new drug targets for lung cancer therapy or prevention.
Materials and methods
Lung tumor histology
Fourteen pairs of lung tumors and matched adjacent normal lung tissues from Chinese patients with lung cancer were analyzed, including 3 ADCs, 5 SCCs, and 6 BACs (Supplementary Table 1). Two samples are from never smokers, and the others are from smokers. None of the patients had a family history of lung cancer. The study protocol was approved by the institutional review boards of the Tianjin Medical University.
The second sample used for MXRA5 mutation screening consisted of tumor-normal pairs of lung tissues extracted from 52 Chinese NSCLC. There are 26 ADC samples, 21 SCC samples, 2 large cell carcinoma samples, and 1 adenosquamous sample (Supplementary Table 1). There are 37 smokers and 13 non-smokers in this sample set. None of the patients had a family history of lung cancer. The study protocol was approved by the institutional review boards of the Tongji Medical College and the Huazhong University of Science and Technology.
Sixty-six pairs of genomic DNA samples for exome sequencing and Sanger sequencing were extracted from lung tumors and matched normal tissues. Primary tumor tissues were sectioned, stained with routine hematoxylin and eosin, and reviewed by pathologists to identify areas of adequate tumor cellularity. Only tumor tissues with at least 70% tumor cells were used in this study. For all of the subjects, macroscopically normal lung tissues, removed at a distance of 2–5cm apart from the tumor, were collected.
DNA library preparation
Paired-end libraries were prepared following the manufacturer’s protocols (Illumina and Agilent). Briefly, 3 µg of genomic DNA was fragmented to 150–200bp using the Covaris E210 sonicator. The ends were repaired, and an “A” base was added to the 3′ ends. Paired-end DNA adaptors (Illumina) with a single “T” base overhang at the 3′ end were ligated, and the resulting constructs were purified using AMPure SPRI beads from Agencourt. The adapter-modified DNA fragments were enriched by four cycles of PCR using PE 1.0 forward and PE 2.0 reverse (Illumina) primers. The concentration and the size distribution of the libraries were determined on an Agilent Bioanalyzer DNA 1000 chip.
Capture of target genome
Whole exome capture was carried out using the protocol for Agilent’s SureSelect Human All Exon kit, targeting 38Mb of sequence from 212 911 exons and their flanking regions in approximately 20 000 genes. Five hundred nanograms of the prepared DNA library was incubated with whole exome biotinylated RNA capture baits supplied in the kit for 24h at 65°C. The captured DNA–RNA hybrids were recovered using Dynabeads MyOne Streptavidin T1 from Dynal. The DNA was eluted from the beads and desalted using Qiagen MinElute PCR purification columns. The purified capture products were then amplified using the SureSelect GA PCR primers (Agilent) for 12 cycles.
Massively parallel sequencing
Sequencing was carried out for the captured libraries with the HiSeq 2000 using 100bp paired-end reads. Libraries were loaded onto paired-end flow cells at concentrations of 4–5 pM (HiSeq 2000) to generate cluster densities of 300 000–500 000/mm2 (HiSeq 2000) following Illumina’s standard protocol using the Illumina cluster station and the Illumina cBot and HiSeq paired-end cluster kit version 1 (HiSeq 2000). To achieve high-level sensitivity and accuracy for detecting all the mutations in the whole exome, each sample was sequenced at the mean depth of 74× (Supplementary Table 2). Image analysis and base calling were carried out by Illumina Software CASAVA with default parameters. All of the sequence runs used in the data analysis passed quality controls with error rates of <2% and Eland alignment rates of >80%. On average, we generated 15 Gb of sequence per sample to obtain a mean sequencing depth of 74-fold. Of the 212 911 exons on targeted regions, 76.3% were covered with more than one read and 65.4% achieved >10× coverage.
Read mapping and alignment and variant analysis
Sequence short reads were aligned to a reference genome (NCBI human genome assembly build 37) using the BWA (Burrows-Wheeler Aligner) (5). Each alignment was assigned a mapping quality score by BWA, which is the Phred-scaled probability that the alignment is incorrect. The PCR duplicates were detected and removed by Picard (http://picard.sourceforge.net). To identify insertions and deletions (indels), the high-quality reads were gap aligned to the reference sequence using BWA (4). We then performed a local realignment of the BWA-aligned reads using the Genome Analysis Toolkit (6), which was also used for base quality score recalibration. Both VarScan (7) and MuTect (https://confluence.broadinstitute.org/display/CGATools/MuTect) were used to call somatic variants based on the local realignment results. For VarScan detection, to minimize false-positives, we set the minimum coverage as 8× in normal and 15× in tumor, the minimum reads of variant allele as 4, and the minimum proportion of variant allele as 15% in tumor. We also filter by a minimum number of two reads supporting the variant allele per strand. After these steps, germ line variants were effectively removed. The lists of SNVs (single nucleotide variants)/indels were then annotated using ANNOVAR (http://www.openbioinformatics.org/annovar/). ANNOVAR is able to use up-to-date information to functionally annotate genetic variants detected from diverse genomes. We first filtered SNVs/indels by various public databases including dbSNP, 1000 genomes, HapMap exome Project, and 200 Danish exome (8). After filtering, the novel SNVs/indels were subject to annotation using NCBI, UCSC, and Ensembl databases.
PCR and Sanger sequencing validation
For validation and screening, we performed PCR reactions and subsequent Sanger sequencing. We designed specific primers around the mutations for PCR and sequencing reaction. The standard PCR was conducted in a 25 μl reaction volume that contains 1–2U Taq DNA polymerase, 10mM Tris–HCl (pH 8.3), 0.25mM dNTPs, 0.2–2mM BSA, 1.5–2.5mM MgCl2, 20 pM of each primer, and ~10ng genomic DNA. The PCR reaction conditions were set as follows: 94°C for the first 5min, followed by 35 cycles of 94°C denaturation for 30 s, 50°C annealing for 30 s, and 72°C extension for 45 s. The PCR products were checked by gel electrophoresis and then sent out to Eton Bioscience Inc. (http://etonbio.com/) for Sanger sequencing. The primers for PCR reaction were used for sequencing reactions. DNA sequences were checked by using the software package Sequencher.
Functional prediction of missense mutations
To infer the functional importance of the identified missense mutations, we used four currently popular algorithms: (i) SIFT (http://sift.jcvi.org/), (ii) Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), (iii) LRT (http://www.genetics.wustl.edu/jflab/lrt_query.html), and (iv) MutationTaster (http://www.mutationtaster.org/) along with a conservation score (PhyloP) (http://compgen.bscb.cornell.edu/phast/help-pages/phyloP.txt) to predict if a non-synonymous SNV affects the function of its associated protein. These four algorithms use different information and are based on different training data; each has its own strength and weakness. The prediction scores from these algorithms were recently compiled in the database dbNSFP (http://sites.google.com/site/jpopgen/dbNSFP). To make a more reliable prediction, we used majority vote standard to identify putative driver mutations. That is, a driver mutation needs to fulfill two requirements: (i) a non-synonymous SNV that is located within a conserved site (i.e., phyloPnew score (from dbNSFP) > 0.95) and (ii) a non-synonymous SNV that is predicted by at least three of four algorithms to affect protein function. In each prediction algorithm, we used default thresholds to define deleterious/damaging non-synonymous SNVs.
Pathway analysis
Pathway and other gene set analyses of the somatic mutations were applied for the gene ontology gene sets obtained through the R/GO.db software package, the Kyoto Encyclopedia of Genes and Genomes pathways obtained through the R/KEGG.db software package, the set of publicly available GeneGo pathways maps, the HGNC gene family database, the chromatin-oriented ChromDB database, and several pathways and gene sets defined by project-specific tumor mutation profiles. Both patient-oriented (9,10) and gene-oriented (11) methods were used. For a given gene set, the patient-oriented algorithm estimates the significance of the distribution of mutations across patients of a per-patient binary gene set statistic that indicate the presence or absence of a somatic mutation in the gene set of question. In the gene-based approach, the significance for each category was obtained by comparing the observed ratios of non-silent to silent somatic mutations for the gene set to the overall ratio for our samples.
Results
Overview of somatic mutation profiles
In total, we identified 3321 high-confidence somatic mutations in the 14 Chinese tumor-normal pairs sequenced (Table I, Supplementary Tables 3 and 4). These 3321 mutations are distributed in various genomic regions, including exonic (48.7%), splicing (1.2%), non-coding RNA (ncRNA; 10.2%), 5′/3′-untranslated regions (UTR) (2.6%), intronic (22.8%), upstream/downstream (0.8%), and intergenic regions (13.7%) (Table I). There are 1659 somatic mutations of potential biological significance as they reside in protein coding regions or splicing sites (Supplementary Table 7). Among them, 447 are silent (synonymous) substitutions, 1038 are missense substitutions causing amino acid changes, 88 are non-sense mutations leading to truncated proteins, 2 are stop-loss mutations causing abnormal protein extension, and 41 SNVs occurred at splicing sites. We also found 39 indels causing frameshift mutations and 4 indels that did not alter reading frame, ranging from 1 to 6bp in length. Non-sense, frameshift indels and splicing mutations generally lead to the inactivation of the protein products. To evaluate missense mutations, we used four algorithms to make a consensus prediction to identify putative driver mutations. These functional prediction algorithms are based on phylogenetics, structural biology, bioinformatics, or population genetics. Of 1038 missense mutations, 91 (8.8%) were predicted to be deleterious by all four algorithms and 216 (20.8%) were predicted by three or four algorithms (Supplementary Table 8).
Table I.
Distribution of somatic SNVs learned from the exome sequencing of 14 Chinese lung cancer patients
| Mean | % | |
|---|---|---|
| Average of total somatic SNVs | 237.2 | |
| Exonic | 115.6 | 48.7 |
| Splicing | 2.9 | 1.2 |
| ncRNA | 24.1 | 10.2 |
| 5′/3′-UTR | 6.1 | 2.6 |
| Intronic | 54.1 | 22.8 |
| Upstream/downstream | 1.9 | 0.8 |
| Intergenic | 32.4 | 13.7 |
| SNVs in protein coding regions | ||
| Frameshift deletion | 1.6 | 1.4 |
| Frameshift insertion | 1.1 | 1.0 |
| Non-frameshift deletion | 0.2 | 0.2 |
| Non-frameshift insertion | 0.1 | 0.1 |
| Nonsense (truncated) | 6.3 | 5.4 |
| Nonsense (extension) | 0.1 | 0.1 |
| Non-synonymous | 74.1 | 64.2 |
| Synonymous | 31.9 | 27.6 |
The somatic mutations occurring in exonic regions or splicing sites are distributed in 1454 genes. A comparison of the 1454 mutated genes with the Catalogue of Somatic Mutations in Cancer (COSMIC) database revealed that 1015 of these genes have been previously archived in COSMIC database to be somatically mutated in human tumors.
We observed, on average, approximately 115 somatic SNVs in exonic regions per Chinese exome with the most frequently altered exome having 493 SNVs. No somatic mutations in exonic regions were identified in two BACs samples. On average, SCC had 243 somatic exonic SNVs, ADC had 111 somatic exonic SNVs, and BAC had only 11 somatic SNVs in exonic regions (Supplementary Figures 1 and 2). The observed ratio of non-synonymous to synonymous change (dN/dS) did not show significant difference across different types of lung tumors (ADC versus BAC versus SCC: 3.3 versus 2 versus 1.9; P > 0.2) (Supplementary Figure 3). Among 3321 SNVs identified by exome sequencing, somatic variants occurred predominantly at G:C base pairs (67.5% in ADC, 70.1% in SCC, and 59.8% in BAC). G:C > T:A and G:C > A:T transversions are the two most prevalent categories in ADC and SCC (32.5% and 23.9% in ADC; 29.8% and 29.1% in SCC for G:C > T:A and G:C > A:T, respectively). For BAC, the top 2 most prevalent mutation types are G:C > A:T and A:T > G:C transversions, with proportions being 30.2% and 19.8%, respectively. When comparing somatic with germ line mutation patterns, G:C to T:A transversions (32.5% versus 7.2% in ADC, 29.8% versus 7.2% in SCC, and 18.4% versus 7.4% in BAC) and A:T to T:A transitions (13.4% versus 5.1% in ADC, 6.5% versus 5.2% in SCC, and 11.3% versus 5.1% in BAC) were strongly enriched for somatic mutations (Supplementary Figure 4).
Fig. 1.
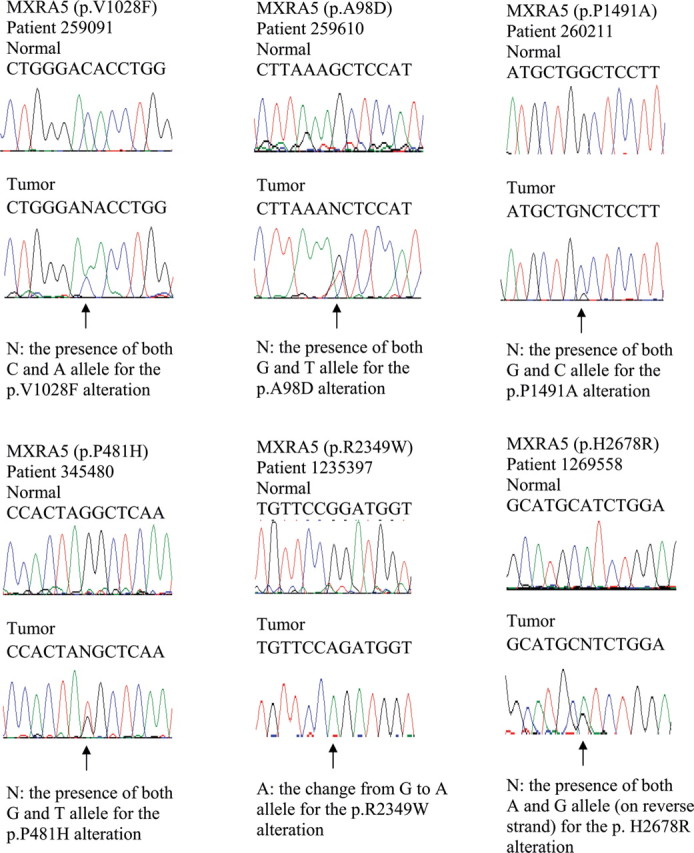
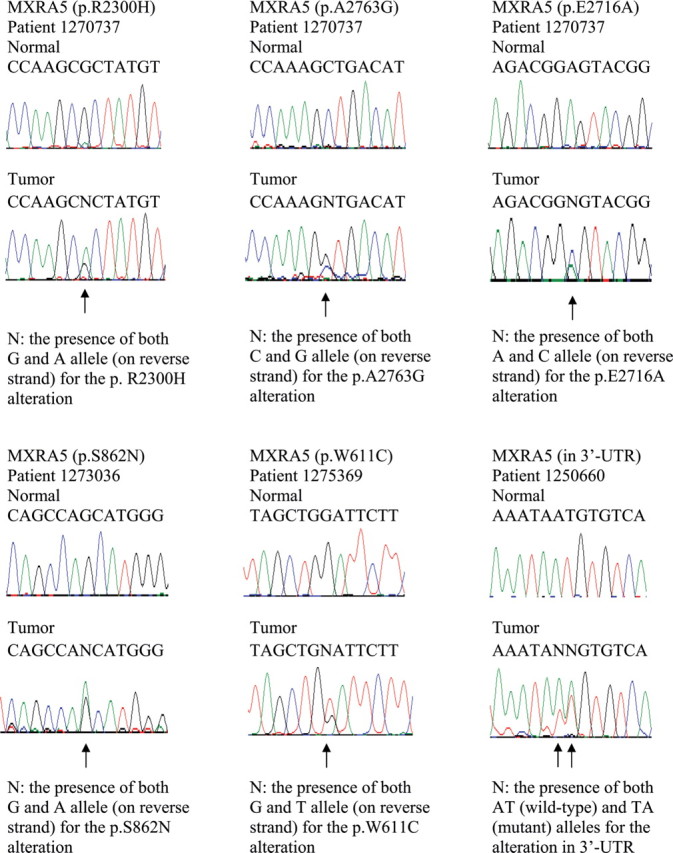
Sanger sequencing chromatograms of somatic MXRA5 mutations in Chinese patients with NSCLC. Mutations are shown in the indicated tumor compared with matched normal DNA and are marked by arrows.
As expected, the number of somatic SNVs identified in never smokers (mean = 63) is much less than in smokers having an average of 219 exonic somatic SNVs per genome (Supplementary Figure 2). In smokers, G:C > T:A transversions (28.4%) were the most common change observed, followed by G:C > A:T (26.5%) and A:T > G:C (17.4%) transversions. In never smokers, G:C > T:A transversions in never smokers (7.8%) are significantly fewer than in smokers (P = 0.0018) (Supplementary Figure 4). These results are consistent with previously documented tobacco exposure-related mutation signatures (2–4).
Somatic variants validation
To evaluate false-positive rates of the identified somatic mutations, we selected a subset of 106 mutations out of our high-confidence somatic mutation list for Sanger sequencing validation. The selected mutations are distributed in two categories of genes: (i) five previously known lung cancer driver genes (TP53, EGFR, KRAS, PIK3CA, and ROS1) that have non-silent somatic mutations or mutations located in ncRNA coding regions; and (b) some novel genes that are predicted to have non-silent somatic mutations in at least 2 tumors in our Chinese cohort. Novel means that these genes remain seldom studied. We validated 98 somatic mutations out of the initial 106 variants, indicating that the false-positive rate of our high-confidence somatic mutations is only approximately 7.5%.
TP53 was the most frequently mutated one, with seven tumor samples (50%) harboring non-silent TP53 mutations (two non-sense and five missense mutations; Supplementary Table 5). Three tumors are SCC, two are ADC, and two are BAC. There are two non-sense mutations causing truncated TP53 protein in one ADC and one BAC tumor (Supplementary Table 5). All of these seven TP53 somatic mutations have been reported in COSMIC, with six (genomic position at chr17: 7576855, 7577120, 7577538, 7577548, 7578455, and 7578534—hg19/GRCh37 coordinates) found in 143 lung tumors (mainly of NSCLC type) according to COSMIC (Supplementary Table 6). EGFR had a five amino acid non-frameshift deletion in one BAC sample at chr7: 55242464. In COSMIC, there is a one amino acid deletion (p.E746del) at this position in a lung ADC sample. We also confirmed two novel somatic mutations in ncRNA coding regions within EGFR, which had not been reported before. The KRAS mutation we identified was a missense mutation in a patient with BAC at chr12:25398284, reported by COSMIC to be existent in 781 lung tumors (most are NSCLC; Supplementary Table 6). Two missense PIK3CA mutations were confirmed in an ADC sample of ours, both of which have been reported by COSMIC as existent in multiple types of tumors. The seven novel somatic mutations in ncRNA coding regions within ROS1 in an SCC sample of ours were validated as well.
As for novel genes, Sanger sequencing validated that 19 genes were mutated in at least 2 of 14 samples, with a mutation rate of >14%, which are as follows: MXRA5, STAG2, ZFHX4, HMCN1, ABCA12, CSMD3, EIF4G3, EVI1/MECOM, FCRL4, FN1, PCSK5, SLC8A1, TSHZ3, KIAA1109, ZBTB41, MUC16, PKHD1L1, USH2A, and XRN1 (Supplementary Table 5). Except for TP53, MXRA5 was the most frequently mutated gene in our sample set, which had four missense mutations in four tumors including one ADC, two SCC and one BAC, with mutation frequency being 28.6% (4 of 14). The genes mutated in three tumors were ZFHX4 and PKHD1L1. FCRL4 was the only gene with somatic missense mutations in two BAC samples (Supplementary Table 5).
Sanger screening for MXRA5 somatic mutations
Because that MXRA5 was seldom studied before and the second most frequently mutated gene in our initial exome sequencing sample, we want to further examine this gene and determine its somatic mutation frequency using larger sample set. Therefore, we performed Sanger sequencing of all the exons and UTRs of this gene using a validation cohort composed of tumor/normal paired DNA samples extracted from another 52 Chinese patients with NSCLC (Supplementary Table 1). Incorporating the original 14 patients with NSCLC analyzed by exome sequencing, we found that 15% of Chinese NSCLCs (10 of 66) contained 12 somatic mutations in MXRA5. Specifically, MXRA5 had mutations in 7 of 29 ADCs (24%), in 2 of 26 SCCs (8%), and in 1 of 6 BAC (17%) (Table II and Figures 2 and 3).
Table II.
MXRA5 (on chromosome X) mutations detected in exome sequencing and screening samples
| Sample ID* | Tumor type | Position | Ref | Var | Region | Function | Seq_change |
|---|---|---|---|---|---|---|---|
| MXRA5 mutations in the initial exome sequencing sample of 14 Chinese NSCLC | |||||||
| 259091 | ADC | 3240644 | C | A | Exonic | Non-synonymous | NM_015419:p.V1028F |
| 259610 | SCC | 3248710 | G | T | Exonic | Non-synonymous | NM_015419:p.A98D |
| 260211 | SCC | 3239255 | G | C | Exonic | Non-synonymous | NM_015419:p.P1491A |
| 345480 | BAC | 3242284 | G | T | Exonic | Non-synonymous | NM_015419:p.P481H |
| MXRA5 mutations in the screening sample of 52 Chinese NSCLC | |||||||
| 1235397 | ADC | 3229199 | G | A | Exonic | Non-synonymous | NM_015419:p.R2349W |
| 1269558 | ADC | 3228211 | T | C | Exonic | Non-synonymous | NM_015419:p.H2678R |
| 1270737 | ADC | 3229345 | C | T | Exonic | Non-synonymous | NM_015419:p.R2300H |
| 1270737 | ADC | 3227956 | G | C | Exonic | Non-synonymous | NM_015419:p.A2763G |
| 1270737 | ADC | 3228097 | T | G | Exonic | Non-synonymous | NM_015419:p.E2716A |
| 1273036 | ADC | 3241141 | C | T | Exonic | Non-synonymous | NM_015419:p.S862N |
| 1275369 | ADC | 3241893 | C | T | Exonic | Non-synonymous | NM_015419:p.W611C |
| 1250660 | ADC | 3227666 | AT | TA | 3ʹ-UTR | Messenger RNA stability | |
*Sample 1269558 is from a male nonsmoker, sample 1273036 is from a female smoker, and all the others are from male smokers.
Fig. 2.

Schematic drawing of MXRA5 gene structure. MXRA5 encoded protein consists of 2824 amino acids. The conserved domains and 11 missense somatic mutations validated in our 9 Chinese NSCLC tumor samples were drawn with MXRA5 structure. Seven missense mutations labeled in red are located in the conserved domains of MXRA5.
Fig. 3.

The GeneGo “Cell Adhesion_ECM remodeling” pathway map. The legend for the symbols of this figure is standard for GeneGo pathway maps and is available at http://www.GeneGo.com. Note that additionally, the golden hexagons with the letter G indicate network objects (e.g. proteins) associated with the genes that are mutated in our samples.
More MXRA5 mutations were seen in smokers, 9 of 48 (18.8%), versus non-smokers, 1 of 16 (6.3%). Furthermore, 9 of 48 (18.8%) men versus 1 of 18 (5.6%) women had mutations in MXRA5. These suggest that smoking and gender may predict somatic MXRA5 mutations in NSCLC, similar to observations on somatic BRAF mutations in colorectal cancer (12). The 12 somatic MXRA5 mutations that we identified in 10 NSCLCs were not present in the dbSNP135 database (http://www.ncbi.nlm.nih.gov/projects/SNP/), which contains approximately 40 million SNPs (single nucleotide polymorphisms) submitted by the 1000 Genomes Project. Similarly, these mutations were not any of the SNVs of MXRA5 identified by the exome sequencing project of the National Heart, Lung and Blood Institute, which sequenced 5400 exomes (http://evs.gs.washington.edu/EVS/). Therefore, our reported somatic mutation loci in the MXRA5 gene were not due to germ line polymorphisms or germ line mutations.
Pathway and gene set analysis
Table III and Supplementary Table 9 shows the most significantly mutated pathways we have identified. The ratios of non-silent to silent somatic (NS/S) mutations across the 10 individuals with at least one non-silent somatic mutations are significantly higher than expected (P ≤ 0.05) in several pathways, including a DNA damage pathway (“GeneGo role of SUMO in p53 regulation,” 8:0), apoptosis-related pathways (“GeneGo antiapoptosis mediated by external signals” and “GeneGo apoptosis stimulation by external signals,” 20:3; “GeneGo role of CDK5 in neuronal death and survival,” 7:0), and AKT signaling (7:0). The “HGNC major cadherins” (9:0) and “HGNC olfactory receptors” (46:9) gene families also showed high NS/S ratios. In addition, several cancer-specific pathways and gene sets had significantly high NS/S ratios (“Stransky 2011 Table S8,” “Liu/You ADC/SCC Mutated,” “Tissue factor signaling in Lung Cancer,” “Cancer Census”).
Table III.
Several most significant pathways for Chinese NSCLC and their associated genes
| Gene | ECM remodeling (GeneGo) | ECM remodeling (Zhang et al. 2009) | Vogelstein Core Cancer Pathways | Other Cancer-Related Pathways | Downstream Pathways |
|---|---|---|---|---|---|
| MXRA5 | Yes | ||||
| FN1 | Yes | Yes | Integrins, TGF-β | Integrins, MAPK, Notch, TGF-β, antiapoptosis, tumor metastasis | α5/β1 integrin |
| NID1 | Yes | ||||
| LAMA3 | Yes | Cell adhesion, integrins | Integrins | ||
| MMP16 | Yes | Invasion | α1/β1 integrin | ||
| COL4A6 | Yes | Yes | Integrins | Integrins, antiapoptosis, cytoskeleton remodeling | α1/β1 integrin |
| CDH13 | Yes | G1/S phase transition, cell adhesion, small GTPases | |||
| NRXN3 | Yes | Cell adhesion (Kyoto Encyclopedia of Genes and Genomes) | |||
| COL4A2 | Yes | Integrins | Integrins, antiapoptosis, cytoskeleton remodeling, tumor metastasis | α1/β1 integrin | |
| VCAN | Yes | EMT, cell adhesion | EGF, ERBB2 family, cytoskeleton remodeling | ||
| LAMA1 | Yes | Cell adhesion, integrins | Integrins |
A second approach for gene set enrichment was used, which measures the presence or absence of at least one alteration for each gene set by sample. Among gene sets with at least two different genes mutated in our samples, the two with the greatest statistical significance (P < 5E-10) in this measure were associated with extracellular matrix (ECM) remodeling (11 of 81 genes mutated in 9 of 10 samples; P = 1e−10). The mutated genes in the most significant ECM remodeling gene set include two genes mutated in multiple samples, MXRA5 (4 samples) and FN1 (2 samples), as well as CDH13, COL4A2, COL4A6, LAMA1, LAMA3, MMP16, NID1, NRXN3, and VCAN (Table III).
Discussion
Similar to the findings in Caucasians (2–4), Chinese smokers have much higher mutation rates than never smokers, and G:C > T:A transversions (28.4%) were the most frequent somatic mutation pattern observed in Chinese smokers with lung cancer. The numbers of somatic mutations were also significantly different across different NSCLC subtypes in Chinese (SCC > ADC > BAC). There exists wide variation in the number of somatic mutations even within the group of patients having the same NSCLC subtype and smoking status (Supplementary Figure 1), suggesting the high heterogeneity in the genetic causes of NSCLC in Chinese. COSMIC database searches for our identified 1454 genes with functionally important somatic mutations revealed that 30% (439) of them were not present in COSMIC, further highlighting the complexity of lung cancer genomes and the strong carcinogenic effects of tobacco exposure on mutagenesis.
We identified recurrently mutated genes in our Chinese patients with NSCLC. In addition to TP53 that has been well established as a tumor suppressor, MXRA5 was the most striking NSCLC candidate gene with frequent somatic mutations in our sample. MXRA5, also known as adlican, encodes an adhesion proteoglycan and belongs to a group of genes involved in ECM remodeling and cell–cell adhesion (13). Although the function of MXRA5 in cancer is unknown, somatic mutations in MXRA5 have been identified in tumors obtained from a variety of tissues such as skin, brain, lung, ovary (reported in the COSMIC database; http://www.sanger.ac.uk/genetics/CGP/cosmic/) and parietal pleura (14). By combining initial discovery cohort and subsequent screening cohort, we identified 11 missense mutations that may affect MXRA5 protein structure and 1 mutation in the 3ʹ-UTR that may conceivably affect messenger RNA stability or micro-RNA interaction. The somatic mutations are distributed throughout the length of the gene. The associations of MXRA5 somatic mutations with lung cancer seem stronger in men because more than 90% (11 of 12) of the mutations were found in tumors from Chinese male patients with NSCLC. This may be partly because men contain a single X chromosome and thus will lose their sole “good” copy of the MXRA5 gene in lung tumors with MXRA5 somatic mutations. Given this and the distribution of mutations throughout the gene, it is reasonable to hypothesize that MXRA5 may act as a tumor suppressor. However, the up-regulation of MXRA5 messenger RNA expression at a modest level was observed in ovarian and esophageal cancer (15,16). Yet, the increased gene expression of MXRA5 in certain cancer may be merely a consequence rather than a cause or a contributing factor of tumorigenesis. Answers remain elusive for the exact role that the ECM structural protein, MXRA5, could play in tumorigenesis.
Among MXRA5 mutations, 64% (7 of 11) are located in highly conserved protein domains. Specifically, one missense mutation, p.A98D, is located within the leucine-rich repeat (LRR) domain, and six missense mutations (p.P481H, p.W611C, p.R2300H, p.H2678R, p.E2716A, and p.A2763G) are within the immunoglobulin (Ig) domains of MXRA5 (Figure 2). The LRR domain mediates protein–ligand interactions, which enables extracellular LRR containing proteins to efficiently regulate intercellular communication and cell–cell adhesion (17). Somatic mutations in the Ig domains of the PTPρ protein, which plays an important role in cell–cell adhesion, are loss-of-function mutations that can promote tumor migration and metastasis (18). In addition, the LKB1 loss of function caused by inactivating mutations promotes NSCLC malignancy through remodeling of the ECM microenvironment (19). Collectively, these findings lead us to hypothesize that the somatic mutations we identified in MXRA5 are inactivating mutations that may well contribute to tumor migration and metastasis via ECM remodeling pathway in a similar way to loss-of-function mutations within PTPρ and LKB1.
Pathway analysis highlighted the importance of ECM remodeling genes in the genetic etiology of Chinese NSCLC. The ECM remodeling pathway was mutated in 9 of the 10 Chinese patients with lung cancer who have at least one non-silent somatic mutation. This pathway is also enriched with genes associated with the wound healing process. Given that smokers constitute the majority of our Chinese sample, exposure to smoking may play a significant role in the development of NSCLC in Chinese smokers by promoting somatic mutations in genes involved in lung injury healing and cell–cell adhesion processes. Many of the genes of this pathway were previously found to have significantly higher expression in tumor fibroblasts than in normal fibroblasts (16) (MXRA5, FN1, CDH13, COL4A6, and NRXN3). The mutated genes of this pathway also belong to multiple specifically defined core cancer pathways (11), including integrin signaling (COL4A2, COL4A6, FN1, LAMA1, and LAMA3), cell adhesion (MXRA5, CDH13, LAMA1, and LAMA3), invasion (MMP16), small GTPases (CDH13), G1/S phase transition (CDH13), transforming growth factor β (TGF-β) signaling (FN1), and other cancer-associated processes such as tumor metastasis, antiapoptosis, and cytoskeleton remodeling (Table III). FN1, which was mutated in two samples of the present study, belongs to integrins, MAPK, Notch, TGF-β, and antiapoptosis signaling pathways and is involved in tumor metastasis. COL4A2, COL4A6, and MMP16 are all upstream of α1/β1 integrin signaling pathway, whereas FN1 is upstream of the α5/β1 integrin signaling pathway (Figure 3). Interestingly, VCAN is upstream of the EGF, ERBB2 family, and cytoskeleton remodeling signaling processes and associated with the epithelial to mesenchymal transition process (Figure 3).
We investigated the potential drugs that may target the most frequently mutated genes and pathways in our Chinese NSCLC sample (Supplementary Table 10). In our gene list, TP53 and several mutated genes in ECM Remodeling category (CDH13, FN1, LAMA1, LAMA3, NID1, and MMP16) may interact with known drugs (20). For example, the gene expression of FN1, which has been reported to be an oncogene (21), is increased in response to the anticancer drug tamoxifen (22) and the anticancer combination of alitretinoin and ascorbic acid (23). Similarly, FN1 activity is associated with chemoresistance to drugs such as doxorubicin (24,25). It has been reported that such chemoresistance affects FN1 in NSCLC work, in part, through the activation of the Akt/mTOR/p70S6K pathway (26). However, the significant drug responses of certain genes do not adequately justify them as feasible drug targets for treating diseases. In this study, an open translational research question then is whether targeting the promising genes identified earlier is effective in preventing recurrence of NSCLC.
We sequenced six BAC samples in this study. BAC is one of four histological distinct subtypes of lung ADC. It possesses unique clinical and pathological features and prognosis and responds to different treatments. FCRL4 is the only gene with missense mutations in two BAC samples. This gene is one of Fc receptor-like glycoproteins and encodes a member of the Ig receptor super family. FCRL4 aberrations were associated with lymphoma and myeloma previously (27,28).
The four truncating mutations we identified in BAC samples occurred in four genes, APC, KIDINS220, KIAA1211, and STAG2. APC is a tumor suppressor gene acting as an antagonist of the Wnt signaling pathway. It is also involved in other processes including cell migration and adhesion, transcriptional activation, and apoptosis. Defects in this gene have been linked to lung cancer before (29,30), consistent with our observation that APC protein is truncated in lung tumor. STAG2 is a gene encoding a subunit of the cohesion complex that regulate the separation of sister chromatids during cell division. STAG2 is located on the X chromosome so a single mutation event like the truncating mutation we identified here in a male patient with BAC (patient 345954) will totally abolish its function. Several very recent papers (31–34) showed that inactivating mutations of STAG2 led to chromatid cohesion defect, which is the cause of aneuploidy that drives mutator phenotypes in human cancer. Our observation of STAG2 truncating mutation in male lung tumor supports the importance of STAG2 in human cancer.
In summary, this study systematically investigated the somatic genetic variants associated with Chinese NSCLC based on whole exome sequencing. The current exome sequencing technology and analytical methods are inefficient to identify certain structural gene changes such as gene fusions. For example, we missed the detection of recently established NSCLC relevant ALK-PTPN3 gene fusion (35) in our samples. However, exome sequencing is especially powerful in detect meaningful point mutations at the genome level, and thus we still made certain novel discoveries. Particularly, we found that the ECM remodeling/cell–cell adhesion gene set is the most significantly altered gene set in Chinese NSCLC. Functionally, ECM modification and remodeling is one of the most frequent cellular events in cancer progression. Before tumor cells leave their sites of origin and become metastatic, they must first detach from neighboring cells and remodel ECM structure and activity to gain the invasion and metastasis phenotypes (36). ECM, on the other hand, is not only a passives structure acting as a physical support/barrier but also a dynamic structure that contains ECM proteins that interact with cell surface receptors to initiate or modulate signal transduction of cancer cells residing on or within it (37). Our findings of frequent MXRA5 somatic mutations in Chinese NSCLCs contribute to the body of evidence implicating altered ECM remodeling in the genetic etiology of NSCLC. Other novel genes identified in this study also provide new clues regarding molecular targets for treating non–small cell lung cancer, especially in Chinese.
Supplementary Data
Supplementary Figure 1. Number of somatic mutations in protein coding regions detected in 14 lung cancer genomes. Samples were organized according to their histology and smoking status. FS, frameshift; indel, insertion and deletion; SMK, smokers; Non-SMK, non-smokers.
Supplementary Figure 2. Number of SNVs across the Chinese lung cancer samples. (A) ADC, BAC, and SCC; (B) smokers and non-smokers.
Supplementary Figure 3. dN/dS across the samples. (A) ADC, BAC, and SCC; (B) smokers and non-smokers.
Supplementary Figure 4. Somatic single-nucleotide mutation trends and patterns in Chinese lung cancer samples. Distribution of specific nucleotide changes among germ line and somatic variations in the lung cancer exome was presented for different histology (ADC, BAC, and SCC) and smoking status (SMK and Non-SMK).
Supplementary Tables
Supplementary Table 1. Clinical information of Chinese patients with NSCLC.
Supplementary Table 2. Sequence reads and coverage among 14 pairs of matched normal and tumor samples.
Supplementary Table 3. Summary of somatic SNVs in 14 Chinese patients with lung cancer.
Supplementary Table 4. All somatic SNVs in 14 Chinese lung cancer genomes.
Supplementary Table 5. Somatic SNVs validated by the Sanger sequencing.
Supplementary Table 6. Our validated somatic SNVs that are reported in the COSMIC database.
Supplementary Table 7. All somatic mutations in protein coding regions or splicing sites in 14 Chinese patients with lung cancer.
Supplementary Table 8. Missense mutations predicted to affect protein function.
Supplementary Table 9. The gene set enrichment analysis results for all of the 14 Chinese
NSCLC tumors in the initial exome sequencing study. The columns of this table contain gene set name (column name: category), patient-oriented permutation P value (p.values.perm.null), non-silent/silent alteration ratio (ratio), number of mutated genes (nG), number of mutated subjects (nS), total number of genes in each gene set (nSet), number of somatic mutations in each gene set (nGS), the mutated genes (geneDstn), the mutated samples (smplDstn), the mutated genes and corresponding samples (genesmplDstn). The significance cutoff is 1E−05.
Supplementary Table 10. Drugs (rows) and their druggable target genes identified in this study (columns). We limit the druggable targets to genes that were mutated in more than two individuals or were part of the most enriched pathway, the ECM remodeling pathway. The green-shaded entries indicate the drug by target interactions (labeled “1”). Entries in black indicate that the corresponding genes are not drug targets. The pink- and orange-shaded drug names indicate drugs that have been applied, at least in clinical trials for treatment of lung cancer (pink) or some other type of cancer (orange).
Acknowledgements
This work was supported in part by the National Institutes of Health (grant nos. 7R01AT003203, 7R01AT005522, 5R01CA134433, 5R01CA134682, 5R01CA113793, and 5R01CA129533) and Advancing a Healthier Wisconsin Fund. The authors thank Gary D. Stoner, Michael James, Haris Vikis, and Jay Tichelaar for their comments on the manuscript.
References
- 1. Houwen L. (2003). State of the art: lung cancer in China. Ann. Thorac. Cardiovasc. Surg. 9 147–148 [PubMed] [Google Scholar]
- 2. Ding L., et al. (2008). Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee W., et al. (2010). The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 465 473–477 [DOI] [PubMed] [Google Scholar]
- 4. Pleasance E.D., et al. (2010). A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463 184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li H. et al. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKenna A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koboldt D.C., et al. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25 2283–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Y., et al. (2010). Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 42 969–972 [DOI] [PubMed] [Google Scholar]
- 9.Boca S.M., et al. Patient-oriented gene set analysis for cancer mutation data. Genome Biol. (2010);11:R112. doi: 10.1186/gb-2010-11-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parsons D.W., et al. (2011). The genetic landscape of the childhood cancer medulloblastoma. Science 331 435–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jones S., et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321 1801–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rozek L.S., et al. (2010). Smoking, gender, and ethnicity predict somatic BRAF mutations in colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 19 838–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rødningen O.K., et al. (2008). Radiation-induced gene expression in human subcutaneous fibroblasts is predictive of radiation-induced fibrosis. Radiother. Oncol. 86 314–320 [DOI] [PubMed] [Google Scholar]
- 14. Sugarbaker D.J., et al. (2008). Transcriptome sequencing of malignant pleural mesothelioma tumors. Proc. Natl. Acad. Sci. U.S.A. 105 3521–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buckanovich R.J., et al. (2007). Tumor vascular proteins as biomarkers in ovarian cancer. J. Clin. Oncol. 25 852–861 [DOI] [PubMed] [Google Scholar]
- 16. Zhang C., et al. (2009). Fibroblast growth factor receptor 2-positive fibroblasts provide a suitable microenvironment for tumor development and progression in esophageal carcinoma. Clin. Cancer Res. 15 4017–4027 [DOI] [PubMed] [Google Scholar]
- 17. de Wit J., et al. (2011). Role of leucine-rich repeat proteins in the development and function of neural circuits. Annu. Rev. Cell Dev. Biol. 27 697–729 [DOI] [PubMed] [Google Scholar]
- 18. Yu J., et al. (2008). Tumor-derived extracellular mutations of PTPRT /PTPrho are defective in cell adhesion. Mol. Cancer Res. 6 1106–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao Y., et al. (2010). LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proc. Natl. Acad. Sci. U.S.A. 107 18892–18897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahmed J., et al. (2011). CancerResource: a comprehensive database of cancer-relevant proteins and compound interactions supported by experimental knowledge. Nucleic Acids Res. 39 D960–D967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bozic I., et al. (2010). Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. U.S.A. 107 18545–18550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horii Y., et al. (2006). The regulatory effect of tamoxifen on fibronectin expression in estrogen-dependent MCF-7 breast carcinoma cells. Oncol. Rep. 15 1191–1195 [PubMed] [Google Scholar]
- 23. Kim K.N., et al. (2006). Retinoic acid and ascorbic acid act synergistically in inhibiting human breast cancer cell proliferation. J. Nutr. Biochem. 17 454–462 [DOI] [PubMed] [Google Scholar]
- 24. Hazlehurst L.A., et al. (2001). Reduction in drug-induced DNA double-strand breaks associated with beta1 integrin-mediated adhesion correlates with drug resistance in U937 cells. Blood 98 1897–1903 [DOI] [PubMed] [Google Scholar]
- 25. Pupa S.M., et al. (2007). Regulation of breast cancer response to chemotherapy by fibulin-1. Cancer Res. 67 4271–4277 [DOI] [PubMed] [Google Scholar]
- 26. Han S., et al. (2006). Fibronectin stimulates non–small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res. 66 315–323 [DOI] [PubMed] [Google Scholar]
- 27. Kazemi T., et al. (2008). Fc receptor-like 1-5 molecules are similarly expressed in progressive and indolent clinical subtypes of B-cell chronic lymphocytic leukemia. Int. J. Cancer 123 2113–2119 [DOI] [PubMed] [Google Scholar]
- 28. Kazemi T., et al. (2009). Low representation of Fc receptor-like 1-5 molecules in leukemic cells from Iranian patients with acute lymphoblastic leukemia. Cancer Immunol. Immunother. 58 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ali A.H., et al. (2011). Aberrant DNA methylation of some tumor suppressor genes in lung cancers from workers with chromate exposure. Mol. Carcinog. 50 89–99 [DOI] [PubMed] [Google Scholar]
- 30. Zhang Y., et al. (2011). Methylation of multiple genes as a candidate biomarker in non–small cell lung cancer. Cancer Lett. 303 21–28 [DOI] [PubMed] [Google Scholar]
- 31.Burgess D.J. Aneuploidy stokes the fire. Nat. Rev. Genet. (2011);12:666. doi: 10.1038/nrg3070. [DOI] [PubMed] [Google Scholar]
- 32. Kolodner R.D., et al. (2011). Cancer. Aneuploidy drives a mutator phenotype in cancer. Science 333 942–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sheltzer J.M., et al. (2011). Aneuploidy drives genomic instability in yeast. Science 333 1026–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Solomon D.A., et al. (2011). Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333 1039–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jung Y., et al. (2012). Discovery of ALK-PTPN3 gene fusion from human non–small cell lung carcinoma cell line using next generation RNA sequencing. Genes. Chromosomes Cancer 51 590–597 [DOI] [PubMed] [Google Scholar]
- 36. Lopez J.I., et al. (2008). Biomechanical regulation of cell orientation and fate. Oncogene 27 6981–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Streuli C. (1999). Extracellular matrix remodelling and cellular differentiation. Curr. Opin. Cell Biol. 11 634–640 [DOI] [PubMed] [Google Scholar]