

Abstract
Bilateral animals are featured by an extremely compact mitochondrial (mt) genome with 37 genes on a single circular chromosome. The human body louse, Pediculus humanus, however, has its mt genes on 20 minichromosomes. We sequenced the mt genomes of two other human lice: the head louse, P. capitis, and the pubic louse, Pthirus pubis. Comparison among the three human lice revealed the presence of fragmented mt genomes in their most recent common ancestor, which lived ∼7 Ma. The head louse has exactly the same set of mt minichromosomes as the body louse, indicating that the number of minichromosomes, and the gene content and gene arrangement in each minichromosome have remained unchanged since the body louse evolved from the head louse ∼107,000 years ago. The pubic louse has the same pattern of one protein-coding or rRNA gene per minichromosome (except one minichromosome with two protein-coding genes, atp6 and atp8) as the head louse and the body louse. This pattern is apparently ancestral to all human lice and has been stable for at least 7 Myr. Most tRNA genes of the pubic louse, however, are on different minichromosomes when compared with their counterparts in the head louse and the body louse. It is evident that rearrangement of four tRNA genes (for leucine, arginine and glycine) was due to gene-identity switch by point mutation at the third anticodon position or by homologous recombination, whereas rearrangement of other tRNA genes was by gene translocation between minichromosomes, likely caused by minichromosome split via gene degeneration and deletion.
Keywords: mitochondrial genome, chromosome evolution, genome fragmentation, human lice
Introduction
Bilateral animals (Bilateria) are featured by an extremely compact mitochondrial genome that consists of 37 genes and a control region on a single circular chromosome, ∼16 kb in size (Boore 1999; Lavrov 2007). This typical mt genome organization is conserved among most of the bilateral animals known, from worms to insects, fish, and humans (Anderson et al. 1981; Lewis et al. 1995; Cao et al. 1998; Lavrov and Brown 2001). On the other hand, however, deviation from the typical mt genome organization has occurred in many bilateral animals. For instance, most nematodes lack atp8 gene and thus have only 36 mt genes (Lavrov and Brown 2001; but see Gissi et al. 2008 and Breton et al. 2010 for an alternative possibility of “missing” atp8). Duplication of mt genes or the control region occurred in many animals, including snakes (Kumazawa et al. 1998), birds (Eberhard et al. 2001), thrips (Shao and Barker 2003), ticks (Shao et al. 2005a), mites (Shao et al. 2005b), and geckos (Fujita et al. 2007). The mt genomes of the rotifer, Brachionus plicatilis, and the booklouse, Liposcelis bostrychophila, consist of two chromosomes (Suga et al. 2008; Wei et al. 2012). The mt genome of the potato cyst nematode, Globodera pallida, has six chromosomes (Gibson et al. 2007).
The most radical departure to date from the typical mt genome organization of bilateral animals is in the human body louse, Pediculus humanus—an exclusive blood sucker and the vector of three infectious diseases of humans: epidemic typhus, relapsing fever, and trench fever (Raoult and Roux 1999). The mt genome of the body louse consists of 20 types of minichromosomes: each minichromosome is 3–4 kb in size and has 1–3 genes and a control region (Shao et al. 2009). Extensive fragmentation of mt genomes also occurred outside bilateral animals in ichthyosporean protists (Burger et al. 2003), diplonemid protists (Marande et al. 2005), and box jellyfish (Smith et al. 2012). In addition, the chloroplast genomes of dinoflagellates also fragmented extensively (Zhang et al. 1999; Howe et al. 2008). Extensive fragmentation of organelle genomes is a fascinating phenomenon; why organelle genomes became fragmented and how they became fragmented, however, are still poorly understood (Zhang et al. 1999; Landweber 2007; Howe et al. 2008; Rand 2009; Shao et al. 2009).
The parasitic lice of humans present an opportunity for investigating how extensively fragmented mt genomes of bilateral animals evolved. In addition to the body louse, P. humanus, humans are also the hosts of two other blood-sucking lice: the head louse, P. capitis, and the pubic louse, Pthirus pubis (Durden and Musser 1994). Humans share the genus Pediculus with chimpanzees, and share the genus Pthirus with gorillas (Durden and Musser 1994). The head louse often infests school children whereas the pubic louse infests primarily adults and is often transmitted via sexual contact (Burgess 2004; Anderson and Chaney 2009). Of the three human lice, the body louse is the youngest: it evolved when humans adopted frequent use of clothing ∼107,000 years ago (Kittler et al. 2003, 2004) [but see Toups et al. (2011) for an alternative dating]. The head louse is much older than the body louse and has been with humans since the ancestors of humans and chimpanzees split ∼5.4 Ma (Chen and Li 2001; Stauffer et al. 2001). The pubic louse was the result of a host switch of gorilla louse to humans 3–4 Ma (Reed et al. 2007). The pubic louse shares the most recent common ancestor (MRCA) with the head louse and the body louse ∼7 Ma before gorillas split from the lineage that led to humans and chimpanzees (Chen and Li 2001; Stauffer et al. 2001). The three human lice, thus, provide a system for understanding how fragmented mt genomes of bilateral animals changed over a period of ∼7 Myr. We sequenced the mt genomes of the head louse and the pubic louse; comparison among the three human lice provided unprecedented insights into the phenomenon of mt genome fragmentation in bilateral animals.
Materials and Methods
Sample Collection, DNA Extraction, Polymerase Chain Reaction Amplification, Cloning, and Capillary Sequencing
Head lice, P. capitis, were collected in Brisbane, Australia (sample B2135), China (B1453), and Iran (B1572A). Pubic lice, P. pubis, were collected in Ipswich and Brisbane, Australia (B2155 and B2182). Total DNA was extracted from individual louse specimens with DNeasy Tissue Kit (QIAGEN). Because of the high sequence similarity between the head louse and the body louse revealed by previous studies (Kittler et al. 2003; Leo et al. 2005; Reed et al. 2007; Herd et al. 2012), we used, initially, the primers designed from the coding regions of the mt minichromosomes of the body louse (Shao et al. 2009) to amplify by polymerase chain reaction (PCR) each mt minichromosome of the head louse. These primers are next to each other with no gap or a small gap (<10 bp) in between; PCRs with these primers amplified 12 entire or near entire mt minichromosomes of the head louse. These amplicons were sequenced directly (without cloning) with AB3730xl 96-capillary sequencers at the Australian Genome Research Facilities (AGRF) in Brisbane, Australia. A pair of head louse–specific primers, PcF and PcR, was designed from conserved noncoding sequences that flank the coding region of each minichromosome (fig. 1A; supplementary table S1, Supplementary Material online). The PCR with PcF and PcR primers produced a mixture of amplicons ranging from 100 bp to 2 kb in size, expected from the coding regions of the whole set of mt minichromosomes of the head louse (fig. 2A); these amplicons were sequenced with a Roche 454 platform at the AGRF.
Fig. 1.—


Alignment of nucleotide sequences in the NCRs of the mitochondrial genomes of the human head louse, Pediculus capitis (A), and the human pubic louse, Pthirus pubis (B).
Fig. 2.—
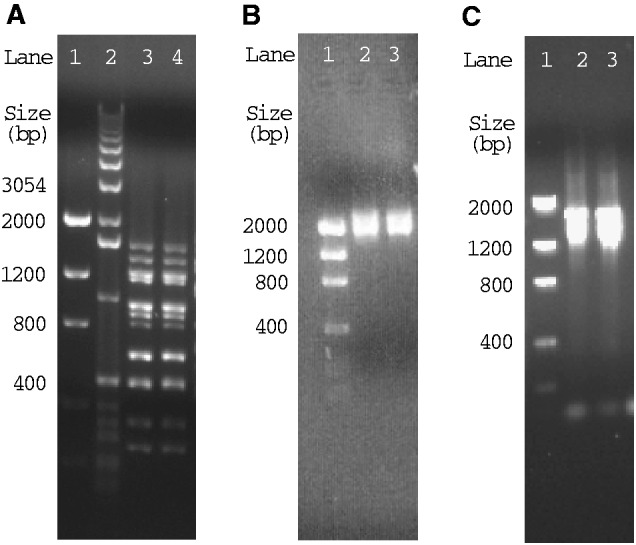
PCR amplicons from the mitochondrial genomes of the human head louse, Pediculus capitis (A) and the human pubic louse, Pthirus pubis (B). (A) Amplicons from PcF and PcR primers that span the entire coding regions of the mt minichromosomes of the head louse. (B) Amplicons from PpF and PpR1 primers that span the entire coding and NCRs of the mt minichromosomes of the pubic louse. (C) Amplicons from PpF and PpR2 primers that span the entire coding regions of the mt minichromosomes of the pubic louse.
For the pubic louse, a 452-bp fragment of mt rrnS gene and a 360-bp fragment of mt rrnL gene were amplified initially by PCR with primer pairs 12SA–12SB and 16SA–16SB (supplementary table S1, Supplementary Material online). These two pairs of primers target sequence motifs that are highly conserved among arthropods. The rrnS and rrnL fragments were sequenced directly; two pairs of pubic louse–specific primers, 12SF–12SR and 16SF–16SR, were designed from rrnS and rrnL genes (supplementary table S1, Supplementary Material online). PCRs with these specific primers amplified a 1.8-kb fragment and a 2.3-kb fragment that spanned nearly the entire trnL1–rrnS minichromosome and trnL2–rrnL minichromosome, respectively. These fragments were cloned and sequenced; two primers, PpF (forward) and PpR1 (reverse), were designed from conserved noncoding sequences adjacent to the 5′-end of the coding regions of trnL1–rrnS minichromosome and trnL2–rrnL minichromosome (fig. 1B). The PCR with the primers PpF and PpR1 produced a mixture of amplicons ranging from 1.6 to 2.7 kb (fig. 2B), expected from the whole set of mt minichromosomes of the pubic louse. PCR amplicons with PpF and PpR1 were cloned and sequenced with AB3730xl 96-capillary sequencers. A reverse primer, PpR2, was also designed from conserved noncoding sequences downstream of the coding regions of minichromosomes (fig. 1B). The PCR with primers PpF and PpR2 generated a mixture of amplicons ranging from 1.2 to 1.9 kb in size (fig. 2C), expected from the coding regions of the whole set of mt minichromosomes of the pubic louse; these amplicons were sequenced with a Roche 454 platform at the AGRF (discussed later).
TaKaRa LA Taq kit was used in all PCR amplifications. Each PCR (25 µL) contained 0.25 µL of LA Taq, 2.5 µL of 10× Buffer, 2.5 µL of MgCl2 (25 mM), 2.0–4.0 µL of dNTP mixture (2.5 mM each), 1.0 µL of forward primer (10 µM), 1.0 µL of reverse primer (10 µM), 1.0 µL of DNA template, and 12.75–14.75 µL of Milli-Q water. PCR cycling conditions were: 94°C for 1 min, 30–40 cycles of 98°C for 10 s, 45–65°C (depending on primers) for 30 s, and 68°C for 1–4 min (depending on target size, ∼1 min/kb), followed by 72°C for 2–6 min. Positive and negative controls were always executed with each PCR experiment to help to detect DNA contamination and false positive amplicons. PCR amplicons were checked by agarose gel (1%) electrophoresis. The sizes of PCR amplicons were estimated by comparison with molecular markers. PCR amplicons used for sequencing were purified with Wizard SV Gel/PCR Clean-up System (Promega); cloning was with pGEM-T Easy Vector System (Promega). Sequence-reads were assembled into contigs with Sequencher 5.0 (Gene Codes); the parameters for assembly were minimum match 90% and minimum overlap 100 bp. tRNA genes were identified with tRNA-Scan (Lowe and Eddy 1997) and ARWEN (Laslett and Canback 2008). Protein-coding genes and rRNA genes were identified with BLAST searches of GenBank (Altschul et al. 1990). Identical sequences shared between genes were identified by Wordmatch (Rice et al. 2000). Sequence alignments were with ClustalX (Larkin et al. 2007).
Roche 454 Sequencing and Verification of Individual Mitochondrial Minichromosomes
The contigs generated from capillary sequencing had low coverage (1 to 3×). To ensure the reliability of sequence analyses, we used a Roche GS-FLX (454) platform to sequence, in high coverage (4 to 2,700×), the amplicons obtained from the mt minichromosomes of the head louse and the pubic louse. Roche 454 sequence reads were assembled and analysed as described earlier. We also verified by PCR the size and the circular organization of each mt minichromosome of the head louse and the pubic louse identified by sequence-read assembly. A pair of outbound primers (forward and reverse) was designed from the coding region of each minichromosome; these two primers were next to each other with a small gap or no gap in between. PCRs with these primers amplify the full or near full length of each minichromosome if it has a circular organization. PCR set-up, cycling conditions, agarose gel electrophoresis and size measurement were the same as described earlier. The nucleotide sequences of the mitochondrial genomes of the human head louse and the pubic louse have been deposited in GenBank under accession numbers EU219988-95, HM241895-8, and JX080388-407.
Results
Mitochondrial Genome of the Human Head Louse, P. capitis
We obtained 9,412 sequence reads from the mt genome of the head louse by capillary sequencing and Roche 454 sequencing (table 1). These sequence-reads range from 100 to 700 bp in length. We assembled these sequence reads into contigs and identified all of the 37 mt genes typical of bilateral animals. The 37 mt genes of the head louse are on 20 minichromosomes; the distribution of these genes among the minichromosomes in the head louse is identical to that in the body louse (Shao et al. 2009). Each minichromosome is 1.5–3.0 kb in size and has a circular organization (fig. 3A). Each minichromosome has a coding region and a noncoding region (NCR). The coding region of each minichromosome contains one to three genes, and varies in size from 65 bp for trnM minichromosome to 1,686 bp for nad5 minichromosome (Note: minichromosomes are named after their genes hereafter). With the intriguing exception of nad1 and trnQ, all mt genes of the head louse have the same orientation of transcription relative to the NCRs (fig. 3A).
Table 1.
Mitochondrial Minichromosomes of the Head Louse, Pediculus capitis, Identified by Capillary Sequencing and Roche 454 Sequencing
| Minichromosome | Size of Coding Region (bp) | Number of Capillary Sequence Reads | Number of Roche 454 Sequence Reads |
|---|---|---|---|
| atp8-atp6 | 846 | 9 | 222 |
| cox1 | 1,572 | 16 | 99 |
| Y-cox2 | 757 | 11 | 222 |
| cox3-A | 875 | 10 | 182 |
| cob | 1,074 | 13 | 125 |
| nad1-Q | 967 | 11 | 70 |
| P-nad2-I | 1,129 | 18 | 89 |
| R-nad3 | 428 | 7 | 683 |
| K-nad4 | 1,419 | 10 | 49 |
| G-nad4L-V | 409 | 0 | 556 |
| nad5 | 1,686 | 11 | 15 |
| F-nad6 | 580 | 0 | 499 |
| L1(tag)/L2(taa)-rrnS-C | 933 | 17 | 411 |
| L1(tag)/L2(taa)-rrnL | 1,220 | 14 | 188 |
| S1(tct)-N-E | 226 | 0 | 1,259 |
| T-D-H | 227 | 0 | 869 |
| M | 65 | 0 | 992 |
| W-S2(tga) | 144 | 0 | 2,735 |
| Total | 14,557 | 147 | 9,265 |
Fig. 3.—

The mitochondrial (mt) genomes of the human head louse, Pediculus capitis (A) and the human pubic louse, Pthirus pubis (B). Each minichromosome has a coding region (with gene name, transcription orientation, and length indicated) and a NCR (in black). The mini-chromosomes are in alphabetical order according to the names of their protein coding and rRNA genes, followed by those with only tRNA genes. Minichromosomes with asterisk symbols (*) are present in all three of the human lice. The gray shading in the trnC minichromosome is the 99 bp unique section that has 90% similarity in sequence to trnL2 and rrnL genes (fig. 4A); the black dot shading is the 751 bp unique section that has 50% similarity in sequence to rrnL gene (fig. 4B). The white dot shading in the trnL2–rrnL minichromosome is the 67 bp unique section that has 59% similarity in sequence to trnC gene (fig. 4C). Protein-coding genes are abbreviated as atp6 and atp8 (for ATP synthase subunits 6 and 8), cox1–3 (for cytochrome c oxidase subunits 1–3), cob (for cytochrome b), and nad1-6 and 4L (for NADH dehydrogenase subunits 1–6 and 4L). rrnL and rrnS are for large and small rRNA subunits. tRNA genes are shown with the single-letter abbreviations of their corresponding amino acids. Courtesy: of Vincent Smith (for the head louse image) and the CDC, USA (for the pubic louse image).
We sequenced the full length NCRs of two mt minichromosomes, cox1 and trnK–nad4, of the head louse (fig. 1A). The six copies of the NCR of the cox1 minichromosome of the head louse are ∼1.4 kb in size, that is, ∼0.5 kb shorter than their counterparts in the body louse (Shao et al. 2009). The NCR of the trnK–nad4 minichromosome of the head louse is 1,905 bp, a similar length to those of the body louse (Shao et al. 2009). The NCRs of the head lice have 72–97% pairwise identity to each other. An AT-rich motif (203 bp, 69% A and T) is present in the NCRs of the head louse before the 5′-end of the coding region, whereas a GC-rich motif (63 bp, 60% C and G) is present after the 3′-end of the coding region (fig. 1A). Between the GC-rich motif and the AT-rich motif, there are numerous conserved sequence motifs ranging from 1 to 103 bp in size, and indels ranging from 1 to 116 bp in size.
Mitochondrial Genome of the Human Pubic Louse, P. pubis
The capillary sequencing and Roche 454 sequencing generated 2,663 sequence reads in total, ranging from 100 to 700 bp in length, from the mt genome of the pubic louse (table 2). We assembled these sequence reads into contigs and identified 34 of the 37 mt genes that are typical of bilateral animals; these 34 genes are on 14 minichromosomes (table 2; fig. 3B). Each minichromosome is circular, 1.8–2.7 kb in size, and consists of a coding region and a NCR. The coding regions contain one to five genes each, and vary in size from 64 bp for trnC minichromosome to 1,647 bp for nad5 minichromosome (table 2; fig. 3B). Each of the 34 mt genes we identified in the pubic louse is present on only one minichromosome; there is no overlap in gene content between different minichromosomes. All of the 34 mt genes, including nad1 and trnQ, have the same orientation of transcription relative to the NCRs (fig. 3B). Intriguingly, we did not find nad4, trnK, and trnN in the pubic louse by either capillary sequencing or Roche 454 sequencing. These three genes are present in the fragmented mt genomes of the head louse (discussed earlier), the body louse (Shao et al. 2009), and the chimpanzee louse, P. schaeffi (Herd K, et al., unpublished data). There could be two possibilities. The minichromosomes that contained these three genes were less abundant than the 14 minichromosomes we identified in the pubic louse, and thus, were missed in our sequencing at the current coverage (table 2). Alternatively, the primers we used to amplify the mt genome of the pubic louse might not be conserved in the minichromosomes that contained nad4, trnK, and trnN genes. The second possibility is more likely, in our view, as the expression of mt genes needs to be coordinated and regulated; a substantially decreased frequency of an mt minichromosome would present serious metabolic challenges to cells (Lane 2005).
Table 2.
Mitochondrial Minichromosomes of the Pubic Louse, Pthirus pubis, Identified by Capillary Sequencing and Roche 454 Sequencing
| Minichromosome | Size of Coding Region (bp) | Number of Clones Sequenced | Number of Capillary Sequence Reads | Number of Roche 454 Sequence Reads |
|---|---|---|---|---|
| atp8-atp6 | 821 | 6 | 12 | 138 |
| cox1 | 1,548 | 3 | 37 | 185 |
| Y-cox2 | 766 | 7 | 21 | 37 |
| cox3-A | 885 | 8 | 22 | 71 |
| cob-S1 | 1,119 | 14 | 37 | 431 |
| nad1-Q | 958 | 6 | 21 | 522 |
| P-nad2-I | 1,126 | 2 | 4 | 41 |
| G-nad3-V-W-S2 | 795 | 9 | 33 | 24 |
| T-D-H-R-nad4L | 597 | 4 | 14 | 45 |
| nad5 | 1,647 | 2 | 8 | 17 |
| F-nad6-E-M | 761 | 2 | 6 | 0 |
| L1(tag)-rrnS | 830 | 9 | 28 | 28 |
| L2(taa)-rrnL | 1,287 | 8 | 25 | 665 |
| C | 64 | 26 | 62 | 129 |
| nad4, K, N | NA | 0 | 0 | 0 |
| Total | 13,204 | 106 | 330 | 2,333 |
Note.—NA, not applicable.
We sequenced the full length NCRs of 10 mt minichromosomes of the pubic louse (fig. 1B). The NCRs of these minichromosomes, with the exception of trnC minichromosome and trnL2–rrnL minichromosome, have similar length (978–1,044 bp) and high sequence similarities to each other (91–99%). A perfectly conserved AT-rich motif (111 bp, 76% A and T) is upstream of the 5′-end of the coding region of each minichromosome; a GC-rich motif (77 bp, 58% G and C) is downstream of the 3′-end of the coding region. In addition, there are numerous highly conserved motifs, ranging from 1 to 87 bp, in the NCRs between the GC-rich motif and the AT-rich motif (fig. 1B).
The NCRs of the trnC minichromosome and the trnL2–rrnL minichromosome of the pubic louse contain unique sections that are not present in the NCRs of other minichromosomes (fig. 3B). One of the unique sections in the NCR of trnC minichromosome is 99 bp long and has 90% similarity in sequence to trnL2 and rrnL genes (fig. 4A); the other unique section is 751 bp long and has 50% similarity in sequence to rrnL gene (fig. 4B). On the other hand, the unique section in the trnL2–rrnL minichromosome is 67 bp long and has 59% similarity in sequence to trnC gene (fig. 4C). The unique sections in the trnC minichromosome are apparently degenerate trnL2 and rrnL genes and the unique section in the trnL2–rrnL minichromosome a degenerate trnC gene.
Fig. 4.—

Alignment of the nucleotide sequences between the two unique sections in the NCR of trnC minichromosome (gray shading and black dot shading in fig. 3B) and the coding region of trnL2–rrnL minichromosome (A and B), and between the unique section in the NCR of trnL2–rrnL minichromosome (white dot shading in fig. 3B) and the coding region of trnC minichromosome of the pubic louse, Pthirus pubis (C).
Discussion
Recombination between Mitochondrial Genes on Different Minichromosomes
Whether and how animal mt DNA recombine has been a controversial issue in the past decade (Rokas et al. 2003; Kraytsberg et al. 2004; Ladoukakis et al. 2011). An extraordinary feature of the fragmented mt genome of the human body louse is the stretches of identical sequences (26–127 bp long) shared between genes on different minichromosomes; this provides unequivocal evidence for inter-minichromosome recombination (Shao et al. 2009; Shao and Barker 2011). As in the body louse, inter-minichromosome recombination also occurs in the head louse and the pubic louse. Six pairs of mt genes in the head louse share identical sequences, 26–127 bp long, which are much longer than expected by chance (table 3). Two pairs of tRNA genes in the pubic louse, trnL1 and trnL2, and trnR and trnG, share identical sequences that are three to five times longer than expected by chance (table 3). Two other pairs of mt genes of the pubic louse, cox1 and nad4L, and trnI and trnT, share identical sequences two to three times longer than expected by chance. It is noteworthy that in the pubic louse, only the genes of the same type, that is, two tRNA genes or two protein-coding genes, share longer than expected identical sequences. In the body louse and the head louse, however, the large subunit rRNA gene, rrnL, shares longer than expected identical sequences, 26 and 99 bp, respectively, with two protein-coding genes, nad2 and nad5 (table 3). The fact that all of the three human lice have longer than expected stretches of identical sequences shared between different mt genes, whereas no other bilateral animals known appear to have this feature indicates that mt genome fragmentation facilitates recombination between mt genes. Indeed, similar evidence for recombination between minichromosomes was also found recently outside bilateral animals in a box jellyfish that has an extensively fragmented mt genome with eight linear minichromosomes (Smith et al. 2012).
Table 3.
The Longest Stretches of Identical Sequence Shared by Mitochondrial Genes of the Three Human Lice, Which Have Fragmented Mitochondrial Genomes, and Six Other Species of Bilateral Animals that Have the Typical Mitochondrial Genomes
| Pairs of gene | The Longest Stretches of Identical Sequence (bp) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pc | Pp | Ph | Bm | Cb | Hm | Dy | Ce | Hs | ||
| nad4 | nad5 | 127, 30 | NA | 127, 30 | 13 | 15 | 15 | 16 | 14 | 11 |
| rrnL | nad5 | 99 | 10 | 99 | 12 | 14 | 13 | 15 | 16 | 10 |
| trnL1 | trnL2 | 33, 32 | 35, 32 | 33, 32 | 7 | 6 | 7 | 10 | 6 | 6 |
| trnR | trnG | 28, 14 | 32, 26 | 28, 14 | 5 | 6 | 7 | 6 | 8 | 6 |
| atp8 | Cob | 26 | 9 | 26 | 10 | 11 | 11 | 12 | NA | 9 |
| rrnL | nad2 | 26 | 10 | 26 | 13 | 11 | 14 | 13 | 12 | 10 |
| cox1 | nad4L | 10 | 29 | 10 | 13 | 11 | 14 | 13 | 12 | 10 |
| trnI | trnT | 6 | 16 | 6 | 6 | 5 | 7 | 7 | 9 | 6 |
Note.—The longest stretches of shared identical sequences are indicated in bold. Pc, Pediculus capititis (human head louse); Pp, Pthirus pubis (human pubic louse); Ph, Pediculus humanus (human body louse); Bm, Bothriometopus macrocnemis (screamer louse); Cb, Campanulotes bidentatus (pigeon louse); Hm, Heterodoxus macropus (wallaby louse); Dy, Drosophila yakuba (fruitfly); Ce, Caenorhabditis elegans (roundworm); Hs, Homo sapiens (human); NA, not applicable.
Unusual Secondary Structures of Mitochondrial tRNAs
Most tRNAs of the human lice have the typical clover leaf–shaped secondary structure; four tRNAs, however, have atypical structures (fig. 5). The anticodon loop (AC-loop) of tRNAs almost always has seven nucleotides; the tRNA–alanine of the pubic louse, however, has nine nucleotides in this loop. tRNA–glutamine lacks the D-arm in all three of the human lice—a feature that is not seen in other species of parasitic lice known (Shao et al. 2001; Covacin et al. 2006; Cameron et al. 2007). As in most bilateral animals, tRNA–serine (UCU) also lacks the D-arm in the three human lice. The genes for tRNA–glutamine and tRNA–serine (UCU) were annotated incorrectly for the body louse due to the atypical structures of these two tRNAs (Shao et al. 2009). Comparison among the head louse, the pubic louse, and the body louse allowed us to correct these misannotations (figs. 3A and 5).
Fig. 5.—


Inferred secondary structures of the mitochondrial tRNAs of the human lice: the body louse, Pediculus humanus (Ph), the head louse, Pediculus capitis (Pc), and the pubic louse, Pthirus pubis (Pp). Shared identical sequences between tRNA genes are in bold (see text in Discussion and table 3).
For tRNAs with the typical structure, the variable loop (V-loop, fig. 5) usually has four or five nucleotides; tRNA–isoleucine of the pubic louse, however, has seven nucleotides in this loop; the extra nucleotides in this loop make this tRNA gene undetectable by the two commonly used tRNA search programs, ARWEN (Laslett and Canback 2008) and tRNAscan–SE (Lowe and Eddy 1997). It is noteworthy that the V-loop of tRNA–isoleucine is part of the 16 nucleotide identical sequence shared between trnI and trnT genes (see above, fig. 5 and table 3), indicating that recombination between tRNA genes may affect the secondary structure of their corresponding tRNAs. This affect is much evident in the secondary structures of four other tRNAs: 1) tRNA–leucine (UAG) and tRNA–leucine (UAA) of the three human lice have identical secondary structures except for a nucleotide variation at the third anticodon position; and 2) tRNA–arginine and tRNA–glycine of the pubic louse also have identical secondary structures except for the nucleotide at the third anticodon position and three other nucleotides of the anticodon arm (AC-arm; fig. 5).
Fragmentation of Mitochondrial Genomes in the Human Lice
The MRCA of the three human lice lived ∼7 Ma on the MRCA of humans, chimpanzees, and gorillas (Chen and Li 2001; Stauffer et al. 2001; Reed et al. 2007). All three of the human lice have fragmented mt genomes and, furthermore, a common pattern for the distribution of protein-coding and rRNA genes among mt minichromosomes, in which atp6 and atp8 are adjacent on one minichromosome, whereas each of the other protein-coding and rRNA genes has its own minichromosome (fig. 3). This pattern of gene distribution and the fragmented mt genomes, thus, have apparently evolved in the MRCA of the three human lice, and have been retained for at least ∼7 Myr.
Eight of the 14 minichromosomes of the pubic louse have identical gene content and gene arrangement to their counterparts in the head louse and the body louse (fig. 3). It is noteworthy that the transcription orientation of nad1–trnQ relative to the NCR differs between the pubic louse on the one hand, and the body louse and the head louse on the other hand. Thus, nad1–trnQ has inverted in either the lineage that led to the pubic louse or the lineage that led to the body louse and the head louse. Six of the 14 minichromosomes of the pubic louse are not present in the body louse and the head louse (fig. 3). The arrangement of tRNA genes in these six minichromosomes distinguishes the pubic louse from the body louse and the head louse. With the only exception of trnC, all other tRNA genes of the pubic louse are either upstream or downstream of a protein-coding or rRNA gene. In the two extreme cases, four tRNA genes in a cluster, trnT–trnD–trnH–trnR, are upstream of nad4L gene, and three other tRNA genes in another cluster, trnV–trnW–trnS2(tga), are downstream of nad3 gene. In the body louse and the head louse, however, the number of tRNA gene upstream or downstream of a protein-coding or rRNA gene does not exceed one; furthermore, nine tRNA genes on four minichromosomes have no connection at all with any protein-coding or rRNA genes (fig. 3A). Clearly, the pace of mt genome fragmentation has been faster in the lineage leading to the body louse and the head louse than in the lineage leading to the pubic louse. In the typical single-chromosome mt genome of bilateral animals, the secondary structure of tRNA sequences serves as punctuation marks in the processing of the polycistronic transcripts into individual mRNAs and rRNAs (Ojala et al. 1981). For the fragmented mt genomes of the human lice, the role of tRNA structures as punctuation marks is likely less important, or not needed, as only the transcript from the atp8–atp6 minichromosome encodes two proteins; all other transcripts encode either only one protein or rRNA, or only tRNAs.
The NCR in the mt genome of bilateral animals contains initiation sites for genome replication and gene transcription and are commonly called the control region (Boore 1999). For instance, in the human mt genome, two of the three transcription-initiation sites and one of the replication-initiation sites are in the NCR (Taanman 1999). In the mt genome of Drosophila melanogaster, two of the five transcription-initiation sites are in the NCR (Berthier et al. 1986). The NCRs of the three human lice vary substantially in both size and nucleotide sequence. Two common features, however, are present in the NCRs of all mt minichromosomes in all three of the human lice: 1) an AT-rich motif (>59% A and T) upstream of each coding region and a CG-rich motif (>60% C and G) downstream of each coding region; and 2) a perfectly conserved motif, ACCAAATAGCTA, between the AT-rich and the CG-rich motifs (fig. 1). The roles of these sequence motifs in the initiation and termination of mt gene transcription and mt minichromosome replication remain to be determined.
Molecular Mechanisms for tRNA Gene Rearrangement between Minichromosomes
Rearrangements of tRNA genes can be powerful phylogenetic markers (Boore et al. 1998). Recognition of tRNA orthologs, however, is not always straightforward. Inferring orthologs solely on anti-codon sequence and deduced secondary structure can be misleading (Rawlings et al. 2003). In the pubic louse, trnR is before nad4L, and trnG is before nad3. In the body louse and the head louse, however, these two genes have different positions: trnR gene is before nad3, and trnG gene is before nad4L (fig. 3). The arrangements, trnR–nad4L and trnG–nad3, in the pubic louse can be inferred to be ancestral to sucking lice (suborder Anoplura, which includes the three human lice) because these arrangements are also present in the screamer louse, Bothriometopus macrocnemis (suborder Ischnocera, sister to the Anoplura + Rhyncopthirina) (Cameron et al. 2007) (fig. 6). Therefore, we infer that trnR and trnG genes had rearranged in the body louse and the head louse. Because of the high sequence similarity between trnR and trnG (fig. 5), the rearrangements of these two tRNA genes in the body louse and the head louse occurred more likely through gene-identity switch, either by point mutation at the third anticodon position or by homologous recombination, than by gene translocation between minichromosomes. In the same way, gene-identity switch between trnL1(tag) and trnL2(taa) explains well the variation in the arrangement of these two tRNA genes between the pubic louse on the one hand, and the body louse and the head louse on the other hand. In the pubic louse, trnL1(tag) is upstream of rrnS gene only, and trnL2(taa) is upstream of rrnL gene only (fig. 3B). In the body louse and the head louse, however, trnL1(tag) is upstream of rrnS gene in one minichromosome and upstream of rrnL gene in another minichromosome; so is trnL2(taa) gene (fig. 3A). Because of the near identical sequence of trnL1(tag) and trnL2(taa) genes in all of the three human lice (fig. 5), their identities can swap relatively easily by either point mutation at the third anticodon position or by homologous recombination. Variation in the arrangement of other 10 tRNA genes, however, between the pubic louse on the one hand, and the body louse and the head louse on the other hand, is very likely due to gene translocation from one minichromosome to another; there is no evidence that gene identity-switch could cause variation in the arrangement of these tRNA genes in the human lice. The degenerate gene sequences present in the trnC minichromosome and the trnL2–rrnL minichromosome of the pubic louse indicates that gene degeneration followed by deletion is likely responsible for the split of one minichromosome into two, and thus for the translocation of tRNA genes between minichromosomes (fig. 7).
Fig. 6.—

Comparison of mitochondrial gene arrangement among the three human lice (suborder Anoplura) and the screamer louse, Bothriometopus macrocnemis (suborder Ischnocera). Genes on the same chromosome are linked by a hyphen (-); different minichromosomes are separated by a comma (,). Genes underlined have an opposite orientation of transcription relative to the NCR, compared with genes on other minichromosomes.
Fig. 7.—
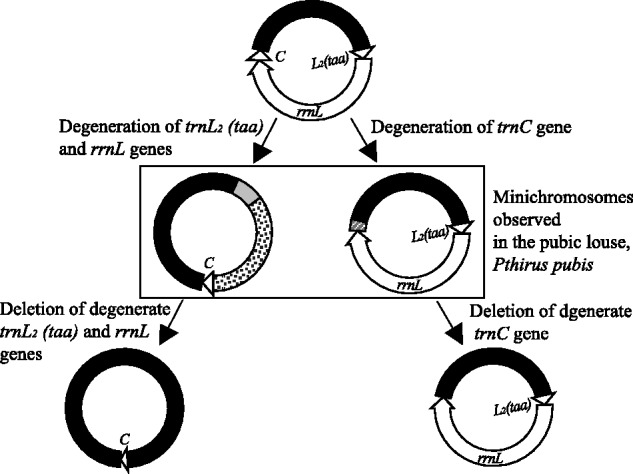
A gene-degeneration-followed-by-deletion model for the split of one mitochondrial minichromosome into two, and thus the translocation of tRNA genes between minichromosomes, based on the degenerate gene sequences present in the trnC and trnL2–rrnL minichromosomes of the human pubic louse, Pthirus pubis.
Conclusion
We sequenced the mt genomes of the human head louse and the human pubic louse. Comparison among the three human lice revealed that fragmented mt genomes were present in the MRCA of these lice ∼7 Ma. The pace of mt genome fragmentation has been unequal; it is faster in the lineage leading to the body louse and the head louse than in the lineage leading to the pubic louse, evidenced by the fact that there are more minichromosomes and thus fewer genes per minichromosome in the body louse and the head louse than in the pubic louse. Despite substantial variation in the sequence and the size of the NCR, the number of mt minichromosomes, and the gene content and gene arrangement in each minichromosome has been perfectly conserved between the head louse and the body louse for ∼107,000 years. The pubic louse has the same pattern as the body louse and the head louse for the distribution of protein-coding genes and rRNA genes among the mt minichromosomes; this pattern, thus, has been stable for at least 7 Myr. The majority of the tRNA genes of the pubic louse are on different minichromosomes when compared with their counterparts in the body louse and the head louse. It is evident that rearrangement of four tRNA genes (for leucine, arginine, and glycine) is due to gene-identity switch caused by either point mutation at the third anticodon position or homologous recombination. Rearrangement of 10 other tRNA genes was by gene translocation between minichromosomes, very likely caused by minichromosome split via gene degeneration and then deletion.
Supplementary Material
Supplementary table S1 is available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Gary Ah-kee, Maryam Ashrafi, Jennifer Cooling, Cath Covacin, Cynthia Pollard, and Chris Whittier for their assistance. The authors also thank the associate editor and the anonymous reviewers for their comments that greatly improved this manuscript. This work was supported by the Australian Research Council (DP0662755 to R.S. and DP120100240 to R.S. and S.C.B). R.S. also acknowledges the funding support from the Australian Government for an Australia-China Science & Research Fund Group Mission visit to China (ACSRF00980).
Literature Cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Anderson AL, Chaney E. Pubic lice (Pthirus pubis): history, biology and treatment vs. knowledge and beliefs of US college students. Int J Environ Res Public Health. 2009;6:592–600. doi: 10.3390/ijerph6020592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Berthier F, Renaud M, Alziari S, Durand R. RNA mapping on Drosophila mitochondrial DNA: precursors and template strands. Nucleic Acids Res. 1986;14:4519–4533. doi: 10.1093/nar/14.11.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL, Lavrov DV, Brown WM. Gene translocation links insects and crustaceans. Nature. 1998;392:667–668. doi: 10.1038/33577. [DOI] [PubMed] [Google Scholar]
- Breton S, Stewart DT, Hoeh WR. Characterization of a mitochondrial ORF from the gender-associated mtDNAs of Mytilus spp. 2010 doi: 10.1016/j.margen.2010.01.001. (Bivalvia: Mytilidae): identification of the “missing” ATPase 8 gene. Marine Genomics 3:11–18. [DOI] [PubMed] [Google Scholar]
- Burger G, Forget L, Zhu Y, Gray MW, Lang BF. Unique mitochondrial genome architecture in unicellular relatives of animals. Proc Natl Acad Sci U S A. 2003;100:892–897. doi: 10.1073/pnas.0336115100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess IF. Human lice and their control. Annu Rev Entomol. 2004;49:457–481. doi: 10.1146/annurev.ento.49.061802.123253. [DOI] [PubMed] [Google Scholar]
- Cameron SL, Johnson KP, Whiting MF. The mitochondrial genome of the screamer louse Bothriometopus (phthiraptera: ischnocera): effects of extensive gene rearrangements on the evolution of the genome. J Mol Evol. 2007;65:589–604. doi: 10.1007/s00239-007-9042-8. [DOI] [PubMed] [Google Scholar]
- Cao Y, Waddell PJ, Okada N, Hasegawa M. The complete mitochondrial DNA sequence of the shark Mustelus manazo: evaluating rooting contradictions to living bony vertebrates. Mol Biol Evol. 1998;15:1637–1646. doi: 10.1093/oxfordjournals.molbev.a025891. [DOI] [PubMed] [Google Scholar]
- Chen FC, Li WH. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet. 2001;68:444–456. doi: 10.1086/318206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacin C, Shao R, Cameron S, Barker SC. Extraordinary number of gene rearrangements in the mitochondrial genomes of lice (Phthiraptera: Insecta) Insect Mol Biol. 2006;15:63–68. doi: 10.1111/j.1365-2583.2005.00608.x. [DOI] [PubMed] [Google Scholar]
- Durden LA, Musser GG. The sucking lice (Insecta, Anoplura) of the World—a taxonomic checklist with records of mammalian hosts and geographical distributions. Bull Am Mus Nat Hist. 1994;218:1–90. [Google Scholar]
- Eberhard JR, Wright TF, Bermingham E. Duplication and concerted evolution of the mitochondrial control region in the parrot genus Amazona. Mol Biol Evol. 2001;18:1330–1342. doi: 10.1093/oxfordjournals.molbev.a003917. [DOI] [PubMed] [Google Scholar]
- Fujita MK, Boore JL, Moritz C. Multiple origins and rapid evolution of duplicated mitochondrial genes in parthenogenetic geckos (Heteronotia binoei; squamata, gekkonidae) Mol Biol Evol. 2007;24:2775–2786. doi: 10.1093/molbev/msm212. [DOI] [PubMed] [Google Scholar]
- Gibson T, Blok VC, Dowton M. Sequence and characterization of six mitochondrial subgenomes from Globodera rostochiensis: multipartite structure is conserved among close nematode relatives. J Mol Evol. 2007;65:308–315. doi: 10.1007/s00239-007-9007-y. [DOI] [PubMed] [Google Scholar]
- Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301–320. doi: 10.1038/hdy.2008.62. [DOI] [PubMed] [Google Scholar]
- Herd K, Barker SC, Shao R. High-level of heteroplasmy in the mitochondrial cox1-minichromosome of the human body louse, Pediculus humanus, and the human head louse, Pediculus capitis. Open Genomics J. 2012;5:14–17. [Google Scholar]
- Howe CJ, Nisbet RER, Barbrook AC. The remarkable chloroplast genome of dinoflagellates. J Exp Bot. 2008;59:1035–1045. doi: 10.1093/jxb/erm292. [DOI] [PubMed] [Google Scholar]
- Kittler R, Kayser M, Stoneking M. Molecular evolution of Pediculus humanus and the origin of clothing. Curr Biol. 2003;13:1414–1417. doi: 10.1016/s0960-9822(03)00507-4. [DOI] [PubMed] [Google Scholar]
- Kittler R, Kayser M, Stoneking M. Molecular evolution of Pediculus humanus and the origin of clothing (Erratum) Curr Biol. 2004;14:2309. doi: 10.1016/s0960-9822(03)00507-4. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, et al. Recombination of human mitochondrial DNA. Science. 2004;304:981. doi: 10.1126/science.1096342. [DOI] [PubMed] [Google Scholar]
- Kumazawa Y, Ota H, Nishida M, Ozawa T. The complete nucleotide sequence of a snake (Dinodon semicarinatus) mitochondrial genome with two identical control regions. Genetics. 1998;150:313–329. doi: 10.1093/genetics/150.1.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoukakis ED, Theologidis I, Rodakis GC, Zouros E. Homologous recombination between highly diverged mitochondrial sequences: examples from maternally and paternally transmitted genomes. Mol Biol Evol. 2011;28:1847–1859. doi: 10.1093/molbev/msr007. [DOI] [PubMed] [Google Scholar]
- Landweber LF. Genetics. Why genomes in pieces? Science. 2007;318:405–407. doi: 10.1126/science.1150280. [DOI] [PubMed] [Google Scholar]
- Lane N. New York: Oxford University Press; 2005. Power, sex, suicide. Mitochondria and the meaning of life. [Google Scholar]
- Larkin MA, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Laslett D, Canback B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–175. doi: 10.1093/bioinformatics/btm573. [DOI] [PubMed] [Google Scholar]
- Lavrov DV. Key transitions in animal evolution: a mitochondrial DNA perspective. Integr Comp Biol. 2007;47:734–743. doi: 10.1093/icb/icm045. [DOI] [PubMed] [Google Scholar]
- Lavrov DV, Brown WM. Trichinella spiralis mtDNA: a nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAS and has a gene arrangement relatable to those of coelomate metazoans. Genetics. 2001;157:621–637. doi: 10.1093/genetics/157.2.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo NP, et al. The head and body lice of humans are genetically distinct (Insecta: Phthiraptera, Pediculidae): evidence from double infestations. Heredity. 2005;95:34–40. doi: 10.1038/sj.hdy.6800663. [DOI] [PubMed] [Google Scholar]
- Lewis DL, Farr CL, Kaguni LS. Drosophila melanogaster mitochondrial DNA: completion of the nucleotide sequence and evolutionary comparisons. Insect Mol Biol. 1995;4:263–278. doi: 10.1111/j.1365-2583.1995.tb00032.x. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marande W, Lukes J, Burger G. Unique mitochondrial genome structure in diplonemids, the sister group of kinetoplastids. Eukaryot Cell. 2005;4:1137–1146. doi: 10.1128/EC.4.6.1137-1146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–474. doi: 10.1038/290470a0. [DOI] [PubMed] [Google Scholar]
- Rand DM. “Why genomes in pieces?” revisited: sucking lice do their own thing in mtDNA circle game. Genome Res. 2009;19:700–702. doi: 10.1101/gr.091132.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoult D, Roux V. The body louse as a vector of reemerging human diseases. Clin Infect Dis. 1999;29:888–911. doi: 10.1086/520454. [DOI] [PubMed] [Google Scholar]
- Rawlings TA, Collins TM, Bieler R. Changing identities: tRNA duplication and remolding within animal mitochondrial genomes. Proc Natl Acad Sci U S A. 2003;100:15700–15705. doi: 10.1073/pnas.2535036100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed DL, Light JE, Allen JM, Kirchman JJ. Pair of lice lost or parasites regained: the evolutionary history of anthropoid primate lice. BMC Biol. 2007;5:7. doi: 10.1186/1741-7007-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rokas A, Ladoukakis E, Zouros E. Animal mitochondrial DNA recombination revisited. Trends Ecol Evol. 2003;18:411–417. [Google Scholar]
- Shao R, Barker SC. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol Biol Evol. 2003;20:362–370. doi: 10.1093/molbev/msg045. [DOI] [PubMed] [Google Scholar]
- Shao R, Barker SC. Chimeric mitochondrial minichromosomes of the human body louse, Pediculus humanus: evidence for homologous and non-homologous recombination. Gene. 2011;473:36–43. doi: 10.1016/j.gene.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Shao R, Barker SC, Mitani H, Aoki Y, Fukunaga M. Evolution of duplicate control regions in the mitochondrial genomes of metazoa: a case study with Australasian Ixodes ticks. Mol Biol Evol. 2005a;22:620–629. doi: 10.1093/molbev/msi047. [DOI] [PubMed] [Google Scholar]
- Shao R, Campbell NJ, Barker SC. Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera) Mol Biol Evol. 2001;18:858–865. doi: 10.1093/oxfordjournals.molbev.a003867. [DOI] [PubMed] [Google Scholar]
- Shao R, Kirkness EF, Barker SC. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009;19:904–912. doi: 10.1101/gr.083188.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R, Mitani H, Barker SC, Takahashi M, Fukunaga M. Novel mitochondrial gene content and gene arrangement indicate illegitimate inter-mtDNA recombination in the chigger mite, Leptotrombidium pallidum. J Mol Evol. 2005b;60:764–773. doi: 10.1007/s00239-004-0226-1. [DOI] [PubMed] [Google Scholar]
- Smith DR, et al. First complete mitochondrial genome sequence from a box jellyfish reveals a highly fragmented linear architecture and insights into telomere evolution. Genome Biol Evol. 2012;4:52–58. doi: 10.1093/gbe/evr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer RL, Walker A, Ryder OA, Lyons-Weiler M, Hedges SB. Human and ape molecular clocks and constraints on paleontological hypotheses. J Hered. 2001;92:469–474. doi: 10.1093/jhered/92.6.469. [DOI] [PubMed] [Google Scholar]
- Suga K, Welch DBM, Tanaka Y, Sakakura Y, Hagiwarak A. Two circular chromosomes of unequal copy number make up the mitochondrial genome of the rotifer Brachionus plicatilis. Mol Biol Evol. 2008;25:1129–1137. doi: 10.1093/molbev/msn058. [DOI] [PubMed] [Google Scholar]
- Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410:103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- Toups MA, Kitchen A, Light JE, Reed DL. Origin of clothing lice indicates early clothing use by anatomically modern humans in Africa. Mol Biol Evol. 2011;28:29–32. doi: 10.1093/molbev/msq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei DD, et al. The multipartite mitochondrial genome of the booklouse: insights into the evolution of mitochondrial genomes in bilateral animals. PLoS One. 2012;7:e33973. doi: 10.1371/journal.pone.0033973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZD, Green BR, Cavalier-Smith T. Single gene circles in dinoflagellate chloroplast genomes. Nature. 1999;400:155–159. doi: 10.1038/22099. [DOI] [PubMed] [Google Scholar]