

Abstract
Genetic variation for plastic phenotypes potentially contributes phenotypic variation to populations that can be selected during adaptation to novel ecological contexts. However, the basis and extent of plastic variation that manifests in diverse environments remains elusive. Here, we characterize copper reaction norms for mRNA abundance among five Saccharomyces cerevisiae strains to 1) describe population variation across the full range of ecologically relevant copper concentrations, from starvation to toxicity, and 2) to test the hypothesis that plastic networks exhibit increased population variation for gene expression. We find that although the vast majority of the variation is small in magnitude (considerably <2-fold), not just some, but most genes demonstrate variable expression across environments, across genetic backgrounds, or both. Plastically expressed genes included both genes regulated directly by copper-binding transcription factors Mac1 and Ace1 and genes indirectly responding to the downstream metabolic consequences of the copper gradient, particularly genes involved in copper, iron, and sulfur homeostasis. Copper-regulated gene networks exhibited more similar behavior within the population in environments where those networks have a large impact on fitness. Nevertheless, expression variation in genes like Cup1, important to surviving copper stress, was linked with variation in mitotic fitness and in the breadth of differential expression across the genome. By revealing a broader and deeper range of population variation, our results provide further evidence for the interconnectedness of genome-wide mRNA levels, their dependence on environmental context and genetic background, and the abundance of variation in gene expression that can contribute to future evolution.
Keywords: gene expression variation, environmental plasticity, copper, mRNA population genetic variation, phenotypic evolution
Introduction
One of the longest standing questions in evolutionary biology is how phenotypic variation is maintained in evolving populations (Bürger 2000). Phenotypic variation within populations comes not only from genetic differences between individuals (G), but also from differences in the environments they experience (E). Moreover, individuals exhibit genetic variation in their degree of response to the same sets of environments (G × E). This population variation for plasticity has important implications for theoretical predictions of phenotypic evolution, introducing context-dependence to both phenotypic variation and selection (Schlichting 2008; Pfennig et al. 2010; Moczek et al. 2011). The nature of polymorphisms inducing plasticity and the molecular mechanisms underlying plasticity are still emerging (Callahan et al. 2008), but genetic variation in plasticity may be assessed early in the chain of events linking genotypes to phenotypes by quantifying the amount of mRNA transcribed for a given gene (Aubin-Horth and Renn 2009). This underlying molecular variation has the potential to illuminate the action of selection and drift on phenotypic plasticity (Hodgins-Davis and Townsend 2009). Two questions fundamental to illuminating this action are addressed by empirical and theoretical studies focusing on variation in mRNA levels: first, how much variation in mRNA abundance arises from genetic differences, environmental context, and their interaction? Second, what does the distribution of this transcriptional variation across plastically and nonplastically expressed components of the genome imply about the forces maintaining it?
Previous empirical data suggest that genes exhibiting plasticity to environmental context also exhibit greater genetic variation within populations, as a joint consequence of both mutational and selective pressure. Genes whose expression is particularly sensitive to mutation were more likely to exhibit perturbed expression when other genes were deleted, and exhibited greater sensitivity to environmental perturbation (Landry et al. 2007). For instance, the presence of a TATA box in the promoter region is associated with high mutational variance in expression (Landry et al. 2007), as well as increased environmental plasticity and expression variation across species (Tirosh et al. 2006). Plastic genes tend to exhibit less stable nucleosome occupancy of the region upstream of their promoters (Tirosh et al. 2009; Weiner et al. 2010), though divergence in nucleosome positioning does not always predict changes in expression levels (Tirosh et al. 2010; Tsui et al. 2011; Huebert et al. 2012). These structural and mutational properties link genes demonstrating plasticity and genetic variation. Mutational variation persists only when not eliminated by drift or purged by natural selection. Accordingly, genes expected to experience relaxed selective constraints, including genes with paralogs in the genome and genes that are dispensable to cell survival in rich conditions, tend to exhibit greater genetic variation for transcriptional plasticity (Landry et al. 2006). These empirical observations suggest genes exhibiting expression plasticity to also exhibit genetic variation within a population and genes exhibiting a high degree of plastic expression variation to be subject to relaxed selection.
The impact of network architecture on transcriptional variation is likely to be complex (Siegal et al. 2007); however, considering population variation in the context of network structure and relevant environmental variation may permit elaboration of further hypotheses. Van Dyken and Wade (2010) have proposed an explanation for the observation that plastically expressed phenotypes exhibit higher genetic variation. Their theoretical model captures a reduction in the strength of selection on plastic phenotypes in direct proportion to the frequency that a population experiences environments inducing the plasticity. Snell-Rood et al. (2010) suggested the internal context of gene networks as well as the external environmental context to shape population variation: modular gene networks that are induced by a subset of environments and demonstrate little pleiotropy might experience relaxed selection. The hypothesis that network structure, network plasticity and environment frequency predict population variation generates testable genomic predictions where these factors are known. Support for this model has already been supplied by the discovery of greater gene expression divergence in genes expressed in discrete environmental contexts, such as quorum-sensing genes in bacteria, male-specific genes in facultatively sexual aphids, and genes specific to alternative morphs in horn-polyphenic beetles, than in constitutively expressed genes (Moczek et al. 2011 and references therein). Whether this framework can be usefully extended to explain population variation for a broader variety of continuous plastic phenotypes remains to be seen. Rarely encountered environments may sometimes impose stronger selection than common environments, limiting population variation at environmental extremes. Alternatively, functional redundancy for production of essential metabolites or gene products might facilitate maintenance of substantial population variation even for genes that are expressed in the most frequently encountered environments.
To assess gene expression variation present in natural populations and the differential expression response to a quantitative environmental variable, we examine the relationship between plasticity and population variation in the model budding yeast Saccharomyces cerevisiae. Gene expression plasticity is easily characterized in S. cerevisiae, and S. cerevisiae transcription factor networks are well studied (Zhu et al. 2008). We examine growth and gene expression responses to a challenging range of concentrations of the metal copper. Copper is an essential cofactor in cellular processes including mitochondrial oxidative phosphorylation, superoxide anion detoxification, and iron metabolism, but extremely toxic when present in free form, to the extent that intracellular copper concentrations are maintained in the range of one free copper molecule per cell (Rae et al. 1999; Lee et al. 2006; Wegner et al. 2011). S. cerevisiae genes differentially regulated in the S288c type strain immediately after an environmental shock have been categorized as belonging to a general environmental stress response (ESR), in which diverse environmental perturbations rapidly shift cell metabolism from investment in functions related to growth to those related to maintaining homeostasis (Gasch et al. 2000; Causton et al. 2001; Gasch and Werner-Washburne 2002). In contrast to the breadth of environments inducing the full ESR, the nutritional copper transcription factor Mac1 and copper toxicity transcription factor Ace1 are highly specific in their induction from initial shock through copper stress-acclimated growth—both are directly bound by copper ions and independently regulated by intracellular copper concentrations (Zhu et al. 1998; Keller et al. 2005). In the absence of copper, Mac1 transcriptionally induces genes involved in copper import, including cell surface metal reductases Fre1 and Fre7 and copper transporters Ctr1 and Ctr3, whereas excess copper permits Ace1 to activate expression of copper-sequestering metallothioneins Cup1 and Crs5 (Zhu et al. 1998; De Freitas et al. 2004; van Bakel et al. 2005; Rustici et al. 2007). Historically, the use of copper as a fungicide in vineyards and in breweries also suggests copper to have been an important selective pressure in those environments (Fernandez-Calvino et al. 2008). Indeed, laboratory selection for copper resistance produces tandem amplifications of CUP1 (Fogel et al. 1983; Welch et al. 1983), mimicking the independent amplifications of CUP1 in the lineages leading to the current wine/European and sake subpopulations (Warringer et al. 2011). Accordingly, substantial population variation exists for resistance to elevated copper concentrations (Warringer et al. 2011), copper-induced colony morphology (Fay et al. 2004) and copper-induced gene expression (Fay et al. 2004).
To ascertain the extent and distribution of transcriptomic variation in G, E, and G × E, we profiled growth and genome-wide gene expression across a gradient of five copper concentrations for five S. cerevisiae strains. We summarized the population distribution of differences among samples varying by genotype and environment and performed analysis of variance (ANOVA) to partition these differences into G, E, and G × E variation. We associated patterns of expression plasticity with copper tolerance in growth rate. Finally, we calculated correlations among strains for transcription factor networks that vary in the breadth of the environmental conditions triggering them, revealing the degree of concordance across genotypes for transcription factor networks with varying centrality and specificity.
Materials and Methods
Strains and Culture Conditions
Strains were selected to sample the genetic, ecological, and phenotypic diversity known for S. cerevisiae (Liti et al. 2009; Warringer et al. 2011), including isolates originally derived from fruit (S288C, UWOPS03-461.4), soil (SK1), and vineyards (L-1528, Y55) and maintained as laboratory cultures for varying durations. Strains were autodiploids, and thus completely homozygotic outside the mating type locus, with exception for S288C, which was haploid. All cultures were grown in synthetic media mimicking wine must (SWM; Bely et al. 1990) to imitate ecologically relevant conditions and for comparison with previous analyses (Landry et al. 2006). A range of copper concentrations in SWM were assayed: 1) copper-deprived: SWM containing no copper at all (–Cu); 2) moderate copper: a base SWM containing the standard 0.04 mM CuSO4 (SWM); and three levels of 3) copper overdose: base SWM supplemented with 0.1 mM CuSO4 (0.1), 4) 0.25 mM CuSO4 (0.25), and 5) 1.0 mM CuSO4 (1.0). Copper concentrations were selected based on pilot experiments to encompass a range producing large genetic variation for growth (supplementary table S1, Supplementary Material online). To mimic natural conditions, copper starvation was induced through successive inoculations in media lacking copper, rather than using a copper chelator. Induction of the copper transporter Ctr1, which is highly expressed under conditions of copper deficiency (Gross et al. 2000; De Freitas et al. 2004), was used as confirmation that cells were starved for copper in the –Cu condition.
Fitness Assays
Strains were subjected to high throughput phenotyping by micro-cultivation (n = 2) as previously described (Warringer et al. 2011). Briefly, strains were inoculated in 350 µL of SWM medium, with normal or zero copper, and incubated for 48 h at 30°C. Pre-cultures were diluted 35× to an OD of 0.03–0.15 in 350 µL of SWM and cultivated (n = 2) at 30.0°C for 72 h in a Bioscreen analyzer C (Oy Growth curves, Finland). Optical density was measured every 20 min using a wide band (450–580 nm) filter. Flocculation, which presents a serious issue to optical density measurement of liquid cultivations of wild yeast cells in higher cultivation volumes, was not observed. The population growth rate (population doubling time, h) was extracted from high-density growth curves and used as a proxy for fitness.
Microarray Experimental Design
Microarray experiments were performed to assay environment-specific gene expression during exponential phase growth for each strain. To ensure that cultures were captured in exponential phase despite differences in growth rate across copper environments, growth profiles of 200-ml test cultures were tracked over several days to identify optical density corresponding to early stationary phase growth. Optical density for harvest during exponential phase was then defined as one-half that of early stationary phase for each strain and environment combination. Replicate 200 ml cultures for RNA harvest were inoculated from copper-matched pre-cultures, grown aerobically to exponential phase (30°C, 150 rpm), then pelleted by centrifugation at room temperature (3,000 g for 4 min), flash frozen, and stored at −80°C. RNA was extracted with hot phenol–chloroform, followed by mRNA purification with oligo (dT)-cellulose columns (Molecular Research Center, Inc. Cincinnati, OH, cat. no. OT125) according to manufacturer’s instructions, and storage at −80°C. Purified mRNA was reverse transcribed, labeled, and hybridized to genome-wide spotted cDNA S. cerevisiae custom microarrays based on the S288C reference sequence (Hudson et al. 1997) as described in Townsend et al. (2003). Samples were co-hybridized to yield a balanced, highly connected modified loop design such that every strain was directly hybridized to two cultures of the same strain in two different copper environments and to two other strains grown in the same copper concentration (supplementary fig. S1, Supplementary Material online).
Gene Expression Analysis
All microarray data were normalized as in Townsend et al. (2003) using the online tool supplied by the Filamentous Fungal Gene Expression Database (Zhang and Townsend 2010). Normalized ratios for well-measured genes were analyzed by BAGEL to incorporate information from both direct and indirect comparisons across the loop design, producing a posterior distribution describing the expression level in each sample (Townsend 2004). The number of genes exhibiting pairwise genetic differences by copper environment and responses to copper by genotype were identified, excluding open reading frames annotated as dubious genes. A highly conservative P < 0.001 was invoked to identify differences in expression. An empirical expectation for the false positive rate was calculated by randomly shuffling the fluorescence ratios across genes for each hybridization and estimating the resulting number of false positives (Meiklejohn and Townsend 2005). For visualization of the data, reaction norms for each strain were plotted as the median of the BAGEL posterior distribution with error bars corresponding to the 95% credible interval. The BAGEL analysis procedure sets the lowest expressed sample to 1 so that the expression units on reaction norm plots represented fold change relative to the lowest measured sample. Power to detect significant differences in pair-wise comparisons was calculated as GEL50, the fold change at which there was an empirical 50% probability of calling a gene differentially expressed between samples (Townsend 2004).
When inferring the distribution of expression differences among pair-wise genetic comparisons of different strains in the same environment, and among pair-wise environmental comparisons of the same strain at different points along the copper reaction norm, we accounted for uncertainty in expression level by drawing 1,000 samples from the posterior distributions of each expression level and calculating the pair-wise difference distributions between all genetic and environmental comparisons. This procedure was performed on all genes, regardless of statistical significance, and also on the subset of genes demonstrating one or more statistically significant pair-wise differences among environmental and genetic comparisons.
To quantify the effects of genotype, environment, and gene-by-environment interaction in the dataset, we performed an ANOVA directly on the normalized fluorescence data using the R package MAANOVA (Wu et al. 2003). As in Landry et al. (2006), two mixed-effects ANOVA models were applied to the normalized data successively. First, a model including Array as a random factor and Dye, Genotype, Environment, and Genotype by Environment as fixed factors was fit for all genes to identify genes demonstrating an interaction effect between strain and copper environment. In the second step, following the approach in Landry et al. (2006), genes that exhibited no significant Genotype × Environment effect were analyzed using a model without any interaction effect to identify genes showing main effects of strain and copper environment alone. Tabulated P values were identified as statistically significant at a conservative P < 0.001. Hierarchical clustering was performed for sets of genes identified via ANOVA as significantly different using the MATLAB clustergram function on the median values from BAGEL posterior distributions. Clusters of all genes showing a difference greater than 3× in any comparison were visualized in G, E, and G × E heat maps, with expression standardized by the population mean for each gene. Functional enrichments (P < 0.01) for gene clusters were identified using the Gene Ontology Term Finder (version 0.83) accessed at SGD in June 2012 (Ashburner et al. 2000). The magnitude of genetic variation for different transcriptional networks or functional categories of genes was assessed by calculating the linear Pearson correlation coefficient between pairs of individuals in each environment. The environment-specific correlation coefficient was calculated for Mac1-regulated genes (n = 7), Ace1-regulated genes (n = 5), Msn2/Msn4-regulated genes (n = 39), ESR-induced genes (n = 282), ESR-repressed genes (n = 585), and genes included in none of these categories (n = 5,075) and plotted as the mean pairwise correlation across strains.
Results
Strains Varied in Their Growth Rate across Copper Concentrations
To map the extent and distribution of transcriptomic variation in environments of diverse copper exposure, five S. cerevisiae strains representing the genetic, ecological, and phenotypic breadth of the species (Liti et al. 2009; Warringer et al. 2011) were cultivated in a range of copper concentrations in a medium mimicking grape must. Isolates showed clearly distinct abilities to maintain mitotic proliferation in a copper gradient, presumably reflecting their diverging evolutionary histories with respect to copper exposure (fig. 1a and b). SK1 and UWOPS03-461.4 exhibited dramatic increases in population doubling time as a consequence of even marginal increases in copper concentration above base SWM, whereas L-1528 and Y55 exhibited more gradual loss of mitotic fitness at elevated copper concentrations. The haploid lab strain S288C was distinct in showing inferior mitotic performance in low copper concentrations, compared with other strains, but notable tolerance to high copper concentrations, resulting in a curiously flat dose-response curve considering the full copper gradient.
Fig. 1.—

Diverging fitness of yeast strains in a gradient of copper concentrations. (a) Population doubling times were estimated from high density growth curves (n = 2) and plotted as a function of copper concentration. Axes are on log2 scales. The x axis is modified to allow visualization of the no copper concentration. (b) Population growth curves (n = 2) of each strain in the five copper concentrations from which mRNA abundance was assayed. Extractions were performed at mid-log phase.
Standing Genetic and Environmental Variation of the Transcriptome
Pairwise Gene Expression Differences Are Extensive
Sampling transcriptomes in mid-exponential mitotic growth phase during exposure to five physiologically distinct concentrations of copper (fig. 1b) yielded 5,450 reaction norms in all strains. A majority of genes in the genome exhibited a significant effect (P < 0.001) of either strain or environment: out of all measured genes, 4,219 genes (77%; 5.8% false positive rate) demonstrated at least one significant difference between any two strains in any one copper concentration, and 3,647 genes (67%; 4.2% false positive rate) demonstrated significant differences between any pair of copper environments for at least one strain (fig. 2). The proportion of genetic differences identified within copper environments was nearly constant, with a small depression in the number of genetic differences identified in the moderate copper environment (fig. 2 and supplementary table S3, Supplementary Material online). The number of genes demonstrating plastic expression in response to copper varied substantially between strains (fig. 2 and supplementary table S2, Supplementary Material online) with SK1, a member of the West African population, showing the highest degree of expression plasticity.
Fig. 2.—

Percentages of genes exhibiting significant pair-wise differences in expression level. (a) Percentage of 4,219 genes exhibiting genetic differences within each copper environment. (b) Percentage of 3,647 genes exhibiting plasticity to copper within each strain. Hatched regions of each bar correspond to the 8% of plastic genes that are plastic across all strains.
The magnitude of variation revealed is in part due to the high power of the study: an analysis of the power to detect differences (Townsend 2004) indicated that our study yielded significant results for most fold-changes of 2× or higher applying the conservative criterion of P < 0.001 (with P < 0.05, GEL50 ∼1.5). Power to detect differences was equally distributed across genetic and environmental comparisons (GEL50: genetic 1.94; environmental 1.95). While 15% of genes exhibited a relative expression difference greater than 5×, the distribution of significant pair-wise expression differences for both genetic and environmental comparisons was highly skewed toward small magnitudes (supplementary tables S4 and S5, Supplementary Material online, and fig. 3). Although the magnitude of differences measured between strains were marginally larger than differences measured across copper reaction norms, the shape of the genetic and environmental distributions of differences were similar in their skew toward smaller magnitudes (supplementary tables S4 and S5, Supplementary Material online). The gene expression differences between excess copper environments (0.1–1.0 mM Cu) were smaller than the gene expression differences between moderate and excess copper or copper depletion and excess (SWM to 0.1 mM, or –Cu to 0.25 mM; supplementary table S4, Supplementary Material online). Excluding comparisons within copper excess, plastic expression differences were similar in magnitude to genetic expression differences (supplementary tables S4 and S5, Supplementary Material online). The distributions of expression differences among strains were more uniform in magnitude, with one interesting exception: differences between copper environments for S288C were one-half of the magnitude of differences for other strains (supplementary table S6, Supplementary Material online), though the size distribution of S288C’s pair-wise genetic differences to other strains remained in the range of other comparisons (supplementary table S5, Supplementary Material online). Thus, overall 1) small differences dominated the expression difference frequency spectrum, 2) environments more similar in copper concentration exhibited smaller expression differences, 3) environments less similar in copper concentration exhibited expression differences approaching the magnitude of expression differences among strains, 4) the shape and skew of the distributions of magnitudes of expression differences for genetic and environmental differences were similar, and 5) S288C was an outlier in terms of its plastic response to copper, presumably reflecting its remarkable ability to maintain proliferation at elevated copper concentrations, but not in the magnitude of expression differences from other strains.
Fig. 3.—
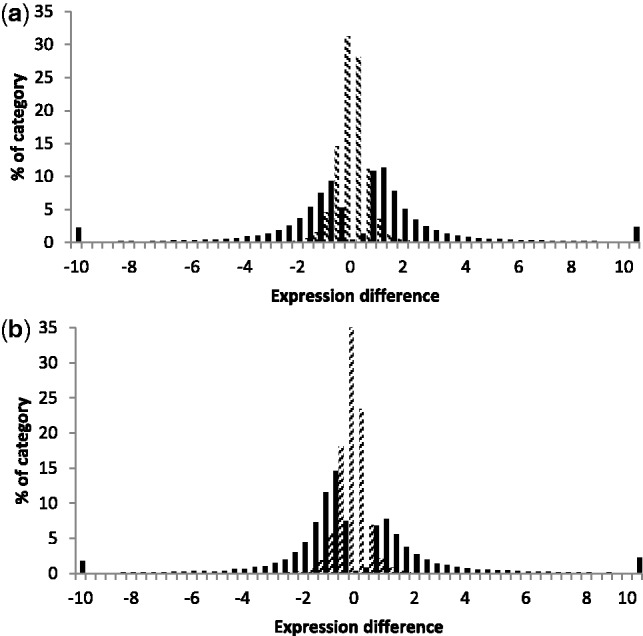
Magnitudes of pair-wise gene expression differences. (a) Magnitudes of significant (solid) and nonsignificant (hatched) genetic differences in expression. (b) Magnitudes of significant (solid) and nonsignificant (hatched) plastic differences in expression.
Gene Expression Reaction Norms Exhibit Diverse Patterns
Overall, G × E effects were rampant. The categorical ANOVA including genotype, environment, and G × E as fixed factors, identified 1,606 genes demonstrating a significant interaction between copper environment and genotype (G × E; P < 0.001). ANOVA without the interaction term, as in Landry et al. (2006), yielded an additional 729 genes with a significant effect of genotype alone (G), 114 genes with an effect of environment alone (E), and 60 genes with independent effects of both genotype and environment (G, E; P < 0.001). G × E effects remained pervasive also when restricting attention to the set of genes that are highly differentially expressed (>3-fold). Among these genes, 413 exhibited a significant effect of genotype alone (G; fig. 4a), 52 exhibited an effect of environment alone (E; fig. 4b), and 42 exhibited independent effects of genotype and environment (G, E, not shown; P < 0.001). Many more genes (874) demonstrated a significant interaction between copper environment and genotype (G × E; P < 0.001; fig. 4c).
Fig. 4.—

Hierarchical clustering of highly expressed genes identified as exhibiting significant genetic, environmental, and gene-by-environment effects. Gene effects were classified by ANOVA. Gene expression levels were estimated by BAGEL and filtered to retain highly differentially expressed genes (>3×fold change), whose annotated status was not dubious. High expression is depicted in red, mean expression across all measured genotypes and conditions is depicted in black, and low expression is depicted in green. Color intensity is scaled by units of gene-specific standard devation. Clusters are annotated at the right with characteristic GO category functional enrichments (supplementary file S1, Supplementary Material online). (a) Genes exhibiting effects of genotype. Expression within each strain is diagrammed in vertical blocks, within which expression is ordered by increasing copper. (b) Genes exhibiting an effect of environment. Expression within each copper concentration is diagrammed in vertical blocks, within which expression is ordered by strain in the same order as panel a. (c) Genes exhibiting gene-by-environment interaction. Expression within each strain is diagrammed in vertical blocks, within which expression is ordered by increasing copper.
Hierarchical clustering of the highly differentially expressed genes with an effect of genotype alone identified clear, strain-specific patterns of expression (fig. 4a and supplementary fig. S2, Supplementary Material online). The associated lists of genes were significantly enriched for numerous specific functional categories (supplementary file S1, Supplementary Material online). The largest cluster (163 genes) exhibited the least plastic response to copper and distinguished S288C (supplementary fig. S2, Supplementary Material online). It was enriched for processes including retrotransposition and reproduction, consistent with S288C’s haploid status and abundance of transposons (Carreto et al. 2008; Liti et al. 2009; Li et al. 2010). The UWOPS strain, SK1, and L-1528 exhibited uniquely high expression for clusters of 33, 20, and 65 genes, respectively, with no functional enrichments (supplementary fig. S2a–c, Supplementary Material online). Sixty-two genes highly expressed by both SK1 and the UWOPS strain exhibited especially high expression in the high copper concentrations; some of these genes were also highly expressed in -Cu by the UWOPS strain. This cluster was enriched for genes functioning in the proteasome complex and assembly (BLM10, ECM29, RPN2, RPT5, RPT4; supplementary fig. S2d and e, Supplementary Material online). Y55 exhibited high expression of a cluster of 77 genes enriched for thiamine biosynthesis, amino acid metabolism, and maltose metabolism (supplementary figs. S2f–h and S7, Supplementary Material online). The 10–60× upregulation of the maltase enzymes IMA1-4, MAL12, and MAL32 and maltose permease MAL31 distinguished Y55 from other strains (supplementary fig. S2f–h, Supplementary Material online), potentially reflecting high gene copy number, commonly observed for MAL loci in domesticated yeasts (Brown et al. 2010).
Hierarchical clustering of the 52 highly differentially expressed genes exhibiting environmental differences, regardless of genotype, identified common responses across strains to specific copper environments (fig. 4b and supplementary fig. S3, Supplementary Material online). This cluster was primarily driven by elevated expression in the absence of copper, as exemplified by upregulation of high affinity copper transporters (fig. 5a and b). A pattern of strain-specific gene expression across moderate copper and excess copper environments was discernible, although not statistically significant by categorical ANOVA. Genes upregulated in the absence of copper for all strains, except S288C, formed a distinct cluster enriched for genes involved in copper ion import and oxidoreductase activity. This cluster divided into two sub-clusters of 15 and 9 genes, each distinguished primarily by induction of either SK1 or UWOPS strain alleles during copper starvation (supplementary fig. S3, Supplementary Material online). The SK1 subcluster was highly enriched for genes involved in amino acid biosynthesis, whereas the UWOPS subcluster was enriched for genes with oxidoreductase activity, including high expression of genes ERG27, FRE1, HMX1, TRR1, and ZWF1.
Fig. 5.—

Expression reaction norms for genes with direct functions in the copper regulon. For each reaction norm, copper concentration is on the x axis, which is scaled by ¼ power to facilitate visualization of all five points on a convenient scale. The y axis is expression level relative to the lowest observed expression. Points plotted are the median of the posterior distribution. Error bars represent the 95% credible interval. Genes involved in copper uptake and import were highly expressed at low copper concentrations, including high affinity copper transporters (a) Ctr1 and (b) Ctr3. Genes involved in mediating copper toxicity were highly expressed at high copper concentrations, including metallothionein-encoding (c) Cup1 and (d) Crs5.
Hierarchical clustering of the 874 highly differentially expressed genes demonstrating a statistically significant G × E effect identified strain-specific responses of functional modules to changes in copper. Cytoplasmic ribosomal genes exhibited coordinated expression across a cluster of 151 genes. The magnitude of differential expression for these genes was small. The highest average expression for a gene in the ribosomal cluster was 4.6× differential expression compared with 10× to 50× differential expression for other G × E clusters. Reaction norms within this cluster were highly stereotyped by strain, consistent with a high degree of co-regulation of these genes, but varied across strains (supplementary fig. S4a and b, Supplementary Material online). High ribosomal gene expression for haploid S288C was consistent with higher ribosomal expression in haploid than autodiploid yeast (Jung et al. 2011). A cluster of 170 genes was highly enriched for functions related to aerobic respiration and the mitochondrial electron transport chain. This cluster also included genes with functions in cell cycle regulation (ASE1, IME1, RIM4, SDS22, SDS23, and YHR122W), protein trafficking and degradation (ATG1, BTN2, FUN19, GYP7, and JID1), and stress response (ACE1, BSD2, CRS5, GAC1, HSP42, RAD54, SKO1, STI, SSA3, UFD4, UBX6, and UBP3). The dominant pattern of strong upregulation in SK1, moderate upregulation in the UWOPS strain, limited expression in L-1528, and constitutively low expression in strains Y55 and S288C exemplified the high degree of G × E in this cluster (supplementary fig. S4c–e, Supplementary Material online). Genes highly expressed in the UWOPS strain in 0.1 mM Cu, were enriched for proteasomal functions, as in the UWOPS03-461.4 and SK1 genetic cluster (supplementary fig. S4f–i, Supplementary Material online). G × E in this cluster included genes having functions in stress response (ADD37, CRH1, CRG1, DAK2, GRE2, HSP10, SLT2, TOS8, and YPS6), the unfolded protein response and membrane maintenance and trafficking (CCT3, CCT8, GET3, KEX2, PRE3, PRE4, PRE9, TCP1, UMP1, and YPS5). L-1528 exhibited abundant expression of a set of 103 genes enriched for processes including sugar transport and carbohydrate metabolism in moderate copper, whereas other strains exhibited variable expression for these genes (supplementary fig. S4j–l, Supplementary Material online). Membrane-localized transport and stress response genes (AIM19, HSP30, INN1, PUT4, SFK1, SOD2, SKN1, SSU1, and WSC4; supplementary figs. S8b and S9c, Supplementary Material online) were similarly upregulated in L-1528 and enriched in this cluster; genes involved in allantoin degradation (AIM19, DUR1,2, and DUR3; supplementary fig. S8a, Supplementary Material online) also exhibited high expression in this cluster. Genes extremely expressed in moderate copper for SK1, constitutively meagerly expressed in S288C, and variably expressed in other strains grouped into a cluster of 102 genes enriched for GO categories including nitrogen compound transport, amino acid catabolism, and thiamine metabolism. Those genes involved in thiamine biosynthesis and metabolism (THI2, THI5, THI11, THI13, THI22, and PET18) and nitrogen compound transport (CAN1, DAL4, DAL5, FCY2, NRT1, THI7, THI72, TPO4, and UGA4) were among the most highly expressed in this study (supplementary figs. S7 and S8, Supplementary Material online). Genes clustered by expression in –Cu for the UWOPS strain, Y55, and L-1528 exhibited strikingly distinctive reaction norms by strain: abundant expression in –Cu for the UWOPS strain, Y55, and L-1528, downregulation in SWM for the UWOPS strain and L-1528, followed by increasing expression for 0.1 mM Cu and above for the UWOPS strain and SK1. Genes with these distinctive reaction norms exhibited dramatic functional enrichments for amino acid biosynthesis, particularly sulfur-containing amino acid biosynthesis (fig. 6), as well as extremely high levels of expression (mean gene expression up to 50-fold). Overall, G × E was abundant and frequently large in magnitude, presenting a pattern in which different genetic backgrounds responded to fluctuations in copper exposure through alterations of distinct transcriptional programs. Moreover, G × E was not restricted to functional categories associated with copper, but extended to functional categories affecting diverse aspects of cellular physiology.
Fig. 6.—

Gene expression reaction norms for genes involved in synthesis of cysteine and glutathione and the methyl cycle. For each reaction norm, copper concentration is on the x axis, which is scaled by ¼ power to facilitate visualization of all five points on a convenient scale. The y axis is expression level relative to the lowest observed expression. Points plotted are the median of the posterior distribution. Error bars represent the 95% credible interval. Genes in thiol redox control and sulfur amino acid biosynthesis pathways are variably upregulated in high and low copper compared with intermediate, including (a) Cys4, (b) Cys3, (c) Gsh1 (rate-limiting), (d) Met6, (e) Sam1, and (f) Sam2.
ESR Genes Exhibit High G × E
ESR genes, defined by their coherent response to a wide variety of environmental shifts in S288C, were particularly prone to exhibit genetic variation in their response to copper across other strains. Of genes with a significant ANOVA effect, ESR genes showed a higher proportion of G × E (ESR induced: 74% and ESR repressed: 78%) than G (25% and12%, respectively) or E effects (4% and 9%).
Standing Genetic and Environmental Variation
The Copper Regulon Responds Consistently Across Strains
Copper regulon genes were highly plastic, consistent with the well-studied molecular biology of the gene expression response of laboratory yeast to copper starvation and overdose (Lee et al. 2006). We observed high expression of genes responding to copper depletion and copper-dependent iron depletion in the –Cu condition, particularly for downstream targets of the nutritional copper transcription factor Mac1 (Jungmann et al. 1993). The well-documented upregulation of copper transporter genes CTR1 and CTR3 by Mac1 in low copper (Labbe et al. 1997) was consistent across strains (fig. 5a and b) with the sole exception of the lack of response in S288C’s CTR3, which is known to possess an inactivating Ty2 transposon (Knight et al. 1998). The cupric reductase genes FRE1 and FRE7, also regulated by Mac1, tended to be more highly expressed in –Cu than SWM, though to varying extent in different genetic backgrounds. These four targets of Mac1 possess two perfect consensus sites in their promoter region for binding of the transcription factor; two other genes, IRC7 and REE1, with one perfect consensus Mac1 binding site each, have previously been reported to respond to low copper (Gross et al. 2000). REE1 was one of the few genes not captured in this study, but expression of the beta lyase IRC7 showed upregulation in –Cu compared to SWM or 0.1 mM Cu for all diploid strains with particularly high expression in SK1. Mac1 has also been shown to be necessary, but not sufficient, for expression of catalase gene CTT1, whose expression was consistently low in –Cu across all strains, likely due to repression of its heme-dependent transcriptional activator Hap1 under iron deprivation (Hausmann et al. 2008), but more variable at higher copper concentrations. Similarly, genes downstream of the transcription factor responding to copper toxicity Ace1 (Thiele 1988) tended to be highly expressed in high copper concentrations (fig. 5c and d). CUP1 was highly upregulated by all strains in at least one high copper environment: across the two probes measuring the message of this duplicated gene, CUP1 exhibited 5- to 33-fold and 35- to 106-fold upregulation in high copper. The metallothionein CRS5, known to be a less effective chelator of copper than CUP1 (Jensen et al. 1996), was less upregulated, but showed significant increases for all strains, except for the UWOPS isolate. The post-transcriptionally regulated gene for superoxide dismutase SOD1 (Brown et al. 2004), exhibited significant increases for all strains except S288C and Y55.
Among these highly and consistently expressed members of the copper regulon, the genetic variance for expression level across strains scaled with the magnitude of expression. Thus, genetic variance in expression level was quite high in some cases even when all strains demonstrated the same qualitative response. However, the direction of regulation for plastic responses was highly uniform for genes regulated by copper-binding transcription factors in the relevant environment (fig. 5) and more variable for genes involved in metabolism (fig. 7). The qualitative similarity of the response of different strains to the same range of copper was captured by the pairwise correlations between strains compared within environments.
Fig. 7.—

Expression reaction norms for selected genes indirectly responding to copper. For each reaction norm, copper concentration is on the x axis, which is scaled by ¼ power to facilitate visualization of all five points on a convenient scale. The y axis is expression level relative to the lowest observed expression. Points plotted are the median of the posterior distribution. Error bars represent the 95% credible interval. Genes involved in copper-independent iron uptake (a) Fit1, encoding a siderophore-iron retaining mannoprotein, and (b) Enb1, encoding a ferric enterobactin transporter, exhibited strong upregulation of expression at low copper concentrations in some strains. (c) Non-essential gene Yah1, which encodes a mitochondrial ferredoxin and requires iron as a cofactor, exhibited marked downregulation in low copper in some strains. (d) Grx6, encoding a monothiol glutaredoxin that helps to cope with oxidative stress, exhibits diverse reaction norms in each strain, whereas oxidative stress gene (e) Ssa3, encoding an Hsp70-family ATPase, and membrane stress gene (f) Ino1, encoding a phospholipid bioynthesis gene, exhibit strong upregulation in high copper in some strains. Nitrogen catabolite repressed genes involved in amino acid biosynthesis are upregulated to differing degrees in differing strains in intermediate copper, including (g) Can1, encoding a plasma membrane arginine permease and (h) Thi20, encoding a protein functioning in thiamine metabolism.
Correlation of Gene Expression across Individuals Is Higher in Environments Relevant to Pathway Function
Within pathways, expression across strains was more correlated in the environments in which the genes had associated function (fig. 8). For example, expression levels of genes regulated by the nutritional copper transcription factor Mac1 (CTR1, CTR3, FRE1, and FRE7) were more correlated across strains in –Cu (mean Pearson’s r = 0.79) than in higher copper concentrations (−0.24 < r < 0.10). In contrast, genes regulated by the copper toxicity transcription factor Ace1 (CUP1-1, CUP1-2, CRS5, and SOD1) were correlated in environments of moderate copper and excess (r ≥ 0.96), but only weakly so in –Cu (r = 0.27). Likewise, expression of genes regulated by ESR-induced general stress responsive transcription factors Msn2/Msn4 exhibited high correlations in copper excess (r > 0.84). Expression of all ESR-induced genes exhibited similarly high correlation in excess copper (r > 0.67) and also a moderate correlation in –Cu (r = 0.59), consistent with the action of the ESR in accommodating stressful conditions. Interestingly, the subset of Msn2/Msn4-regulated genes exhibited lower correlation in expression in the putatively stressful copper-depleted environment (r = 0.34). The expression of genes repressed in the ESR was less correlated among strains in copper stress (0.44 < r < 0.53) and was approximately equal in magnitude to the mean genome-wide correlations among genes not involved in the copper regulon or ESR (mean copper stress r = 0.42). Overall, gene expression within these functional categories was most similar among strains for environments most relevant to their function.
Fig. 8.—

Copper-regulated and stress-responsive gene networks exhibit correlated expression among strains in environments where the networks are functionally important and highly expressed. Mean network correlation was calculated by averaging the gene-specific Pearson correlations among individuals for Mac1-regulated genes (n = 7), Ace1-regulated genes (n = 5), Msn2/Msn4-regulated genes (n = 39), ESR-induced genes (n = 282), ESR-repressed genes (n = 585), and genes included in none of these categories (n = 5,075). Mean correlations are plotted across environments, with copper concentration increasing from left to right for each network.
Reaction Norms Exhibit Common Responses for Copper and Stress-Regulated Genes and Diverse Responses for Genes Indirectly Affected by Copper Environment
While strains exhibited correlated and consistent Mac1, Ace1, and Msn2/Msn4 regulation, they showed uncorrelated, but consistent, functional responses across pathways. For example, in –Cu strains responded to the iron deprivation triggered by copper deficiency by regulating expression to conserve iron in varied ways. In 0.1 mM Cu and above, strains responded to the oxidative stress of excess copper by upregulating diverse oxidative stress response and cell wall repair genes. In SWM, strains responded to the challenge of fast growth with diverse regulation of amino acid catabolism and thiamine synthesis and import genes.
Copper Starvation Leads to Iron Deprivation, Which Is Dealt With Differently Across Strains
Induction of Mac1-regulated genes indicated that copper depletion triggered copper starvation in all strains (fig. 5a and b). Major shifts in iron metabolism indicated complex, strain-specific patterns of iron conservation in response to this copper starvation with correlated changes in energy metabolism and patterns of amino acid synthesis. While copper-dependent iron import genes FTR1 and FET3 tended to be uniformly downregulated under copper deprivation, strains exhibited variable upregulation of genes functioning in copper-independent iron import via acquisition and retention of siderophore-bound iron chelates (ARN2, ARN3, ENB1, and FIT1; fig. 7a and b). Under copper deprivation, strains also demonstrated individualized metabolic re-routing away from nonessential enzymes possessing iron–sulfur clusters (fig. 7c and d and supplementary table S7a, Supplementary Material online) and preferentially expressed alternative metabolic pathways requiring less iron for glutamate synthesis (supplementary fig. S5a vs. supplementary fig. S5b and c, Supplementary Material online), consistent with previous descriptions of expression responses to iron deprivation (Puig et al. 2005; van Bakel et al. 2005; Shakoury-Elizeh et al. 2010). Genes encoding heme dependent or heme biosynthetic functions were also variably expressed across strains, but tended to be significantly downregulated in –Cu (supplementary table S7b and fig. S6, Supplementary Material online). Likewise, ACO1, KGD1, KGD2, SDH4, and RIP1 genes involved in the iron-demanding TCA cycle and oxidative respiration were differentially expressed across strains but tended to be downregulated with decreasing copper (fig. 8b and c). In parallel, expression of fermentation-specific isozymes ALD6, GDH1, GND1, HXK2, and TKL1 in –Cu for strains L-1528, SK1, and Y55 suggested these strains conserved iron in part by transitioning to fermentation-dominated respiration (supplementary fig. S10, Supplementary Material online). These patterns of functional similarities in iron conservation across strains with variation in the magnitude and identity of genes responding by strain for genes involved in energy metabolism were paralleled by variable upregulation of many amino acid synthesis genes in –Cu (supplementary table S8, Supplementary Material online), particularly induction of FeS-containing essential amino acid synthesis genes LEU1, MET5, and MET8 (supplementary fig. S5d–f, Supplementary Material online).
Copper Overdose Leads to Oxidative and Cell Wall Stress, Which Is Dealt with Differently Across Strains
In addition to the canonical Ace1-regulated copper response common to all isolates, strains also responded to increasing copper toxicity by strain-specific upregulation of genes maintaining homeostasis under oxidative stress (supplementary fig. S9, Supplementary Material online). The number of genes upregulated and their magnitude of upregulation varied across strains; for instance, SK1 exhibited particularly abundant expression of genes involved in coping with the consequences of copper toxicity. SK1 is also homozygous for the CUP1 “low” allele, which is functional but lacks the tandem duplications found in many strains from the wine/European and Sake populations (Winzeler et al. 2003; Warringer et al. 2011). The upregulated stress response genes, particularly highly expressed in SK1, included heat shock proteins, chaperones, and oxidative and osmotic stress response genes (supplementary fig. S9, Supplementary Material online). At the two highest copper concentrations, this variable upregulation extended to genes with roles in cell wall maintenance and resistance to pH stress (supplementary fig. S9e–g, Supplementary Material online).
Even in Moderate Copper, Strains Exhibit Differences in Expression
In SWM, strains directed expression toward growth rather than stress response, including investment in acquisition of necessary nitrogen and thiamine for fast growth, through largely individualized regulatory responses. Y55 showed the highest expression of thiamine biosynthesis genes in SWM and neighboring environments, whereas SK1 demonstrated abundant expression of thiamine import genes (supplementary fig. S7, Supplementary Material online). Strains SK1, UWOPS03-461.4, and L-1528 indicated exhaustion of assimilable nitrogen by upregulating expression of nitrogen-catabolism-repressed (NCR) and allantoin degradation genes in SWM, the latter despite the absence of allantoin in the medium, whereas S288C and Y55 showed less dynamic regulation of these nitrogen acquisition pathways (supplementary figs. S5g and S8, Supplementary Material online). Among these strains dynamically regulating nitrogen, SK1 dramatically upregulated expression of allantoin import and degradation genes, particularly early in the pathway, whereas L-1528 showed abundant expression of urea amidolyase Dur1,2 (supplementary fig. S8a, Supplementary Material online). These strains also differentially regulated NCR genes at different positions in the proline and arginine utilization pathways (supplementary fig. S8b and c, Supplementary Material online). Finally, strain-specific variation was evident in genes of the Ehrlich pathway, which degrades the reaction products of branched chain amino acid catabolism (supplementary fig. S11, Supplementary Material online). Overall, these alternate genetic responses to a rapid-growth environment demonstrated that genetic variation was not limited to copper stress conditions.
Discussion
We have examined relative mRNA abundance for a cross-section of S. cerevisiae population variation across a finer gradient of copper than previously measured and revealed more expression plasticity to copper than previously reported (Gross et al. 2000; Fay et al. 2004; van Bakel et al. 2005; Jin et al. 2008). The vast majority of plastic responses across copper gene expression reaction norms exhibited genetic variation (G × E: 29% of the transcriptome vs. E alone: 2%). Likewise, more than twice as many genes showed genetic variation in copper response as showed genetic variation independent of copper (G × E: 29% of the transcriptome vs. G alone: 13%) in agreement with previous observations of fermentation expression reaction norms (Landry et al. 2006). Pairwise differences were even more extensive: 77% of the measured transcriptome exhibited a significant difference among at least two strains measured in a common copper environment, whereas 67% exhibited a response to copper across the reaction norm of at least one strain. The extensive population variation captured here compared with previous copper gene expression studies results from a highly powered assay of multiple points along the population reaction norm and represents both direct expression responses to the copper gradient mediated by genetic background and indirect expression responses resulting from differential growth effects of copper across strains. In capturing both copper-specific and growth-related differences among strains, these results reflect a range of phenotypic variation expressed by natural populations encountering a common set of environmental challenges.
The distribution of genetic and environmental differences measured across these isolates adds another observation to the general pattern that intraspecific population variation for mRNA abundance is dominated by differences that are small in magnitude. In this work, slightly fewer differences were observed among copper environments overall than between genotypes, and differences between samples from the same genotypes compared among high copper treatments particularly showed fewer and smaller differences (supplementary tables S2 and S4, Supplementary Material online), consistent with greater similarity in functional response or intracellular context among cells all experiencing copper overdose. The intraspecific abundance of small magnitude variation could be a consequence of a skewed mutational distribution, selection against large expression differences, or homogenization of population variation by genetic drift. A high frequency of small gene expression differences is consistent with the observation of abundant intraspecific trans regulatory variation, which is, on average, more mutationally available, smaller in magnitude, more frequently recessive, and associated with plasticity than cis regulatory variation (Landry et al. 2006; Li et al. 2006; Wittkopp et al. 2008; Gruber et al. 2012).
Gene deletion experiments and functional assays predict that copper regulator Ace1 is necessary in high copper concentrations (Fogel et al. 1983; Winzeler et al. 1999; Hillenmeyer et al. 2008). If proliferation during exposure to stressful environments such as high copper concentrations is critical to persistence of S. cerevisiae isolates, even rare exposure might be expected to select for common expression responses within the population for adaptively plastic genes like the copper regulon. In agreement with this prediction, we observe higher correlations among strains for expression of stress-regulated gene networks in the environments where those networks are predicted to have the greatest impact on fitness. For example, Mac1 targets exhibited correlated expression across strains in the copper-starved environments, but not in copper-sufficient environments, whereas Ace1 targets exhibited correlated behavior among strains in moderate to high copper environments, but not in low copper (fig. 8). The ESR as a whole also showed more similar expression in the two environments of copper stress than it did in moderate copper conditions. In contrast, genes with no stress-response annotation showed a higher degree of population consistency in moderate copper. These observations suggest that the strength of selection as well as the frequency with which plastic gene networks are induced is likely to influence the degree of population variation maintained for plastic traits.
Though copper stress-responsive genes were more correlated among strains in their inducing than noninducing environments, they still exhibited variation in the magnitude of expression for these genes that was potentially linked with their difference in growth. Growth variation among isolates implied some to be better able to maintain mitotic proliferation under fluctuating copper exposure than others. Variation in expression of genes important for reproduction in nonoptimal conditions may be particularly likely to contribute to population G × E as genetic backgrounds lacking sufficient stress protection may experience the same environmental context as a more severe stress. For example, in high copper concentrations strains UWOPS03-461.4 and SK1 exhibited the lowest expression of the metallothionein CUP1, the principle cellular copper-sequestering mechanism, of the diploid strains (fig. 5c and d), likely because they possess the fewest copies of this tandemly duplicated gene (Warringer et al. 2011). These CUP1-poor strains exhibited increasing expression of genes characteristic of oxidative respiration in high copper concentrations, including genes involved in electron transport, aerobic respiration, general mitochondrial activity, and response to oxidative stress. Recent expression studies of auxotrophic mutants growing under nutrient limitation suggests induction of these genes increases survival under oxidative stress, but that nonrespiratory functions of the regulatory cascade confers most of the survival benefit (Petti et al. 2011). Thus, the lower relative induction of CUP1 in UWOPS03-461.4 and SK1 compared with Y55 and L-1528 in high copper may have necessitated an extensive, protective transition to slower growth and largely oxidative respiration for these strains, even in the presence of high glucose concentrations. Conversely, diploid strains Y55 and L-1528 exhibited higher CUP1 expression and less severe growth inhibition in high copper concentrations. These two strains also exhibited fewer plastic genes in response to copper than SK1 or UWOPS03-461.4, potentially as the product of a less perturbed genome-wide state. These observations support the idea that expression variation for plastic genes that are key to survival, even in a restricted range of environments, can have broad-ranging impacts on both genome-wide gene expression and growth rate.
Population variation for plasticity included many genes other than CUP1, both those that responded directly to copper stresses and those that were indirectly influenced by the cascading downstream effects of changed copper. Some genes exhibiting differential plasticity among strains appeared to reflect parallel functional responses by different genetic backgrounds to the same form of stress. In other cases, strains appear to exhibit functionally different responses to the same range of copper environments. Below, we summarize these broad patterns of plasticity, the metabolic responses to copper they imply, and the extensive differences between strains in plastic gene expression.
Copper Environment Influences Diverse Aspects of Cellular Redox Balance and Metabolism
Gene expression in environments corresponding to copper starvation, fast growth in wine must, and copper toxicity were distinguished not only by changes in the expression of copper homeostasis genes but also by changes in the expression of genes that maintain oxidoreductive balance and regulate nutrient flux. Among the genes known to be directly regulated by binding of copper ions to copper-responsive transcription factors, the increase of copper from absence to toxicity caused a well-characterized shift from a transcriptional program of extracellular copper scavenging by copper transporters to one of intracellular copper sequestration by metallothioneins (Gross et al. 2000; van Bakel et al. 2005). Interestingly, at both extremes of copper stress, sulfur metabolism genes manifested a pattern of high expression, suggesting that thiol redox activity helped to maintain cellular homeostasis in both copper starvation and excess. Beyond sulfur metabolism, the two copper extremes exhibited broad differences in expression profiles of genes involved in amino acid biosynthesis, oxidative stress, and cellular respiration. Moderate copper was largely characterized by patterns of nitrogen and thiamine depletion during fast growth, reflecting the relative scarcity of nitrogen relative to carbon in wine must.
The ensemble of sulfur amino acid synthesis reaction norms suggests that genome-wide patterns of gene expression are influenced by a complex interplay among copper, sulfur, and iron homeostasis that is strongly dependent on environmental context. The abundant expression of sulfur metabolism genes in high copper concentrations in our study is consistent with more than 50 years of experimental observations (Ashida and Nakamura 1959). In contrast, the implication of sulfur assimilation and amino acid biosynthesis genes in response to copper starvation is largely unique to this study (though see the unusual case of IRC7 in (Gross et al. 2000). The extremely high expression of sulfur metabolism genes here (>80× in some cases) may be due in part to the sparse supply of sulfur-containing amino acids cysteine and methionine in SWM compared with the more standard YPD (Bely et al. 1990). However, the role of the cysteine-derived antioxidant glutathione in iron–sulfur cluster maturation (Lill and Muehlenhoff 2008; Muehlenhoff et al. 2010; Sharma et al. 2010; Kumar et al. 2011), and more generally the role of thiols in regulating redox balance in the cytosol (Lopez-Mirabal and Winther 2008), suggests a mechanism by which limiting copper leads to a higher demand for sulfur amino acid metabolism to maintain oxidoreductive balance. For high copper environments, the upregulation of sulfur amino acid metabolism (Gross et al. 2000; De Freitas et al. 2004; Fay et al. 2004) also seems likely to function in increasing production of glutathione (Mendoza-Cozatl et al. 2005; Delalande et al. 2010) and controlling redox inactivation of iron–sulfur cluster containing proteins (Macomber and Imlay 2009; Py et al. 2011; Reeder et al. 2011).
The link between expression responses to changing copper and sulfur amino acid metabolism, which may be mediated by iron–sulfur cluster proteins at both extremes, may reflect a broader genomic coupling of copper and iron homeostasis. Altered copper levels have been known to influence mRNA levels linked to iron availability in the yeast (Gross et al. 2000), in particular through a shift from copper-dependent to copper-independent mechanisms of iron import (van Bakel et al. 2005). Accordingly, deletion of MAC1, encoding the nutritional copper transcription factor, produces transcriptional conservation of iron through downregulation of proteins containing iron–sulfur clusters (De Freitas et al. 2004). In this study, diploid population isolates cultured in the absence of copper showed expression patterns consistent with even wider iron-sparing responses as previously observed under strict iron deprivation (Kaplan et al. 2006; Shakoury-Elizeh et al. 2010).
In addition to illustrating the direct impact of copper depletion on metalloprotein regulation, expression profiles from the environment of limiting copper also illustrated the broad-reaching downstream effects of copper absence on core metabolism. In particular, limitation in the availability of this single micronutrient appears to have affected major shifts in energy metabolism and amino acid biosynthesis, particularly those related to iron depletion. Expression of genes involved in fermentation for strains L-1528, SK1, and Y55 in –Cu indicates that these strains were performing mainly fermentation in the absence of copper (supplementary fig. S10, Supplementary Material online), as expected under iron limitation (van Bakel et al. 2005; Kaplan et al. 2006). Additionally, the upregulation of amino acid biosynthesis genes in –Cu paired with the requirement for Fe–S clusters in several essential biosynthetic enzymes (ACO1, ILV3, LEU1, LYS1 and MET5) suggested that iron starvation triggered a shift in the general control of amino acid synthesis (as reviewed in Philpott et al. 2012). In the course of this well-characterized regulatory shift, general protein translation is slowed, whereas translation of the transcriptional activator Gcn4 is de-repressed, leading to globally increased expression of amino acid biosynthesis genes under conditions of nutrient starvation (Natarajan et al. 2001; Hinnebusch 2005). Upregulation of Gcn4 targets was evident in our dataset in the –Cu environment including upregulation of arginine, cysteine, histidine, leucine, lysine, methionine and serine biosynthesis genes, suggesting that copper starvation induced this altered regulatory state. Overall, strains varied widely in which iron-containing and amino acid biosynthesis genes were strongly regulated, but the global shift toward an iron-limited metabolism in which nonessential iron-requiring protein synthesis was suppressed, whereas fermentation and essential amino acid synthesis were induced was largely consistent across diploid strains (supplementary table S7 and S8, Supplementary Material online).
In contrast, high concentrations of copper consistently stimulated an expression program characterized by copper stress responses extending from copper sequestration to shifts in energy metabolism and amino acid usage. SK1 and UWOPS03-461.4 exhibited increasing expression of genes involved in oxidative respiration and the electron transport chain with increasing copper, whereas L-1528 expressed carbohydrate transport and catabolism genes enriched for the pentose phosphate shunt (fig. 3c and supplementary fig. S10, Supplementary Material online). Genes involved in the heat shock and oxidative stress responses as well as sulfur metabolism were also highly expressed across most diploid strains with some genetic variation in the magnitude of expression and reaction norm shape (figs. 6 and 7), consistent with a transition to largely oxidative respiration in high copper.
The dual roles of copper as both an essential micronutrient and as a toxic catalyst of undesirable reactions may contribute to the breadth of its effects on cellular metabolism. Nevertheless, copper is unlikely to be unique among environmental stimuli in the degree of plasticity it provokes. The breadth of the effects of copper concentration on genes functioning in processes from metal import and regulation to carbohydrate metabolism to amino acid usage in this study is consistent with copper concentration shifting the reductive and metabolic balance of yeast cells along a more universal continuum from fast fermentation to slower, stress-resistant oxidative respiration in a manner dependent on genetic background. Among the 65% of genes in the genome that were differentially expressed with changing copper, many genes likely responded indirectly to changes in growth rate or redox state rather than directly to the presence of copper ions, given the diversity of their functional annotations. Accordingly, a number of recent studies of growth rate and expression suggest that yeast cells integrate diverse signals from their internal and external environment to regulate their growth along this fermentation-respiration continuum to avoid nutrient starvation and survive environmental stresses (Brauer et al. 2008; Gresham et al. 2011; Slavov and Botstein 2011). However, two complexities to this simple model are introduced by our results. The first is that transitions between fast growth and stress protection are subject to the particular constraints of the inducing environmental stress. For instance, in diploid strains L-1528, SK1, and Y55, the limited iron available in copper-starved conditions apparently produced a fermentative stress response in the lowest copper condition. Second, very few reaction norms demonstrated identical plasticity and magnitude of expression across strains: we observed significant genetic variation in the nature of the regulatory programs induced by changing copper and in the magnitude of their growth-associated shifts in metabolism due to copper exposure. Thus, a full account of population variation for gene expression is captured only by assaying a cross-section of both relevant environments and genetic variation.
Strains Exhibit Individualized Responses to Environmental Stresses
The population variation for gene expression suggested that differing genetic backgrounds respond to the same environmental stress via regulation of different molecular pathways. Reaction norms exhibiting strain-specific patterns of expression identified these functional differences in the responses of different strains to copper concentration. For example, West African strain UWOPS03-461.4 exhibited uniquely high expression of proteasome genes at intermediate copper concentrations, suggesting an upregulation of protein degradation as one response to the dramatic decrease in growth it exhibited to increasing copper (fig. 4a and c). UWOPS03-461.4 also induced a number of enzyme isoforms characteristic of respiratory growth (ALD4, GLK1, GND2 and HXK1), as well as the glycolysis genes GND1 and TKL1 in the lowest copper environment suggesting a mix of respirofermentative growth for this strain even in the presence of iron limitation (supplementary fig. S10, Supplementary Material online). In contrast, Y55 exhibited high expression of genes induced during fermentation for copper environments from –Cu to 0.25 mM Cu, suggesting a later transition to oxidative growth than other diploid strains, if at all (supplementary fig. S10, Supplementary Material online). Y55 also demonstrated the consistently highest expression of amino acid biosynthesis genes among strains in –Cu, suggesting a more dramatic iron deprivation than exhibited by other strains (fig. 6). L-1528 uniquely induced a large panel of sugar transport and carbohydrate catabolism genes, including genes enriched for roles in the pentose pathway (fig. 3c), suggesting that L-1528 preferentially used the pentose pathway to reduce NADP under the oxidative stress of high copper (Kruger et al. 2011). SK1 exhibited highest expression of genes responding to oxidative respiration and the electron transport chain in 1.0 mM Cu (fig. 4c and supplementary fig. S10, Supplementary Material online) and also highest expression of several stress responsive genes with diverse functions (including BCY1, HSP26, HSP42, SSA3, SKO1, and SBA1; supplementary fig. S9a–d, Supplementary Material online), potentially correlated with the strong growth inhibition exhibited by SK1 in high copper. Such intraspecies variation in response to stress may presage the accumulation of between-species differences in plasticity (Tirosh et al. 2011).
Environment-specific genetic variation was not confined to the extremes of copper stress. We observed substantial variability among strains in their responses to nitrogen and thiamine shortages imposed by exponential growth in SWM media. Population transcriptomic responses to growth in SWM were characteristic of nutrient limitation for thiamine and assimilable nitrogen, two commonly limiting components in grape must strongly influencing the kinetics of wine fermentation (Bataillon et al. 1996; Bell and Henschke 2005; Ericsson et al. 2008). These strains solved their nitrogen and thiamine needs using a slightly variable set of metabolic genes, including different members of the NCR genes (ter Schure et al. 2000) (fig. 7g and supplementary fig. S8, Supplementary Material online), the Ehrlich pathway (Hazelwood et al. 2008) (supplementary fig. S11, Supplementary Material online) and genes contributing to either thiamine import or synthesis (fig. 7h and supplementary fig. S7, Supplementary Material online).
Population Transcriptomic Variation Also Includes Fixed Genetic Differences
Reaction norms exhibiting strain-specific patterns of expression also showed functional differences among the strains that were fixed with respect to copper. Most dramatic were the expression differences between haploid S288C and other diploids: S288C exhibited constant and high expression of 174 genes enriched for functions including retrotransposition and mating, characteristic of haploidy (Li et al. 2010) and S288C’s high transposon content (Carreto et al. 2008; Liti et al. 2009). S288C was also distinct in its high expression of ribosomal genes in intermediate copper, consistent with their higher expression in haploids than diploids (Jung et al. 2011). Other reports demonstrate that the laboratory strain S288C diverges dramatically from other strains independently from ploidy status (Kvitek et al. 2008; Warringer et al. 2011), and thus interpretation of the comparatively low expression and plasticity of S288C is challenging. High expression of isomaltases IMA1-4 and maltose catabolism genes MAL12, MAL31, and MAL32 distinguished Y55 from other strains. The high rate of gene duplication and rapid divergence documented among subtelomeric MAL gene families suggests that the Y55 lineage examined might have recently acquired additional gene copies of these genes as previous analyses of MAL gene copy number among the five strains we studied would not have suggested the pattern of genetic differences we observed (Brown et al. 2010). Other genes highly expressed in Y55 compared with all other genotoypes included a group of genes functioning in thiamine metabolism.
By studying a fine gradient of copper at a high experimental power in multiple genetic contexts, we have captured a more fine-grained view of transcriptomic variation in S. cerevisiae in response to copper than previously available. We have shown different S. cerevisiae isolates to be distinct metabolic systems dynamically and differentially responding to the constraints of both the external environment and genetic context. The tremendous degree of genetic and environmental variability in expression and plasticity available from this restricted sample of S. cerevisiae diversity suggests a substantial potential for phenotypic drift in expression under neutral regimes and adaptive evolution under selective regimes in natural populations.
Supplementary Material
Supplementary file S1, figures S1–S11, and tables S1–S8 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
The raw and processed microarray data reported in this study are publically available at the Filamentous Fungal Gene Expression Database (FFGED Expt 45 http://bioinfo.townsend.yale.edu/).
Acknowledgments
The authors thank Robert Bjornsen for his assistance implementing MAANOVA via parallel computing on the Yale Bulldog cluster. They thank Carl Schlichting for his close reading of earlier versions of the manuscript, and Antonia Monteiro and Emilie Snell-Rood for their thoughtful reflections on this work. This work was in part supported by the National Institute of Health predoctoral Genetics Training grant (T32 GM007499) to A.H.D.
Literature Cited
- Ashburner M, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashida J, Nakamura H. Role of sulfur metabolism in copper resistance of yeast. Plant Cell Physiol. 1959;1:71–79. [Google Scholar]
- Aubin-Horth N, Renn SCP. Genomic reaction norms: using integrative biology to understand molecular mechanisms of phenotypic plasticity. Mol Ecol. 2009;18:3763–3780. doi: 10.1111/j.1365-294X.2009.04313.x. [DOI] [PubMed] [Google Scholar]
- Bataillon M, Rico A, Sablayrolles JM, Salmon JM, Barre P. Early thiamin assimilation by yeasts under enological conditions: impact on alcoholic fermentation kinetics. J Ferment Bioeng. 1996;82:145–150. [Google Scholar]
- Bell SJ, Henschke PA. Implications of nitrogen nutrition for grapes, fermentation and wine. Aust J Grape Wine Res. 2005;11:242–295. [Google Scholar]
- Bely M, Sablayrolles JM, Barre P. Description of alcoholic fermentation kinetics: its variability and significance. Am J Enol Vitic. 1990;40:319–324. [Google Scholar]
- Brauer MJ, et al. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CA, Murray AW, Verstrepen KJ. Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr Biol. 2010;20:895–903. doi: 10.1016/j.cub.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NM, Torres AS, Doan PE, O'Halloran TV. Oxygen and the copper chaperone CCS regulate posttranslational activation of Cu, Zn superoxide dismutase. Proc Natl Acad Sci U S A. 2004;101:5518–5523. doi: 10.1073/pnas.0401175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürger R. Chichester (United Kingdom): Wiley; 2000. The mathematical theory of selection, recombination and mutation. [Google Scholar]
- Callahan H, Maughan H, Steiner U. Phenotypic plasticity, costs of phenotypes, and costs of plasticity: toward an integrative view. Ann NY Acad Sci. 2008;1133:44–66. doi: 10.1196/annals.1438.008. [DOI] [PubMed] [Google Scholar]
- Carreto L, et al. Comparative genomics of wild type yeast strains unveils important genome diversity. BMC Genomics. 2008;9:17. doi: 10.1186/1471-2164-9-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causton H, et al. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Freitas J, et al. Exploratory and confirmatory gene expression profiling of mac1delta. J Biol Chem. 2004;279:4450–4458. doi: 10.1074/jbc.M212308200. [DOI] [PubMed] [Google Scholar]
- Delalande O, et al. Cadmium-glutathione solution structures provide new insights into heavy metal detoxification. FEBS J. 2010;277:5086–5096. doi: 10.1111/j.1742-4658.2010.07913.x. [DOI] [PubMed] [Google Scholar]
- Ericsson A, Mojzita D, Schmidt H, Hohmann S. Case study in systematic modelling: thiamine uptake in the yeast Saccharomyces cerevisiae. Essays Biochem. 2008;45:135–146. doi: 10.1042/BSE0450135. [DOI] [PubMed] [Google Scholar]
- Fay J, McCullough H, Sniegowski P, Eisen M. Population genetic variation in gene expression is associated with phenotypic variation in Saccharomyces cerevisiae. Genome Biol. 2004;5:R26. doi: 10.1186/gb-2004-5-4-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Calvino D, Pateiro-Moure M, Lopez-Periago E, Arias-Estevez M, Novoa-Munoz J. Copper distribution and acid-base mobilization in vineyard soils and sediments from Galicia (NW Spain) Eur J Soil Sci. 2008;59:315–326. [Google Scholar]
- Fogel S, Welch J, Cathala G, Karin M. Gene amplification in yeast—CUP1 copy number regulates copper resistance. Curr Genet. 1983;7:347–355. doi: 10.1007/BF00445874. [DOI] [PubMed] [Google Scholar]
- Gasch A, et al. Genomic expression programs in the response of yeast cells to environmental change. Mol Cell Biol. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch A, Werner-Washburne M. The genomics of yeast responses to environmental stress and starvation. Funct Integr Genomics. 2002;2:181–192. doi: 10.1007/s10142-002-0058-2. [DOI] [PubMed] [Google Scholar]
- Gresham D, et al. System-level analysis of genes and functions affecting survival during nutrient starvation in Saccharomyces cerevisiae. Genetics. 2011;187:299–317. doi: 10.1534/genetics.110.120766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Kelleher M, Iyer V, Brown P, Winge D. Identification of the copper regulon in Saccharomyces cerevisiae by DNA microarrays. J Biol Chem. 2000;275:32310–32316. doi: 10.1074/jbc.M005946200. [DOI] [PubMed] [Google Scholar]
- Gruber JD, Vogel K, Kalay G, Wittkopp PJ. Contrasting properties of gene-specific regulatory, coding, and copy number mutations in Saccharomyces cerevisiae: frequency, effects, and dominance. PLoS Genet. 2012;8:e1002497. doi: 10.1371/journal.pgen.1002497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann A, Samans B, Lill R, Muehlenhoff U. Cellular and mitochondrial remodeling upon defects in iron-sulfur protein biogenesis. J Biol Chem. 2008;283:8318–8330. doi: 10.1074/jbc.M705570200. [DOI] [PubMed] [Google Scholar]
- Hazelwood LA, Daran J-M, van Maris AJA, Pronk JT, Dickinson JR. The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Appl Environ Microbiol. 2008;74:3920–3920. doi: 10.1128/AEM.02625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenmeyer M, et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008;320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- Hodgins-Davis A, Townsend JP. Evolving gene expression: from G to E to G x E. Trends Ecol Evol. 2009;24:649–658. doi: 10.1016/j.tree.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson JR, et al. The complete set of predicted genes from Saccharomyces cerevisiae in a readily usable form. Genome Res. 1997;7:1169–1173. doi: 10.1101/gr.7.12.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebert DJ, Kuan PF, Keles S, Gasch AP. Dynamic changes in nucleosome occupancy are not predictive of gene expression dynamics but are linked to transcription and chromatin regulators. Mol Cell Biol. 2012;32:1645–1653. doi: 10.1128/MCB.06170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LT, Howard WR, Strain JJ, Winge DR, Culotta VC. Enhanced effectiveness of copper ion buffering by CUP1 metallothionein compared with CRS5 metallothionein in Saccharomyces cerevisiae. J Biol Chem. 1996;271:18514–18519. doi: 10.1074/jbc.271.31.18514. [DOI] [PubMed] [Google Scholar]
- Jin Y, et al. Global transcriptome and deletome profiles of yeast exposed to transition metals. PLoS Genet. 2008;4:e1000053. doi: 10.1371/journal.pgen.1000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung PP, et al. Ploidy influences cellular responses to gross chromosomal rearrangements in Saccharomyces cerevisiae. BMC Genomics. 2011;12:331. doi: 10.1186/1471-2164-12-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann J, et al. MAC1, a nuclear regulatory protein related to Cu-dependent transcription factors is involved in Cu/Fe utilization and stress resistance in yeast. EMBO J. 1993;12:5051–5056. doi: 10.1002/j.1460-2075.1993.tb06198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J, Ward DM, Crisp RJ, Philpott CC. Iron-dependent metabolic remodeling in S. cerevisiae. Biochim Biophys Acta Mol Cell Res. 2006;1763:646–651. doi: 10.1016/j.bbamcr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Keller G, Bird A, Winge D. Independent metalloregulation of Ace1 and Mac1 in Saccharomyces cerevisiae. Eukaryot Cell. 2005;4:1863–1871. doi: 10.1128/EC.4.11.1863-1871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SAB, Labbe S, Kwon LF, Kosman DJ, Thiele DJ. A widespread transposable element masks expression of a yeast copper transport gene. Genes Dev. 1998;10:1917–1929. doi: 10.1101/gad.10.15.1917. [DOI] [PubMed] [Google Scholar]
- Kruger A, et al. The pentose phosphate pathway is a metabolic redox sensor and regulates transcription during the antioxidant response. Antioxid Redox Signal. 2011;15:311–324. doi: 10.1089/ars.2010.3797. [DOI] [PubMed] [Google Scholar]
- Kumar C, et al. Glutathione revisited: a vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011;30:2044–2056. doi: 10.1038/emboj.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitek DJ, Will JL, Gasch AP. Variations in stress sensitivity and genomic expression in diverse S. cerevisiae isolates. PLoS Genet. 2008;4: e1000223. doi: 10.1371/journal.pgen.1000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe S, Zhu ZW, Thiele DJ. Copper-specific transcriptional repression of yeast genes encoding critical components in the copper transport pathway. J Biol Chem. 1997;272:15951–15958. doi: 10.1074/jbc.272.25.15951. [DOI] [PubMed] [Google Scholar]
- Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL. Genetic properties influencing the evolvability of gene expression. Science. 2007;317:118–121. doi: 10.1126/science.1140247. [DOI] [PubMed] [Google Scholar]
- Landry CR, Oh J, Hartl DL, Cavalieri D. Genome-wide scan reveals that genetic variation for transcriptional plasticity in yeast is biased towards multi-copy and dispensible genes. Gene. 2006;366:343–351. doi: 10.1016/j.gene.2005.10.042. [DOI] [PubMed] [Google Scholar]
- Lee J, Adle D, Kim H. Molecular mechanisms of copper homeostasis in yeast. In: Tamás MJ, Martinoia E, editors. Molecular biology of metal homeostasis and detoxification: from microbes to man. Germany: Springer Verlag; 2006. pp. 1–37. [Google Scholar]
- Li BZ, Cheng JS, Ding MZ, Yuan YJ. Transcriptome analysis of differential responses of diploid and haploid yeast to ethanol stress. J Biotechnol. 2010;148:194–203. doi: 10.1016/j.jbiotec.2010.06.013. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. Mapping determinants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet. 2006;2:2155–2161. doi: 10.1371/journal.pgen.0020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill R, Muehlenhoff U. Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu Rev Biochem. 2008;77:669–700. doi: 10.1146/annurev.biochem.76.052705.162653. [DOI] [PubMed] [Google Scholar]
- Liti G, et al. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mirabal HR, Winther JR. Redox characteristics of the eukaryotic cytosol. Biochim Biophys Acta Mol Cell Res. 2008;1783:629–640. doi: 10.1016/j.bbamcr.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Macomber L, Imlay JA. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad Sci U S A. 2009;106:8344–8349. doi: 10.1073/pnas.0812808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Townsend JP. A Bayesian method for analysing spotted microarray data. Brief Bioinform. 2005;6:318–330. doi: 10.1093/bib/6.4.318. [DOI] [PubMed] [Google Scholar]
- Mendoza-Cozatl D, Loza-Tavera H, Hernandez-Navarro A, Moreno-Sanchez R. Sulfur assimilation and glutathione metabolism under cadmium stress in yeast, protists and plants. FEMS Microbiol Rev. 2005;29:653–671. doi: 10.1016/j.femsre.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Moczek AP, et al. The role of developmental plasticity in evolutionary innovation. Proc Biol Sci. 2011;278:2705–2713. doi: 10.1098/rspb.2011.0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muehlenhoff U, et al. Cytosolic monothiol glutaredoxins function in intracellular iron sensing and trafficking via their bound iron-sulfur cluster. Cell Metab. 2010;12:373–385. doi: 10.1016/j.cmet.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan K, et al. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol. 2001;21:4347–4368. doi: 10.1128/MCB.21.13.4347-4368.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petti AA, Crutchfield CA, Rabinowitz JD, Botstein D. Survival of starving yeast is correlated with oxidative stress response and nonrespiratory mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:E1089–E1098. doi: 10.1073/pnas.1101494108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfennig DW, et al. Phenotypic plasticity's impacts on diversification and speciation. Trends Ecol Evol. 2010;25:459–467. doi: 10.1016/j.tree.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Philpott CC, Leidgens S, Frey AG. Metabolic remodeling in iron-deficient fungi. Biochim Biophys Acta. 2012;1823:1509–1520. doi: 10.1016/j.bbamcr.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig S, Askeland E, Thiele D. Coordinated remodeling of cellular metabolism during iron deficiency through targeted mRNA degradation. Cell. 2005;120:99–110. doi: 10.1016/j.cell.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Py B, Moreau PL, Barras F. Fe-S clusters, fragile sentinels of the cell. Curr Opin Microbiol. 2011;14:218–223. doi: 10.1016/j.mib.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- Reeder NL, et al. Zinc pyrithione inhibits yeast growth through copper influx and inactivation of iron-sulfur proteins. Antimicrob Agents Chemother. 2011;55:5753–5760. doi: 10.1128/AAC.00724-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustici G, et al. Global transcriptional responses of fission and budding yeast to changes in copper and iron levels: a comparative study. Genome Biol. 2007;8:R73. doi: 10.1186/gb-2007-8-5-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichting CD. Hidden reaction norms, cryptic genetic variation, and evolvability. Ann N Y Acad Sci. 2008;1133:187–203. doi: 10.1196/annals.1438.010. [DOI] [PubMed] [Google Scholar]
- Shakoury-Elizeh M, et al. Metabolic response to iron deficiency in Saccharomyces cerevisiae. J Biol Chem. 2010;285:14823–14833. doi: 10.1074/jbc.M109.091710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AK, Pallesen LJ, Spang RJ, Walden WE. Cytosolic iron-sulfur cluster assembly (CIA) system: factors, mechanism, and relevance to cellular iron regulation. J Biol Chem. 2010;285:26745–26751. doi: 10.1074/jbc.R110.122218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal ML, Promislow DEL, Bergman A. Functional and evolutionary inference in gene networks: does topology matter? Genetica. 2007;129:83–103. doi: 10.1007/s10709-006-0035-0. [DOI] [PubMed] [Google Scholar]
- Slavov N, Botstein D. Coupling among growth rate response, metabolic cycle, and cell division cycle in yeast. Mol Biol Cell. 2011;22:1997–2009. doi: 10.1091/mbc.E11-02-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell-Rood EC, Van Dyken JD, Cruickshank T, Wade MJ, Moczek AP. Toward a population genetic framework of developmental evolution: the costs, limits, and consequences of phenotypic plasticity. Bioessays. 2010;32:71–81. doi: 10.1002/bies.200900132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Schure EG, van Riel NAW, Verrips CT. The role of ammonia metabolism in nitrogen catabolite repression in Saccharomyces cerevisiae. FEMS Microbiol Rev. 2000;24:67–83. doi: 10.1111/j.1574-6976.2000.tb00533.x. [DOI] [PubMed] [Google Scholar]
- Thiele D. ACE1 regulates expression of the Saccharomyces cerevisiae metallothionein gene. Mol Cell Biol. 1988;8:2745–2752. doi: 10.1128/mcb.8.7.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Barkai N, Verstrepen KJ. Promoter architecture and the evolvability of gene expression. J Biol. 2009;8:95. doi: 10.1186/jbiol204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Sigal N, Barkai N. Divergence of nucleosome positioning between two closely related yeast species: genetic basis and functional consequences. Mol Syst Biol. 2010;6:365. doi: 10.1038/msb.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Weinberger A, Carmi M, Barkai N. A genetic signature of interspecies variations in gene expression. Nat Genet. 2006;38:830–834. doi: 10.1038/ng1819. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Wong KH, Barkai N, Struhl K. Extensive divergence of yeast stress responses through transitions between induced and constitutive activation. Proc Natl Acad Sci U S A. 2011;108:16693–16698. doi: 10.1073/pnas.1113718108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend J, Cavalieri D, Hartl D. Population genetic variation in genome-wide gene expression. Mol Cell Biol. 2003;20:955–963. doi: 10.1093/molbev/msg106. [DOI] [PubMed] [Google Scholar]
- Townsend JP. Resolution of large and small differences in gene expression using models for the Bayesian analysis of gene expression levels and spotted DNA microarrays. BMC Bioinformatics. 2004;5:5–54. doi: 10.1186/1471-2105-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui K, et al. Evolution of nucleosome occupancy: conservation of global properties and divergence of gene-specific patterns. Mol Cell Biol. 2011;31:4348–4355. doi: 10.1128/MCB.05276-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bakel H, Strengman E, Wijmenga C, Holstege F. Gene expression profiling and phenotype analyses of S. cerevisiae in response to changing copper reveals six genes with new roles in copper and iron metabolism. Physiol Genomics. 2005;22:356–367. doi: 10.1152/physiolgenomics.00055.2005. [DOI] [PubMed] [Google Scholar]
- van Dyken JD, Wade MJ. The genetic signature of conditional expression. Genetics. 2010;184:557–570. doi: 10.1534/genetics.109.110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warringer J, et al. Trait variation in yeast is defined by population history. PLoS Genet. 2011;7:e1002111. doi: 10.1371/journal.pgen.1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner SV, Sun F, Hernandez N, He CA. The tightly regulated copper window in yeast. Chem Commun. 2011;47:2571–2573. doi: 10.1039/c0cc04292g. [DOI] [PubMed] [Google Scholar]
- Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res. 2010;20:90–100. doi: 10.1101/gr.098509.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch J, Fogel S, Cathala G, Karin M. Industrial yeasts display tandem gene iteration at the CUP1 region. Mol Cell Biol. 1983;3:1353–1361. doi: 10.1128/mcb.3.8.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler E, et al. Genetic diversity in yeast assessed with whole-genome oligonucleotide arrays. Genetics. 2003;163:79–89. doi: 10.1093/genetics/163.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Wittkopp P, Haerum B, Clark A. Regulatory changes underlying expression differences within and between Drosophila species. Nat Genet. 2008;40:346–350. doi: 10.1038/ng.77. [DOI] [PubMed] [Google Scholar]
- Wu H, Kerr M, Cui X, Churchill G. MAANOVA: a software package for the analysis of spotted cDNA microarray experiments. In: Parmigiani G, Garrett ES, Irizarry RA, Zeger SL, editors. The analysis of gene expression data. New York: Springer; 2003. pp. 313–341. [Google Scholar]
- Zhang Z, Townsend J. The filamentous fungi gene expression database (FFGED) Fungal Genet Biol. 2010;47:199–204. doi: 10.1016/j.fgb.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet. 2008;40:854–861. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Labbe S, Pena M, Thiele D. Copper differentially regulates the activity and degradation of yeast Mac1 transcription factor. J Biol Chem. 1998;273:1277–1280. doi: 10.1074/jbc.273.3.1277. [DOI] [PubMed] [Google Scholar]