

Abstract
Functional modification of regulatory proteins can affect hundreds of genes throughout the genome, and is therefore thought to be almost universally deleterious. This belief, however, has recently been challenged. A potential example comes from transcription factor SP1, for which statistical evidence indicates that motif preferences were altered in eutherian mammals. Here, we set out to discover possible structural and theoretical explanations, evaluate the role of selection in SP1 evolution, and discover effects on coregulatory proteins. We show that SP1 motif preferences were convergently altered in birds as well as mammals, inducing coevolutionary changes in over 800 regulatory regions. Structural and phylogenic evidence implicates a single causative amino acid replacement at the same SP1 position along both lineages. Furthermore, paralogs SP3 and SP4, which coregulate SP1 target genes through competitive binding to the same sites, have accumulated convergent replacements at the homologous position multiple times during eutherian and bird evolution, presumably to preserve competitive binding. To determine plausibility, we developed and implemented a simple model of transcription factor and binding site coevolution. This model predicts that, in contrast to prevailing beliefs, even small selective benefits per locus can drive concurrent fixation of transcription factor and binding site mutants under a broad range of conditions. Novel binding sites tend to arise de novo, rather than by mutation from ancestral sites, a prediction substantiated by SP1-binding site alignments. Thus, multiple lines of evidence indicate that selection has driven convergent evolution of transcription factors along with their binding sites and coregulatory proteins.
Keywords: cis-regulatory element evolution, SP1, zinc finger proteins, GC box
Introduction
Gene regulation is a major contributor to species morphology and development (Wray et al. 2003; Wray 2007; Li and Johnson 2010). A central point of control for gene expression is at the level of transcription, which is mediated by transcription factor proteins that bind to commonly occurring DNA sequence motifs (Pedersen et al. 1999; Li and Johnson 2010). Such regulatory proteins often bind specifically to hundreds of sites across the genome, and are therefore believed to rarely change motif preferences. This belief has been argued for on the basis of first principles (Stern 2000; Prud'homme et al. 2007; Peter and Davidson 2011), because altered motif preferences seem likely to cause widespread binding incompatibilities across an extensive number of loci throughout the genome. Although there are a few known cases in which the binding specificity of a transcription factor has become modified during evolution (Gasch et al. 2004; Kuo et al. 2010; Baker et al. 2011), the observed changes have generally been limited to regulatory elements targeting a small set of genes with highly specific functions. Such cases are generally thought to be the exception rather than the rule, and the paradigm remains that changes in a single protein trans-factor are not feasible in cases where the downstream effects, such as those induced by changes in binding specificity, are widespread across loci and biological function (Peter and Davidson 2011).
A recent statistical analysis of trends in binding motif frequencies among vertebrates, however, suggests that cis-regulatory motif preferences can vary across species (Yokoyama et al. 2010). The binding element for transcription factor SP1 was one of the more clear examples, with marsupials, monotremes, and amphibians significantly deviating from the well-studied “GC box“ consensus (Philipsen and Suske 1999; Bouwman and Philipsen 2002) in humans and other eutherian mammals (Yokoyama et al. 2010). SP1 is one of the most universal transcriptional regulators (FitzGerald et al. 2004; Fukue et al. 2005), and activates expression in a large and diverse set of genes (Fukue et al. 2005), including housekeeping genes as well as those involved in cell differentiation, signal transduction and apoptosis (Philipsen and Suske 1999; Kaczynski et al. 2003; Zhao and Meng 2005). If a universal regulator such as SP1 can change motif-binding preferences, the theory of conservation of transcription factor functional evolution should be re-evaluated. The coevolution of SP1 and its binding sites is therefore an important example that should be carefully studied to gain insight into how transcription factors can, in fact, functionally evolve.
In this study, we set out to find evidence for involvement of SP1 in binding site evolution, to obtain better evidence for the role of selection in SP1 changes, and to consider how, in principle, functional evolution of transcription factors such as SP1 might occur. We also analyzed a more densely sampled set of vertebrate genomes to find out whether SP1-binding site consensus sequences appear to have changed in other lineages. We show that SP1-binding preferences not only appeared to change in birds as well as eutherian mammals, but also that the apparent change in preference was convergent, with both evolving to prefer the GC box. We then analyzed the function of converted binding sites, and showed that they are enriched for known SP1 function. We then developed an analytical model to look at the tempo and mode of birth and death of binding site sequences and to understand how these changes occurred over evolutionary time. In conjunction with this, we applied a simple model of functional transcription factor evolution to determine how much selection is required to drive coevolutionary change in transcription factors and hundreds of binding sites.
In addition, we analyzed evolutionary changes in SP1 that might have driven its altered binding preferences, predicting the effects of amino acid replacements along the phylogeny on the structure of SP1. Both phylogenic and structural evidence suggests that the observed genome-wide shifts in cis-regulatory composition in eutherian mammals and birds originate from a single amino acid change in the binding module of SP1 along both lineages. The importance of adaptation and the complexity of events is further illustrated by SP3 and SP4, two paralogs of SP1 that bind competitively to SP1-binding sites (Philipsen and Suske 1999; Bouwman and Philipsen 2002). These paralogs coregulate SP1 target genes through competitive binding to the same sites, and both accumulated convergent replacements at the homologous amino acid position multiple times during eutherian and avian evolution. The multiple convergent changes were presumably driven by the need for these coregulatory transcription factors to better recognize the newly modified binding sites. The results of these analyses strongly reinforce each other and compel us to revise our understanding of how selection can modify complex multigenic regulatory systems.
Materials and Methods
Assessing SP1-Binding Motif Preferences
Cross-species comparisons were conducted across 46 vertebrate species using the multiz46way (hg19) alignments at the UCSC Genome Browser (genome.ucsc.edu) (Karolchik et al. 2003, 2007). Columns corresponding to the functional GC box (−130, −30) region (Yokoyama et al. 2009, 2010) were extracted using human RefSeq transcription start site annotations (Maglott et al. 2000; Pruitt and Maglott 2001). Occurrences of the human GC box core motif (GGGCGG [Kriwacki et al. 1992; Marco et al. 2003; Yokoyama et al. 2010]) were tabulated on both strands, allowing for a single mismatch at any given consensus site. The preferred binding consensus for each species was then taken to be the most commonly occurring 6-mer.
The Birth–Death-Binding Site Model
To assess the rates of change for SP1-binding sites along the phylogeny, we used a quasi-birth–death-binding site model, similar to the M/M/c model described in (Wagner et al. 2007; Otto et al. 2009). At a single nucleotide position within the target region, we let the birth rate 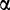 represent the probability (per year) that a new binding site appears when no binding site is present and death rate
represent the probability (per year) that a new binding site appears when no binding site is present and death rate 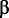 represent the probability per year that an existing binding site is lost. As discussed in more detail in the supplementary material, Supplementary Material online, the probability that a single position is occupied by a given binding site is
represent the probability per year that an existing binding site is lost. As discussed in more detail in the supplementary material, Supplementary Material online, the probability that a single position is occupied by a given binding site is
| (1) |
when a binding site originally existed at 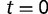 , or
, or
| (2) |
when the site is originally unoccupied. If we assume that 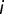 binding sites exist at
binding sites exist at 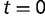 , the probability
, the probability 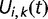 that
that 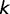 of these original binding sites remain after time
of these original binding sites remain after time 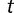 follows the binomial distribution, with number of trials
follows the binomial distribution, with number of trials 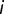 and probability of success
and probability of success 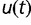 [i.e., Ui,k(t)∼Binom (i, u(t))]. Similarly, if
[i.e., Ui,k(t)∼Binom (i, u(t))]. Similarly, if 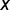 is the number of nucleotide positions in the target region (here,
is the number of nucleotide positions in the target region (here, 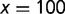 ), and again
), and again 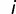 is the number of binding sites at time
is the number of binding sites at time  , the probability
, the probability 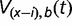 that
that 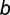 binding sites are “born” among the
binding sites are “born” among the  unoccupied sites also follows the binomial distribution, where V(x−i),b(t)∼Binom(x − i, v(t)). Thus, the overall probability Pij (t) that
unoccupied sites also follows the binomial distribution, where V(x−i),b(t)∼Binom(x − i, v(t)). Thus, the overall probability Pij (t) that 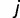 binding sites exist at time
binding sites exist at time  is
is
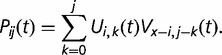 |
(3) |
Birth and death rates 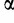 and
and 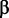 can thus be estimated according to the number of observed binding sites along the leaves of the phylogeny using maximum likelihood, using Felsenstein’s pruning algorithm (Felsenstein 1973) (supplementary material, Supplementary Material online).
can thus be estimated according to the number of observed binding sites along the leaves of the phylogeny using maximum likelihood, using Felsenstein’s pruning algorithm (Felsenstein 1973) (supplementary material, Supplementary Material online).
Population Genetics of Adaptive Changes in Binding Preference
In our population genetics model, we suppose that a transcription factor protein (SP) has two variants, SPA and SPC, which represent the wild-type and mutant alleles, respectively. We model only genes for which binding of the SP protein is beneficial, where the wild-type variant SPA binds to a promoter containing its cognate binding site (BOXA) at relative fitness 1. Binding of mutant SPC to BOXC has an adaptive advantage, with relative fitness 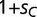 (where
(where 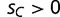 ). We assume both transcription factors can bind weakly to the alternative sequence, although loci with only weak binding interactions have a decrease in fitness, given by
). We assume both transcription factors can bind weakly to the alternative sequence, although loci with only weak binding interactions have a decrease in fitness, given by 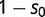 . In our analyses, we set
. In our analyses, we set 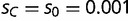 . Loci without either binding motif are considered recessive lethal, and have a fitness of zero.
. Loci without either binding motif are considered recessive lethal, and have a fitness of zero.
We allow both BOXA and BOXC to be present in the same promoter, each either present or absent at a given gene. We then consider total fitness of the individual to be multiplicative across loci. Given this deterministic model, we can trace the frequency (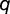 ) of the transcription factor allele as well as the binding locus haplotypes within the population over time (supplementary material, Supplementary Material online).
) of the transcription factor allele as well as the binding locus haplotypes within the population over time (supplementary material, Supplementary Material online).
Initial population frequencies of both BOXA and BOXC were obtained by determining the equilibrium binding site frequencies prior to the introduction of mutant SPC into the population. At generation  , we introduce mutant factor SPC into the population at a frequency of
, we introduce mutant factor SPC into the population at a frequency of 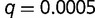 , equivalent to a single heterozygous mutation in one individual at a population size of n= 1,000.
, equivalent to a single heterozygous mutation in one individual at a population size of n= 1,000.
Modeling Free Energy Structures in zf2
To determine the effects of amino acid replacements on the structure of zinc finger 2, we used a simple free energy model adapted from that used in the program Rosetta (Havranek et al. 2004). Determining the lowest free energy structure involves minimizing the free energy function 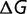 , which is given by
, which is given by
| (4) |
This model considers Van der Waals interactions using a standard 12-6 Lennard–Jones potential energy function (term 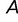 and
and 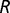 ) and rotational torsions of the phi- and psi-angles of each amino acid (term
) and rotational torsions of the phi- and psi-angles of each amino acid (term 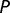 ), as in Havranek et al. (2004). In addition, due to the importance of zinc ion bonding within the zinc finger structure (Dhanasekaran et al. 2006), we added a zinc ion bonding term (term
), as in Havranek et al. (2004). In addition, due to the importance of zinc ion bonding within the zinc finger structure (Dhanasekaran et al. 2006), we added a zinc ion bonding term (term 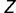 ). W values Watr, Wrep, WZ, and Wϕψ represent weights applied to each of the terms to control the relative contribution of each part of the model, and were set to values empirically determined in Havernek et al. Details of the model are given in the supplementary material, Supplementary Material online.
). W values Watr, Wrep, WZ, and Wϕψ represent weights applied to each of the terms to control the relative contribution of each part of the model, and were set to values empirically determined in Havernek et al. Details of the model are given in the supplementary material, Supplementary Material online.
To determine structural effects of amino acid substitutions on the zf2 peptide, we started with the known structure of human zf2 (Oka et al. 2004), introducing amino acid replacements found along the phylogeny, initially preserving phi- and psi-angles at each position. The free energy function was minimized by iteratively altering the phi- and psi-angles individually for each residue, keeping changes that lowered the free energy and discarding changes that increased the free energy. The process was continued until there were no changes that lowered the free energy function, up to a resolution of 0.165 degrees.
Convergent SP1-Binding Site Evolution in Humans and Birds
Convergent SP1 binding site conversions in human and birds (table 1) were assessed according to the amount of overlap in ancestral GA box- and GC box-containing genes at the root of mammals. To accurately assess convergent cis-regulatory conversions without the effects of sequence conservation, genes inferred to contain both a GA box and a GC box at the root of mammals were discarded. We determined genes with a GA box or a GC box in humans and birds, where a motif was considered to be present in birds if it occurred in either chicken or zebrafinch. For every three-way combination of GA/GC box cooccurrences in humans, birds, and the common mammalian ancestor, we determined the subset of these genes (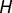 ) containing a GC/GA box in humans and the subset of genes (
) containing a GC/GA box in humans and the subset of genes (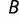 ) containing a GC/GA box in birds. We let
) containing a GC/GA box in birds. We let 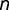 be the total number of GA/GC box-containing genes at the common mammalian ancestor, and
be the total number of GA/GC box-containing genes at the common mammalian ancestor, and 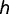 and
and 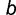 are the number of genes in
are the number of genes in 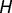 and
and 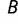 , respectively. If
, respectively. If 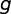 is the number of genes that overlap across
is the number of genes that overlap across 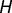 and
and 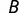 , P values representing the significance of enrichment in each category were determined using Fisher’s exact test, where
, P values representing the significance of enrichment in each category were determined using Fisher’s exact test, where
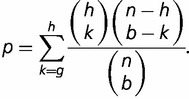 |
(5) |
Table 1.
Convergence of SP1 Cis-Regulatory Conversions within Birds and Placental Mammals
| Human GC Box |
Human GA Box |
|||||
|---|---|---|---|---|---|---|
| Obs.a | Expb | Pc | Obsa | Expb | Pc | |
| Mammal root: GA boxd | ||||||
| Bird GC box | 63 | 43 | 5.7e−5e | 55 | 55 | 0.32 |
| Bird GA box | 47 | 48 | 0.39 | 83 | 61 | 1.6e−5 |
| Mammal root: GC boxd | ||||||
| Bird GC box | 109 | 90 | 2.6e−4 | 37 | 34 | 0.17 |
| Bird GA box | 51 | 45 | 0.06 | 27 | 17 | 3.0e−4 |
aThe observed numbers of GA/GC box co-occurrences across orthologous genes.
bThe expected number of GA/GC box co-occurrences, assuming a random distribution of motifs without regards to gene orthology.
cP values representing the significance of enrichment using Fisher’s exact test.
dGenes are separated according to the motif inferred in the common mammalian ancestor; genes inferred to contain both motifs at the root of mammals have been excluded.
eGenes in this category have gained a GC box independently along the human and bird lineages; no GC box was present at the root of mammals.
Molecular Functions of SP1 Target Genes
Enrichments of human GC box target genes for biological processes and molecular functions were conducted using annotations from the gene ontology (GO) database (www.geneontology.org) (Ashburner et al. 2000). We determined the amount of enrichment for each GO category in the total set of GC box-containing genes. P values representing the statistical significance of enrichment were determined using Fisher’s exact test.
Similar assessments were used to determine the enrichment of CpG islands within GC box-containing gene promoters and the under-enrichment of methylated promoters in GC box containing genes. Promoters containing CpG islands within the (−200, −1) region of each gene were determined using CpG island annotations from the UCSC Table Browser (Gardiner-Garden and Frommer 1987) and genes methylated in the (−200, −1) region were determined using Methyl-seq ENCODE data (Brunner et al. 2009).
Results
SP1-Binding Motif Preferences Evolved Convergently in Birds and Eutherian Mammals
The core SP1-binding motif (“GC box”) appears in humans, and most mammals, as the 6-mer consensus GGGCGG (Kriwacki et al. 1992; Marco et al. 2003), which occurs commonly on both strands in the upstream proximal promoter. The GC box is found highly overrepresented specifically within the (−130, −30) window relative to the transcription start site (TSS) (Yokoyama et al. 2009, 2010) where it is known to function, and is often present in multiple copies within the same promoter (Kriwacki et al. 1992). This location of overrepresentation is highly conserved across vertebrates, from human to zebrafish (Yokoyama et al. 2010).
Previous comparisons of nine mammals and two nonmammalian vertebrates suggested that the SP1 binding element in opossum, platypus, lizard, and frog differ from the well-studied GC box consensus (Yokoyama et al. 2010). To trace the evolutionary history of the SP1 binding motif in more detail, we searched across an expanded set of 46 vertebrate genomes for cross-species differences in nucleotide preferences. Orthologous promoter regions were extracted from multiple alignments available at the UCSC Genome Browser (http://genome.ucsc.edu) (Karolchik et al. 2003; Karolchik et al. 2007), using known RefSeq TSS annotations in humans (Maglott et al. 2000; Pruitt et al. 2000). Comparing nucleotide frequencies at each site of the SP1 binding sequence within its functional region, we found that the classical GC box 6-mer consensus was the preferred binding sequence in all eutherian mammals as well as in birds (chicken and zebrafinch). However, in other vertebrates, ranging from fish to marsupials, an adenine-containing version of this motif (GGGAGG, or “GA box”) appears more frequently than the GC box. The only exception was tetraodon, which had a preferred SP1 motif that differed from the GC box at two nucleotide sites, most commonly appearing as a variant similar to the GA box (AGGAGG). Parsimony arguments therefore suggest that the GA box preference represents the ancestral state, with two independent convergent shifts to the GC box preference in birds and placental mammals (fig. 1).
Fig. 1.—

Evolution of SP transcription factors. (A) SP1 binds preferentially to the GC box in placental mammals and birds (red) and to the ancestral GA box consensus in other vertebrates (black). Modifications in binding motif preferences along the phylogeny are denoted by red-filled circles. “Variable regions” in zinc finger 2 (zf2-VR), containing all nonconserved sites in zinc finger 2 within vertebrates, are shown for SP1, SP3, and SP4. Site –13 (highlighted) is putatively responsible for the change in SP1 binding preferences. (B) Zinc finger 2 (zf2) of human SP1, SP3, and SP4. Each zinc finger contains an alpha-helix and two beta sheets (Philipsen and Suske 1999; Dhanasekaran et al. 2006). Red and gray columns denote sites nonconserved across vertebrates; all are contained in the boxed variable region (zf2-VR), comprising sites −13 to −8. Site +3 binds directly to the convergent A/C fourth site of the GC box. (C) SP1 binds to the DNA via zinc fingers 1–3 (zf1-zf3), where zf2 binds to the three central nucleotides of the GC box (GGGCGG) (Philipsen and Suske 1999; Bouwman and Philipsen 2002; Dhanasekaran et al. 2006). Site −13 (red) is only 9.5 Å from site +3 (green) and directly contacts the neighboring site (site +4) (Bouwman and Philipsen 2002; Oka et al. 2004; Dhanasekaran et al. 2006).
Bird and Mammal SP1 Binding Sites Were Preferentially Converted in Orthologous and Functionally Related Genes
It is our expectation that genes with GC boxes in their regulatory target regions may have experienced varying degrees of selection for SP1 binding. It is reasonable to hypothesize that genes most strongly selected for SP1 binding are most likely to have experienced binding site conversion to maintain SP1 regulation across the phylogeny. Such genes should rapidly convert whenever binding preferences change, and thus should be congruent in both birds and placental mammals. In support of this hypothesis, we found strong patterns of convergence within the set of promoters that underwent a GA-to-GC box conversion in eutherian mammals and those containing the same conversion in birds. That is, genes that have a GA box in the common mammalian ancestor and a GC box in humans tend to contain a GC box in birds significantly more often than expected by chance, even when discarding genes containing a GC box at the root of mammals (table 1). Because these genes do not contain a GC box at the root of mammals, this correlation cannot be explained by conservation, and therefore these genes represent convergent binding site conversions occurring in eutherian mammals independently of birds. No such pattern was found in genes containing only a GC box at the root of mammals, supporting the functional role of the ancestral GA box motifs in the common mammalian ancestor.
Also in support of this hypothesis, we observed a consistent pattern of cis-regulatory conversions in genes involved in specific functional categories. Overall, GC box-containing genes in humans are enriched for protein/nucleotide binding categories and molecular signaling mechanisms (table 2), consistent with the role of SP1 in cell proliferation, differentiation, and cancer progression (Philipsen and Suske 1999; Kaczynski et al. 2003; Zhao and Meng 2005). These GO category enrichments were consistently stronger among human GC box target genes inferred to contain a GA box in the mammalian ancestor than those that were not (table 2). In addition, promoter methylation, which is inhibited by SP1 binding (Brandeis et al. 1994; Macleod et al. 1994), is much more rare in human GC box promoters inferred to contain a GA box in the common mammalian ancestor. These patterns of over- and under-enrichment suggest that SP1 activation is particularly critical in these functionally related target genes, which have maintained SP1 binding throughout the phylogeny via the observed cis-regulatory sequence conversions. We note also that these observations appear to rule out mutational bias as an explanation for the changes in SP1 binding sites, as a neutral mutational model cannot explain the significant relative enrichment of SP1 function in converted promoters.
Table 2.
Enrichment of Functional Categories in SP1 Target Genes
| Human GC Box Promoters |
Anc GA |
Anc No GA |
Anc GA |
||||
|---|---|---|---|---|---|---|---|
| Pa | Obsb | Expc | Ratiod | Hum GCe | Hum GCf | Hum No GCg | |
| Protein binding | 2e−27 | 2,354 | 2,034 | 1.16× | 1.27× | 1.10× | 1.04× |
| Transferase activity | 9e−9 | 450 | 364 | 1.24× | 1.35× | 1.23× | 0.91× |
| Protein amino acid phosphorylation | 8e−8 | 208 | 154 | 1.35× | 1.43× | 1.32× | 0.92× |
| Protein serine/threonine activity | 4e−7 | 148 | 107 | 1.39× | 1.52× | 1.35× | 0.97× |
| Nucleotide binding | 9e−7 | 696 | 606 | 1.15× | 1.19× | 1.18× | 0.98× |
| Purine nucleotide biosynthetic process | 1e−5 | 12 | 4 | 2.76× | 4.40× | 2.10× | 0.0 |
| CpG island promoters | 0.0 | 3,583 | 2,405 | 1.49× | 1.49× | 1.49× | 1.07× |
| Methylation: BG02ES (human embryonic stem cells) | 6e−4h | 29 | 45 | 0.64× | 0.23× | 0.88× | 0.69× |
| Methylation: H1hESC (human embryonic stem cells) | 8e−4h | 15 | 28 | 0.54× | 0.19× | 0.75× | 0.83× |
| Methylation: HAL (human adult liver) | 7e−3h | 15 | 24 | 0.62× | 0.22× | 0.86× | 0.94× |
aP values represent the significance of enrichment according to Fisher’s exact test.
bThe observed number of genes in each category.
cThe expected number of genes in each category.
dThe observed-to-expected ratio.
eObserved-to-expected ratios for human GC box target genes with a GA box in the ancestor. Note the consistent over-enrichment of GO categories and under-enrichment of promoter methylation.
fObserved-to-expected ratios for human GC box target genes without a GA box in the ancestor.
gObserved-to-expected ratios for genes with a GA box in the ancestor and without a GC box in humans.
hUnder-enrichment for methylated promoters.
The Timing of Birth and Death of SP1 Cis-Regulatory Elements
The GC box comprises one of the most prevalent cis-regulatory elements in mammalian promoters (FitzGerald et al. 2004; Fukue et al. 2005). In humans, the core GC box 6-mer motif is present in the target region of 6,092 promoters (31% of all genes), whereas the GA box motif is present in only 4,559 promoters (supplementary table S1, Supplementary Material online). Thus, the global shift from the ancestral GA box preference to the newly acquired GC box preference represents promoter conversions across an extensive number of loci, motivating questions into the mechanisms by which such modifications can occur.
Inspection of the cross-species alignments showed many gains and losses of SP1-binding sequences, even within relatively closely related species. It is common to find multiple SP1-binding sites in a single promoter (Kriwacki et al. 1992), and the number of binding sites often varies across lineages. For example, 39% of promoters containing at least one GC box in both human and mouse differ in the number of GC boxes present. These observations suggest that binding site turnover may play an important role in SP1 binding site evolution.
Thus, to determine the rates of cis-regulatory conversions within the phylogeny, we modeled binding site evolution as a generalized birth–death process. In this approach, we estimate the birth rate α at which a new binding site may appear at a given position (per year), and a death rate β, at which an existing site is lost per year. This framework is formally known as a quasi-birth–death process, similar to an M/M/c model used in queuing theory, which has previously been used to study binding site turnover (Wagner et al. 2007; Otto et al. 2009). Although transitions between functional motifs (i.e., from a GA box to a GC box, and vice versa) can, of course, occur in nature, birth and death rates were modeled separately for different motifs, allowing us to assess the timing of binding site evolution without any assumptions as to the origin or mechanisms by which they occur.
Ancestral reconstruction according to this framework showed that exactly 1,800 genes inferred to contain a GA box in the common mammalian ancestor (the preferred binding motif at the root of mammals) now contain a GC box in humans. Although it is common to observe GA and GC boxes in the same promoter, at least 826 of these genes did not originally contain a GC box at the root of mammals, providing a lower-bound estimate of the number of ancestral SP1 target genes gaining a GC box along the human lineage.
To directly assess the timing of these cis-regulatory modifications, we inspected the birth–death rates along various parts of the mammal phylogeny. There was a 45-fold increase in the GC box birth/death-rate ratio along the eutherian branch immediately following the split with marsupials (fig. 2). Notably, the birth/death rate returned to approximately the ancestral rates near the time of eutherian radiation, indicating that the pronounced rise in GC box frequency was rapid, occurring primarily within a span of ∼60 million years directly after the split with marsupials (Hedges et al. 2006).
Fig. 2.—

Birth-death rates of the SP1-binding motif in mammals. Birth rates (α) denote the probability (per year) that an unoccupied position will gain a binding site; death rates (β) give the probability (per year) that an existing binding site is lost. Branches in the mammalian phylogeny were partitioned into three groups: early eutherian mammals (red), late eutherian mammals (black), and GA box-preferring noneutherian mammals (blue). Birth and death rates of each group were estimated for the GC box (GGGCGG), GA box (GGGAGG), and the nonfunctional motif GGGTGG (Letovsky and Dynan 1989; Wierstra 2008).
In contrast to the rapid birth of new GC boxes, the decline of GA boxes occurred gradually throughout eutherian evolution. There was not a rapid rise in death rates of GA boxes prior to the eutherian radiation. Instead, GA box frequencies decreased slowly following the eutherian radiation, with a 41% decrease in birth rates and little change in the death rate. Since GA→GC box mutations would eliminate GA boxes in concert with the birth of GC boxes, these results suggest that the rapid accumulation of GC boxes was instead caused by regulatory element duplication or de novo mutation from previously nonfunctional sequence. As a negative control for the birth–death model, we also considered the birth and death rates of GGGTGG, which is close in sequence to the GA and GC boxes, but which binds SP1 with low affinity (Letovsky and Dynan 1989; Wierstra 2008). We detected no change in birth or death rates of this motif on any branch of the phylogeny (fig. 2).
Small Selective Pressures Can Alter Transcription Factor Binding Preferences
One might expect that the cumulative selective effect of mismatched binding preferences at hundreds of loci would outweigh any adaptive gains from transcription factor modification, yet the observed SP1 cis-regulatory modifications contradict this assumption. To address the theoretical feasibility of selective change affecting hundreds of loci, we analyzed regulatory element evolution in diploids using a simple deterministic model. In this model, deleterious effects of regulatory mismatch were initially considered recessive. This is likely to be realistic in many cases, as we would not expect the existence of a weak binding site to adversely affect transcription factor binding at a stronger site. Because there may be some reduction in fitness at a weaker-binding locus due to cis-acting effects, this assumption was relaxed in later implementations.
We consider the case in which transcription factor protein (SP) has two variants, the ancestral variant (SPA) and an adaptive mutant variant (SPC), which occur at frequencies 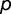 and
and 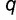 within the population, respectively (where
within the population, respectively (where 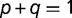 ). These variants recognize different binding sequences, BOXA and BOXC, respectively. Our framework assumes only modest benefits per regulatory locus for adaptive interactions and identical deleterious costs per locus for maladaptive binding events. More specifically, a promoter containing BOXA in the presence of SPA is given a relative fitness of 1, whereas promoters containing BOXC in the presence of SPC are assumed to have an adaptive advantage, with a relative fitness of
). These variants recognize different binding sequences, BOXA and BOXC, respectively. Our framework assumes only modest benefits per regulatory locus for adaptive interactions and identical deleterious costs per locus for maladaptive binding events. More specifically, a promoter containing BOXA in the presence of SPA is given a relative fitness of 1, whereas promoters containing BOXC in the presence of SPC are assumed to have an adaptive advantage, with a relative fitness of 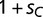 . Maladaptive binding events, where BOXA occurs only in the presence of SPC, or where BOXC occurs in only the presence of SPA, are assumed to have a decrease in fitness (
. Maladaptive binding events, where BOXA occurs only in the presence of SPC, or where BOXC occurs in only the presence of SPA, are assumed to have a decrease in fitness ( ). In our application, we set
). In our application, we set 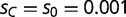 , which corresponds to a nearly neutral selective advantage for the novel binding event per promoter. We then assume multiplicative fitness across promoters for each individual, starting at an initial equilibrium state in which only the wild-type transcription factor variant exists within the population (
, which corresponds to a nearly neutral selective advantage for the novel binding event per promoter. We then assume multiplicative fitness across promoters for each individual, starting at an initial equilibrium state in which only the wild-type transcription factor variant exists within the population (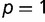 ). We then introduce the adaptive transcription factor variant in a single heterozygous individual at generation
). We then introduce the adaptive transcription factor variant in a single heterozygous individual at generation  , tracking the frequencies of each transcription factor variant and binding loci within the population over time.
, tracking the frequencies of each transcription factor variant and binding loci within the population over time.
Simulations using this model show that transcription factor variants with novel binding preferences will rapidly rise in frequency, reaching a temporary overdominant steady state in which a large fraction of the population is heterozygous for the mutant transcription factor allele (fig. 3A). Ancestral binding sites as well as novel binding sites are recognized by the heterozygous population, which therefore experience no deleterious effects from mismatch binding, and at the same time benefit from improved binding with the novel variant. This puts selective pressure on all regulated loci to increase the frequency of the novel cognate binding site. The frequency of the novel transcription factor subsequently rises in concert with the accumulation of its cognate binding sites at multiple loci, and the novel transcription factor and its cognate binding sites eventually become fixed due to selective advantage. In contrast to the rapid increase of novel binding sites, the ancestral wild-type binding sites decrease in frequency gradually over time via neutral decay.
Fig. 3.—

Population frequencies of an adaptive mutant transcription factor and its binding sites. (A) Shown are the population frequencies of the adaptive mutant transcription factor allele (blue), which first occurs in a single heterozygous individual at generation  (population size: n = 1,000). The total population frequency of the novel binding consensus (BOXC) and the initial wild-type binding motif (BOXA) are shown in red and black, respectively. We assume a small adaptive benefit for the adaptive transcription factor SPC binding to BOXC (relative fitness
(population size: n = 1,000). The total population frequency of the novel binding consensus (BOXC) and the initial wild-type binding motif (BOXA) are shown in red and black, respectively. We assume a small adaptive benefit for the adaptive transcription factor SPC binding to BOXC (relative fitness 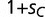 , where
, where 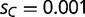 ) over the wild-type transcription factor and its motif (relative fitness 1). Maladaptive binding events (SPC binding to BOXA or the wild-type transcription factor binding to BOXC) have reduced fitness (
) over the wild-type transcription factor and its motif (relative fitness 1). Maladaptive binding events (SPC binding to BOXA or the wild-type transcription factor binding to BOXC) have reduced fitness (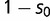 , where
, where 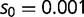 ). Population frequencies of SPC, BOXA, and BOXC are given on the left for the first 20,000 generations and on the right for 150,000 generations. (B) Evolution of the adaptive trans-factor and binding sites under a semi-dominant model. SPC binding to BOXC is assigned relative fitness
). Population frequencies of SPC, BOXA, and BOXC are given on the left for the first 20,000 generations and on the right for 150,000 generations. (B) Evolution of the adaptive trans-factor and binding sites under a semi-dominant model. SPC binding to BOXC is assigned relative fitness 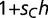 for individuals heterozygous for the transcription factor genotype (
for individuals heterozygous for the transcription factor genotype (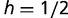 ) and
) and 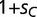 for individuals homozygous for the mutant transcription factor. (C) The single binding site locus model. In contrast to the previous model, each locus is restricted to no more than one binding motif (either BOXA or BOXC).
for individuals homozygous for the mutant transcription factor. (C) The single binding site locus model. In contrast to the previous model, each locus is restricted to no more than one binding motif (either BOXA or BOXC).
Although the model presented here is a simplistic representation of cis- and trans-element coevolution, experimenting with the general framework of the model under various conditions suggests that many of the details of the model are nonessential for the outcome of fixation. For instance, we can generalize the degree of dominance of the transcription factor genotype, modeling novel binding interactions at a relative fitness of 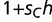 in individuals heterozygous for the mutant transcription factor (where
in individuals heterozygous for the mutant transcription factor (where 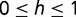 ). In such cases, the adaptive trans-factor will still go to fixation for most values of
). In such cases, the adaptive trans-factor will still go to fixation for most values of 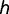 , including the semi-dominant case in which
, including the semi-dominant case in which 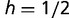 , although the rate of fixation is decreased (fig. 3B). Likewise, although we normally expect that more than one binding site can be present within a given target region (or regulatory module), the adaptive transcription factor and its binding sites can also rise to fixation when only one binding site is allowed per locus (fig. 3C). In separate work, we have also implemented a stochastic rather than a deterministic analysis of this model, and find that although some details concerning the timing of fixation change, the general result is not altered. Thus, the feasibility of mutant trans-factor fixation appears to be largely unaffected by details of the model and implementation, suggesting that adaptive changes in binding preference are plausible in diploid organisms under varying conditions with only a small selective advantage per binding locus for the novel interaction.
, although the rate of fixation is decreased (fig. 3B). Likewise, although we normally expect that more than one binding site can be present within a given target region (or regulatory module), the adaptive transcription factor and its binding sites can also rise to fixation when only one binding site is allowed per locus (fig. 3C). In separate work, we have also implemented a stochastic rather than a deterministic analysis of this model, and find that although some details concerning the timing of fixation change, the general result is not altered. Thus, the feasibility of mutant trans-factor fixation appears to be largely unaffected by details of the model and implementation, suggesting that adaptive changes in binding preference are plausible in diploid organisms under varying conditions with only a small selective advantage per binding locus for the novel interaction.
The SP1-Binding Site Quasi-Birth–Death Process Matches Theoretical Predictions
An important result from analysis of the theoretical model is that it provides an explanation for otherwise unexplained results from the quasi-birth–death analyses, which suggest that GC boxes arise rapidly by duplication or de novo mutation rather than by mutation from ancestral GA boxes. The frequencies of novel binding sites at target promoters are predicted to initially increase rapidly due to selective pressure, but will then reach an unstable steady state. This is because sites that are homozygous for GC boxes and lack GA boxes are deleterious in organisms that are homozygous for the GA-box-binding SP1. Such promoters are thus at a competitive disadvantage with those that have created a new GC box without losing the GA box. The model thus predicts that both the GC and GA boxes should coexist together in the same target promoters in the initial stages of evolution, a prediction that is consistent with our results in the quasi-birth–death analysis. In addition, the theoretical model also predicts that ancestral binding site frequencies should decrease much more gradually over time through neutral decay rather than selection (fig. 3A, right), and this also corresponds with results from the quasi-birth-death analysis.
A useful aspect of the quasi-birth–death analysis was that the numbers of GA boxes and GC boxes could be obtained separately, without the need for alignment or creating a complex model of functional conversion between motifs. Because the mode of GC box creation is an important prediction of the theoretical model, however, we evaluated the details of the quasi-birth–death process and interconversion by inspecting multiple alignments of GC box target regions in human promoters that had orthologous regions in marsupials. As predicted, a small fraction (3.8%) of all GC boxes in humans aligned to GA boxes in marsupials. This is substantially lower than the number of human GC boxes that are conserved within marsupials (14.5%). In most cases, GC boxes in eutherian mammals were not consistently aligned to any particular sequence element, but instead appeared to arise de novo from nonfunctional DNA. Several sequence motifs that do not bind SP1 (Letovsky and Dynan 1989; Wierstra 2008) converted to the GC box at a much higher rate than the GA box (table 3). The rate of conversion of such motifs to the GC box was often higher in genes containing a GA box in marsupials (i.e., in genes that are likely to have been regulated by SP1 throughout mammalian evolution). This is consistent with the hypothesis that such conversions are due to selection, and also the idea (predicted by the model) that although a GA box was present in these genes, it was more advantageous to gain a GC box de novo from the surrounding nonfunctional DNA than through direct conversion of the ancestral GA boxes.
Table 3.
Motifs in Opossum that Frequently Align with the GC Box in Humans
| Motif | All Promoters |
Promoters Containing a GA Box in Opossum |
|||||
|---|---|---|---|---|---|---|---|
| Aligneda | Totalb | Fractionc (%) | Aligneda | Totalb | Fractionc (%) | ||
| 1 | GGGCGG | 971 | 4,035 | 24.1 | 436 | 1,837 | 23.7 |
| 2 | GGGAGG | 228 | 5,007 | 4.6 | 228 | 5,007 | 4.6 |
| 3 | AGGCGG | 216 | 1,795 | 12.0d | 111 | 853 | 13.0d |
| 4 | GGGTGG | 203 | 2,102 | 9.7d | 99 | 942 | 10.5d |
| 5 | GGGCAG | 105 | 1,660 | 6.3d | 42 | 705 | 6.0d |
| 6 | GGGCTG | 77 | 1,803 | 4.3 | 35 | 743 | 4.7d |
| 7 | GGGGGG | 59 | 3,402 | 1.7 | 34 | 1,921 | 1.8 |
| 8 | GGGCCG | 52 | 1,493 | 3.5 | 30 | 610 | 4.9d |
| 9 | GGGCGT | 45 | 640 | 7.0d | 18 | 233 | 7.7d |
| 10 | GAGCGG | 40 | 990 | 4.0 | 18 | 436 | 4.1 |
| 11 | GGACGG | 36 | 618 | 5.8d | 18 | 284 | 6.3d |
| 12 | GGCCGG | 34 | 1,647 | 2.1 | 19 | 716 | 2.7 |
| 13 | GGGCGA | 32 | 596 | 5.4d | 15 | 248 | 6.0d |
| 14 | GGGCGC | 29 | 1,287 | 2.3 | 15 | 535 | 2.8 |
| 15 | TGGCGG | 27 | 887 | 3.0 | 6 | 315 | 1.9 |
aThe number of sites in opossum aligned to a human GC box.
bThe total number of motifs in opossum.
cThe fraction of each motif that align to human GC boxes.
dMotifs that have higher rates of conversion to the GC box than the GA box.
In contrast to the rapid birth of new GC boxes along the ancestral eutherian branch, GA boxes appear to have been lost throughout eutherian mammal evolution, often independently along the various descendant eutherian lineages. Notably, almost a quarter (24%) of all GA boxes in marsupials were conserved in at least one of the eutherian mammal lineages, suggesting that a large fraction of the ancestral GA boxes were present in the common eutherian ancestor. The independent loss of GA boxes along the remaining lineages supports the theoretical prediction that they evolved neutrally once the new SP1 variant and its binding sites became fixed, and were then lost gradually over the course of evolution due to drift.
Evidence Is Inconsistent with a Mutational Bias Explanation for Shifts in Binding Site Composition
Many of the previous results (as well as results presented later) suggest that the observed changes in binding composition were induced by changes in SP1-binding preferences. For instance, the greater enrichment for SP1 functional categories among human SP1 target promoters containing an ancestral GA box is consistent with a functional shift in motif preferences, rather than a mutation-driven change in sequence content. However, it is worth further considering whether the observed changes might have resulted from other factors such as shifts in dinucleotide content. Indeed, GC content has increased in placental mammals and birds relative to both marsupials and many cold-blooded vertebrates (Bernardi 1993; Kai et al. 2011). It is thus plausible that an increase in CpG dinucleotides might have shifted GC box frequencies.
This alternative hypothesis, however, was not well supported by the data. For instance, although placental mammals have a higher GC content than marsupials and amphibians (Bernardi 1993; Kai et al. 2011), two GA box-preferring lineages, fugu and medaka, have previously been shown to have higher genome-wide GC content than eutherian mammals and birds (Aparicio et al. 2002; Kai et al. 2011). In the SP1 target region, GC dinucleotide content was also high in certain fish species (e.g., 18.5% in tetraodon and 17.0% in medaka) despite their preference for the GA box. This is similar to human GC content (17.6%) and significantly higher than the SP1 target region GC content of opossum (13.7%), frog (13.7%), and zebrafish (10.8%). This analysis indicates that shifts in GC content do not deterministically cause a shift to GC box usage.
More direct evidence against a mutation- or dinucleotide-driven explanation is found in the observation that the majority of GC boxes in the target region originates de novo from nonfunctional DNA and only rarely arises from GA boxes. Because GC boxes preferentially occur in promoters containing a GA box in the ancestor (which is predictive of a functional SP1 binding site), the only possible neutral mutational explanation is a bias favoring direct GA→GC box mutations in the eutherian ancestor. Such a bias is improbable a priori because the A to C mutation is a transversion, which is relatively uncommon. Indeed, the observed rate of conversion to the GC box is substantially lower for the ancestral GA boxes than for many similar motifs that do not bind SP1 (table 3), despite the preference for GC boxes to arise in promoters containing a GA box in the ancestor. This observation is not consistent with a mutation-driven explanation, because GC boxes rarely arise from point mutations from the ancestral GA boxes, with less than 4% of all GC boxes in the target region derived from point mutations of an ancestral GA box. It is also not consistent with a dinucleotide-bias explanation, because the creation of new GC boxes only rarely led to an increase in CpG dinucleotides. Most GC boxes have been created through changes at other nucleotide positions, and often contain a CpG dinucleotide in the ancestral sequence. Indeed, the coordinated birth of new GC boxes from a large array of nonfunctional ancestral motifs strongly supports the dominant role of changes in trans-factor SP1 over a neutral mutational explanation.
Convergent Amino Acid Replacements at the Same Position in SP1 Coincide with Altered Motif Preferences
Human SP1 binds to the classical GC box three times stronger than the GA box (Letovsky and Dynan 1989; Wierstra 2008), and thus the ancestral preference for the GA box strongly suggests evolutionary changes in binding affinity. Given this, it is reasonable to expect that the global cis-regulatory changes might have been driven by the trans-acting SP1 protein.
SP1 binds to the DNA via three zinc finger modules (zf1-zf3) that are sufficient for sequence-specific recognition of the DNA in the absence of the rest of the protein (Kriwacki et al. 1992; Marco et al. 2003; Dhanasekaran et al. 2006). These three modules occur in tandem between amino acid sites 625–709 in human SP1 and are highly conserved across vertebrates. Seventy-two of the 85 sites show strong amounts of conservation, differing in two or fewer lineages. In particular, zf2 is alone responsible for sequence-specific recognition of the three central nucleotides of the SP1 binding site (GGGCGG) (Bouwman and Philipsen 2002; Dhanasekaran et al. 2006), and contains only four variable sites. At three of these variables sites (sites −11, −9, and −8), we observed a mixed representation of amino acid residues among GA box- and GC box-preferring lineages, with common eutherian residues also found present in GA box-preferring lineages.
In contrast, the remaining variable site, site −13, consistently contains a conserved ancestral valine residue in all GA box-preferring lineages and a methionine in all eutherian mammals, except for shrew, which contains a leucine (fig. 1). Notably, a similar replacement to isoleucine was found at site −13 in birds, representing a one-to-one correspondence between substitutions at this site and shifts in SP1 motif preferences. This correspondence was unique to site −13; no other amino acid position in any of the three zinc fingers, either alone or in combination, coincided with the shifts observed in SP1 motif preferences in both lineages. Given the small amount of variation in the zinc finger, it is highly improbable that similar replacements would have occurred at the same position in the correct zinc finger and on the correct two branches by chance. Instead this is consistent with the idea that SP1 evolution drove changes in binding site preferences.
The Convergent SP1 Replacements Are Predicted to Modify DNA-Binding Interactions at the Evolving Consensus Nucleotide
The position that differs between the GA box and the GC box is the fourth position of the SP1 binding site (GGGCGG). Zinc finger 2 binds to this position via site +3 within the alpha-helix (Bouwman and Philipsen 2002; Dhanasekaran et al. 2006). Although site −13 is far removed from this residue in the primary amino acid sequence, it is notably close (9.5 Å) to site +3 in the three-dimensional structure, and directly contacts the neighboring residue (site +4) (Oka et al. 2004). To directly assess the possible structural impact of replacements at site −13 relative to other sites, we determined the lowest free energy structures of the human zf2 peptide following amino acid replacements at the four variable sites as observed within the phylogeny. Comparisons of these lowest free energy peptide structures predicted that replacements at the three other variable sites would have no substantial impact on structure (RMSD < 0.048), either individually or in combination. In contrast, replacements at site −13 were predicted to have a notable impact on structure (table 4). Comparisons between the lowest free energy structure following replacements to valine (M-13V) produced an RMSD value of 0.135 with native human peptide (−13M) and an RMSD value of 0.174 with the bird isoleucine-containing peptide (M-13I). Interestingly, the native human peptide and the isoleucine-containing peptide were more similar to each other (RMSD = 0.091) than either was to the valine-containing peptide. This is consistent with the hypothesis that independent replacements to methionine and isoleucine at this position have convergently modified SP1 binding preferences in eutherian mammals and birds. Moreover, these results suggest that site −13 is alone responsible for changes in SP1-binding preferences, because replacements at the remaining three variable sites do not appear to have any notable effect on peptide structure.
Table 4.
Effects of Amino Acid Replacements on SP1 Zinc Finger 2 (zf2) Structure
| Peptidea | Human zf2 (-13M)b | M-13V peptidec |
|---|---|---|
| M-13V | 0.135 | 0 |
| M-13I | 0.091 | 0.174 |
| T-11S | 0.048 | 0.131 |
| S-9M | 0 | 0.135 |
| S-9V | 0.045 | 0.130 |
| Y-8F | 0 | 0.135 |
| S-9L | 0 | 0.135 |
| T-11S/S-9L | 0.039 | 0.137 |
| S-9V/Y-8F | 0.045 | 0.130 |
| T-11S/S-9M | 0.039 | 0.137 |
| T-11S/Y-8F | 0.039 | 0.137 |
aLowest free energy peptide structures are labeled according to amino acid replacements relative to the human zf2 peptide.
bRMSD values of each peptide compared with the lowest free energy human zf2 structure.
cRMSD values compared with the M-13V peptide.
To consider possible structural mechanisms by which replacements at site −13 may have altered binding specificity, we inspected the location of predicted structural modifications within the zf2 peptide following amino acid replacements at this position. Structural alignments indicated a nonuniform effect on peptide structure following replacements at site −13. For both human zf2 and the M-13I peptide, structural alignments with the M-13V peptide each contained a well-aligned region toward the 3′-end of the alpha-helix and a structurally displaced region prior to the alpha-helix. This reflects structural modifications beginning in the intermediate region at the 5′-end of the alpha-helix. Notably, this intermediate region includes site +3, which binds to the A/C evolving site of the SP1 motif.
In addition to whole-peptide alignments, this predicted effect on peptide structure was also observed in structural alignments conducted using only residues on the 5′-end of the peptide (residues −16 to −12). Although these alignments were created according to the residues at the 5′-end of the zinc finger, residues at the 3′-end of the zinc finger (sites +6 to +10) remained consistently well-aligned across all three peptides (fig. 4). In contrast, the central amino acid residues, comprising the 5′-end of the alpha-helix and residues prior to the alpha-helix, were significantly displaced in both the native human −13M peptide and the M-13I peptide relative to the M-13V peptide (fig. 4). Thus, replacements at site −13 are predicted to selectively alter the zinc finger peptide within and prior to the 5′-end of the alpha-helix, where the peptide contacts the A/C evolving fourth nucleotide of the binding motif, but not at the 3′-end of the helix, which contacts the neighboring (third) nucleotide of the GC box (Bouwman and Philipsen 2002; Dhanasekaran et al. 2006).
Fig. 4.—

Structural changes of SP1 zinc finger 2 (zf2) following replacements at site −13. (Top) Comparisons of predicted lowest-energy zf2 structures between the native human peptide (−13M), and peptides following replacements to the ancestral valine (M-13V) and bird isoleucine (M-13I) at site −13. Structural alignments were conducted according to residues on the 5′-end of the peptide (residues −16 to −12). Both −13M and M-13I peptides showed displacement of residues 5′ to the DNA-contacting alpha-helix (sites −6 to −1) compared with the ancestral valine peptide. No such displacement was seen between −13M and M-13I. All three peptides aligned closely at the 3′-end of the alpha-helix (sites +6 to +10), reflecting structural modifications at the 5′-end of the alpha-helix. (Bottom) Distances between alpha carbons prior to and within the alpha-helix (blue and orange, respectively). Comparisons between the native human peptide and M-13V (left) and between M-13I and M-13V (center) show closely aligned residues at the 3′-end of the alpha-helix and increasing displacement toward the 5′-end. These modifications begin around site +3, which directly contacts the A/C evolving site of the SP1-binding motif (Philipsen and Suske 1999; Bouwman and Philipsen 2002; Dhanasekaran et al. 2006). No such region-specific displacement between −13M and M-13I was observed between −13M and M-13I (right).
Notably, no region-specific differences in structure were predicted at this location between the human −13M peptide and the M-13I peptide (fig. 4). As with the M-13V peptide alignments, the 3′-end of the alpha-helix remained well aligned, although there was no displacement within or prior to the 5′-end of the helix in the −13M/M-13I peptide alignment, unlike alignments with the ancestral valine-containing peptide. Thus, the independent replacement to isoleucine at site −13 in birds is predicted to alter peptide structure in a similar manner as the methionine replacement in eutherian mammals.
Although the fine details of all structural predictions should be considered preliminary in the absence of X-ray crystal or nuclear magnetic resonance structures, the striking correspondence between the structural predictions and the phylogenetic evidence is noteworthy, because only the putative causative amino acid is predicted to cause a structural change. Furthermore, the predicted effect upon structure is focused at exactly the part of the peptide contacting the evolving nucleotide of the binding site, strongly supporting the hypothesis that these replacements have convergently altered binding interactions along these two lineages.
Coregulatory Paralogs SP3 and SP4 Have Convergently Evolved Multiple Times at Site −13 in Eutherian Mammal and Bird Evolution
The SP protein family contains at least eight paralogous transcription factor proteins in addition to SP1 (SP2–SP9) (Wierstra 2008). All members of this family contain the three highly conserved zinc finger DNA-binding domains (Philipsen and Suske 1999; Bouwman and Philipsen 2002; Wierstra 2008). Two of these paralogs, SP3 and SP4, bind with similar affinities to the SP1 binding consensus, regulating transcription in a complex manner through competitive binding to the same sites (Philipsen and Suske 1999; Suske 1999; Bouwman and Philipsen 2002; Wierstra 2008). Coregulation of these target genes requires similar binding preferences for all three proteins, and it is therefore reasonable to expect that changes in SP1 binding affinity might affect the evolution of both SP3 and SP4.
In both SP3 and SP4, 28 out of the 30 residues in zf2 are highly conserved across vertebrates; only site −13 is variable in both proteins. We found a striking difference in the evolution of site −13 between GA box- and GC box-preferring lineages. With the single exception of lizard, the ancestral valine at site −13 was completely conserved in SP3 and SP4 in all GA box-preferring lineages. Fish do not possess SP3, but they do possess SP4, and the ancestral valine residue in fish SP4 was consistently conserved at site −13, indicating that it was conserved over at least 450 million years of evolution (fig. 1) (Hedges et al. 2006). Similarly, the ancestral valine residue at site −13 in SP3 was completely conserved over at least 360 million years of evolution along the amphibian lineage (Hedges et al. 2006). These patterns of conservation match those seen in other members of the SP protein family, with the ancestral valine at site −13 consistently conserved in SP2 and SP5–SP9 throughout the entire vertebrate phylogeny (with the single exception of xenopus SP5, which is present in two copies in frog [Zhao and Meng 2005]). Such strong patterns of conservation are particularly notable, because all six of these paralogs arose from duplication events prior to the teleost and tetrapod lineages and are present in all vertebrates, ranging from human to fish (Zhao and Meng 2005).
In contrast, 21 eutherian mammals (66%), as well as one of the two bird lineages, contain a replacement to isoleucine at this site in SP3 or SP4. These convergent replacements have thus occurred at least three times in bird/eutherian mammal SP3 and at least five times in SP4. Such a large number of convergent replacements at the same previously conserved site strongly indicate adaptive pressure (Castoe et al. 2009). Notably, these replacements are identical to the replacement that appears to have caused the ancestral bird SP1 to recognize the GC box. This further supports the idea that the convergent replacements in SP3 and SP4 were caused by positive selective pressure, and is consistent with the idea that SP1-binding site conversions have driven subsequent replacements at this homologous position in these paralogous proteins to allow competitive binding, preserving the roles of SP3 and SP4 in gene regulation.
Discussion
We have demonstrated massive convergent shifts in the SP1 regulatory system in birds and eutherian mammals, including global cis-regulatory conversions across hundreds of promoters and multiple convergent replacements in paralogs SP3 and SP4. The combined phylogenic, functional, and structural evidence suggests that this extensive series of events originated from a single adaptive amino acid replacement occurring independently at the same position in bird and eutherian SP1, inducing convergent structural modifications in the DNA-binding domain. Furthermore, we developed a simple model to show that, rather than requiring prohibitively strong selection to effect a change in binding preference, it is reasonable in many cases to expect that such a change can occur with only small selective benefits for the modified interactions.
Although divergence in trans-factor binding preferences have recently been reported in fungi (Gasch et al. 2004; Kuo et al. 2010; Baker et al. 2011), the role of selective pressure in cis- and trans-element coevolution has remained largely unclear. Many regulatory systems are extremely complex, with multiple trans-factors collectively regulating each gene, and each transcription factor controlling a large number of loci (Wray et al. 2003; Wray 2007; Peter and Davidson 2011). Functional changes in such trans-regulatory elements therefore would often affect multiple processes in the cell, and are assumed to be highly deleterious and therefore quite rare (Stern 2000; Prud'homme et al. 2007; Peter and Davidson 2011). In this context, transcription factors that regulate a limited number of genes are expected to be less constrained, and thus it has previously been possible to attribute compensatory coevolution of such factors and their binding sites to a relative lack of constraint rather than to selection.
Although both intuitive and logical, this popular line of reasoning cannot explain the findings presented here. SP1 is one of the most universal transcriptional regulators (Fukue et al. 2005), involved in the activation of a large set of genes with diverse functions (Philipsen and Suske 1999; Kaczynski et al. 2003; Zhao and Meng 2005). Many of the functional categories enriched among SP1 target genes (table 2) involve the most basic biological processes essential for cell survival. It is therefore difficult to imagine that changes in SP1-binding preferences, and the subsequent shifts in cis-regulatory composition across the genome, could be simply due to stochastic drift or a lack of constraint. In addition to the magnitude of the resulting cis-regulatory modifications, several independent observations provide strong corroborating support for the role of selection in driving the evolution of the SP1 regulatory system. The amino acid replacements in SP1 occurred independently at the same position along the bird and eutherian mammal lineages, driving convergent modification of the zf2 structure as well as the binding site composition of its target genes. Convergently modified binding sites are over-enriched for SP1 functional categories, which cannot be explained by neutral processes, such as genetic drift, changes in dinucleotide composition, or lineage-specific changes in mutational bias. New GC boxes strongly tended to be created from diverse sequences rather than the more prevalent GA boxes, an observation predicted by our theoretical model as being caused by selection, but which also cannot be explained by neutral mutational processes. And finally, convergent replacements at the previously highly conserved homologous position in the coregulatory paralogs SP3 and SP4 occurred repeatedly during bird and eutherian evolution, and none of the densely-sampled eutherian lineages have reverted back to the ancestral state in SP1. Jointly, these observations overwhelmingly support the role of positive selection in the evolution of the SP1 regulatory system.
Despite the magnitude of the shifts in cis-regulatory composition, however, several observations make these findings perhaps less implausible than previously anticipated. First, although human SP1 binds primarily to the GC box, it also recognizes the ancestral GA box consensus, although at much weaker affinity (Letovsky and Dynan 1989; Wierstra 2008). In addition, SP1 target promoters are frequently located within CpG islands (Bouwman and Philipsen 2002), facilitating the creation of new GC box elements through high GC content. It is therefore possible that functional changes in SP1 and shifts in cis-regulatory composition were not necessarily disruptive, but aided both by weak recognition of the ancestral sites and efficient creation of novel GC boxes in early eutherian evolution.
It remains an open question whether these adaptive changes have altered the regulatory topology of the SP1 regulatory system. This is relevant because much of the literature regarding the evolution of gene regulation focuses on changes in regulatory network topologies (Gasch et al. 2004; Doniger and Fay 2007; Wray 2007; Carroll 2008; Peter and Davidson 2011). The bulk of our evidence, however, indicates that the topology of the system was largely maintained. If so, the large-scale genome-wide modifications that occurred are still in sharp contrast to the current dogma that it is difficult to functionally modify transcription factors because the functional changes will affect many regulated genes at once (Stern 2000; Prud'homme et al. 2007; Peter and Davidson 2011). Nevertheless, the observation that genes with a human GC box but not an ancestral GA box (table 2) are enriched for SP1-like GO terms is consistent with recruitment of new genes to the SP1 regulatory system. The overwhelming support for selection-driven convergence in SP1 and its binding sites shows that a large multi-gene regulatory system can indeed undergo biologically relevant change, whether or not the functional changes in SP1 involved changes in regulatory topology.
The population genetic model we developed shows that it is possible for a general transcription factor such as SP1 to functionally evolve. This model suggests that such adaptability may not be a property of SP1 alone, but can be generalized to many transcription factors, despite the initial fitness penalty induced by suboptimal binding events between a new transcription factor variant and the ancestral binding sites. The essential feature of this model is that positive selective effects for a regulated locus outweigh negative effects in individuals that are jointly heterozygous for the binding site and the transcription factor locus. Note that the positive balance can apply to either duplicated or converted binding site loci, or both. In such situations, heterozygous individuals experience a gain of fitness from interactions with the adaptive transcription factor allele, producing a rapid accumulation of novel binding sites in the population. Although details of the underlying mechanisms controlling cis- and trans-regulatory evolution in some systems may affect the conditions under which beneficial selective change may occur, many of the details of the model and its implementation (such as the degree of dominance of the trans-factor and number of binding sites per locus) are nonessential to the ultimate outcome of fixation. Although the general framework of the model can be extended to be arbitrarily complex, this simple model provides an explanation for the findings presented here and a theoretical framework to counter the intuitive conclusion that such evolution is always difficult.
These results are consistent with studies emphasizing the role of binding site turnover in regulatory evolution (Dermitzakis and Clark 2002; Odom et al. 2007; Hare et al. 2008), which appears to be the major mechanism driving shifts in cis-regulatory composition. One implication is that changes in binding motif preferences are likely to be undetectable using alignment-based approaches, as the novel and ancestral binding sites generally do not align, and thus we expect that the prevalence of changes in trans-factor binding preferences is likely to have been previously underestimated.
Although the correspondence between site −13 in SP1 and the convergent genome-wide shifts in cis-regulatory element frequencies provides strong evidence that this site is causative, structural analyses suggest a physical mechanism and make it improbable that replacements at other positions were involved. In contrast to site −13, replacements at all three other variable sites in zf2 were predicted to have little effect on peptide structure, whether alone or in combination. This reflects a lack of inter-residue atomic interactions at these positions, whose side-chains are freely separated from the rest of the peptide (fig. 1C) and thus are predicted to only minimally affect structure when altered. Site −13, however, is located in a beta-sheet of the zinc finger domain that plays a significant role in DNA recognition (Dhanasekaran et al. 2006, 2007), and which would be expected to affect binding preferences even in the absence of phylogenic data. The main advantage to this approach is that we were able to predict the structural mechanisms by which binding preferences were altered. Predicted structural modifications following replacements at site −13 in birds and eutherian mammals were localized near the 5′-end of the alpha-helix, thus potentially explaining the change in binding preference from adenine to cytosine at the evolving nucleotide. In contrast, the preservation of structure at the 3′-end of the helix explains the conservation of nucleotide preferences at adjacent sites of the binding motif.
The observation that a transcription factor and its binding sites can adaptively coevolve raises questions regarding the mechanistic impetus for the selective advantage gained. Although it is likely that selective changes in a transcription factor must precede the subsequent evolution of the binding sites, we do not intend to argue that such adaptive changes are inherent to the protein outside of external factors. On the contrary, selective forces acting upon such systems may well be specific to a given lineage or environment. We leave the details to future study, but our results indicate that even a weak selective benefit is sufficient to drive the evolution of the system, and the impetus might thus be quite subtle. Potential causes range from physiological to genetic to biophysical adaptation. Plausible physiological adaptations shared by birds and mammals compared to other vertebrates include high metabolic rates, large brains, complex social networks and extreme endothermy (Schmidt-Nielsen 1997; Burish et al. 2004; Nagy 2005; Northcutt 2011). Physiological explanations are not necessarily exclusive from biophysical explanations; for instance, DNA bending is substantially promoted specifically in GC-rich sequences in warm-blooded animals relative to cold-blooded animals (Vinogradov 2001). As DNA conformation and bending is crucial for protein recognition (Pabo and Sauer 1992; Allemann and Egli 1997), it is possible that selective benefits for GC box recognition are biophysical, yet specific to endothermic vertebrates. Another possible (but difficult to elucidate) explanation might be coevolutionary interactions within SP1 that made a previously deleterious amino acid replacement acceptable. An alternative explanation might involve complex tradeoffs between binding affinity and specificity (Havranek et al. 2004; Ashworth and Baker 2009) that could be elucidated using recently developed high-throughput approaches (Zhao and Meng 2009; Nutiu et al. 2011; Pollock et al. 2011). Previous binding affinity studies have determined that SP1 binds three times better to the GC box than to the ancestrally preferred binding box (Letovsky and Dynan 1989; Wierstra 2008), but interpretation of biophysical results is complicated by a lack of clear prior expectations as to what changes should be selectively advantageous. The frequencies of alternative binding motifs in different lineages may also have an impact. Finally, enrichment for SP1-like GO terms in genes that have recently evolved an SP1 target sequence is consistent with recruitment of genes to the SP1 regulatory system. If so, genetics (i.e., alterations in regulatory topology) may have provided part of the adaptive impetus, but it will be difficult to determine whether it was a driver or an incidental side effect.
The findings presented here demonstrate and explain a previously unrecognized means of adapting the design of complex regulatory systems via selective changes in trans-regulatory elements. Knowledge regarding the nature by which such regulatory systems evolve requires an understanding of the complex relationships between multiple cis- and trans-regulatory elements in the system, and further work is recommended to elucidate the generality of these findings in other regulatory systems.
Supplementary Material
Supplementary material and table S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Jeffrey Thorne, Todd Castoe, Suzi Bryan, Jason de Koning, and two anonymous reviewers for their comments on this work. This research was supported by National Institutes of Health grant R01 GM083127 and Computational Bioscience Program Training grant NIH 5T15LM009451.
Literature Cited
- Allemann RK, Egli M. DNA recognition and bending. Chem Biol. 1997;4:643–650. doi: 10.1016/s1074-5521(97)90218-0. [DOI] [PubMed] [Google Scholar]
- Aparicio S, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–1310. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth J, Baker D. Assessment of the optimization of affinity and specificity at protein-DNA interfaces. Nucleic Acids Res. 2009;37:e73. doi: 10.1093/nar/gkp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CR, Tuch BB, Johnson AD. Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proc Natl Acad Sci U S A. 2011;108:7493–7498. doi: 10.1073/pnas.1019177108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi G. The vertebrate genome: isochores and evolution. Mol Biol Evol. 1993;10:186–204. doi: 10.1093/oxfordjournals.molbev.a039994. [DOI] [PubMed] [Google Scholar]
- Bouwman P, Philipsen S. Regulation of the activity of Sp1-related transcription factors. Mol Cell Endocrinol. 2002;195:27–38. doi: 10.1016/s0303-7207(02)00221-6. [DOI] [PubMed] [Google Scholar]
- Brandeis M, et al. Sp1 elements protect a CpG island from de novo methylation. Nature. 1994;371:435–438. doi: 10.1038/371435a0. [DOI] [PubMed] [Google Scholar]
- Brunner AL, et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009;19:1044–1056. doi: 10.1101/gr.088773.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burish MJ, Kueh HY, Wang SS. Brain architecture and social complexity in modern and ancient birds. Brain Behav Evol. 2004;63:107–124. doi: 10.1159/000075674. [DOI] [PubMed] [Google Scholar]
- Carroll SB. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell. 2008;134:25–36. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- Castoe TA, et al. Evidence for an ancient adaptive episode of convergent molecular evolution. Proc Natl Acad Sci U S A. 2009;106:8986–8991. doi: 10.1073/pnas.0900233106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Clark AG. Evolution of transcription factor binding sites in mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol. 2002;19:1114–1121. doi: 10.1093/oxfordjournals.molbev.a004169. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran M, Negi S, Imanishi M, Sugiura Y. DNA-binding ability of GAGA zinc finger depends on the nature of amino acids present in the beta-hairpin. Biochemistry. 2007;46:7506–7513. doi: 10.1021/bi700009q. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran M, Negi S, Sugiura Y. Designer zinc finger proteins: tools for creating artificial DNA-binding functional proteins. Acc Chem Res. 2006;39:45–52. doi: 10.1021/ar050158u. [DOI] [PubMed] [Google Scholar]
- Doniger SW, Fay JC. Frequent gain and loss of functional transcription factor binding sites. PLoS Comput Biol. 2007;3:e99. doi: 10.1371/journal.pcbi.0030099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Maximum-likelihood estimation of evolutionary trees from continuous characters. Am J Hum Genet. 1973;25:471–492. [PMC free article] [PubMed] [Google Scholar]
- FitzGerald PC, Shlyakhtenko A, Mir AA, Vinson C. Clustering of DNA sequences in human promoters. Genome Res. 2004;14:1562–1574. doi: 10.1101/gr.1953904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukue Y, Sumida N, Tanase J, Ohyama T. A highly distinctive mechanical property found in the majority of human promoters and its transcriptional relevance. Nucleic Acids Res. 2005;33:3821–3827. doi: 10.1093/nar/gki700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Gasch AP, et al. Conservation and evolution of cis-regulatory systems in ascomycete fungi. PLoS Biol. 2004;2:e398. doi: 10.1371/journal.pbio.0020398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare EE, Peterson BK, Iyer VN, Meier R, Eisen MB. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 2008;4:e1000106. doi: 10.1371/journal.pgen.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havranek JJ, Duarte CM, Baker D. A simple physical model for the prediction and design of protein-DNA interactions. J Mol Biol. 2004;344:59–70. doi: 10.1016/j.jmb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22:2971–2972. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- Kaczynski J, Cook T, Urrutia R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003;4:206. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai W, et al. Integration of the genetic map and genome assembly of fugu facilitates insights into distinct features of genome evolution in teleosts and mammals. Genome Biol Evol. 2011;3:424–442. doi: 10.1093/gbe/evr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, et al. Comparative genomic analysis using the UCSC genome browser. Methods Mol Biol. 2007;395:17–34. doi: 10.1007/978-1-59745-514-5_2. [DOI] [PubMed] [Google Scholar]
- Kriwacki RW, Schultz SC, Steitz TA, Caradonna JP. Sequence-specific recognition of DNA by zinc-finger peptides derived from the transcription factor Sp1. Proc Natl Acad Sci U S A. 1992;89:9759–9763. doi: 10.1073/pnas.89.20.9759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo D, et al. Coevolution within a transcriptional network by compensatory trans and cis mutations. Genome Res. 2010;20:1672–1678. doi: 10.1101/gr.111765.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letovsky J, Dynan WS. Measurement of the binding of transcription factor Sp1 to a single GC box recognition sequence. Nucleic Acids Res. 1989;17:2639–2653. doi: 10.1093/nar/17.7.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Johnson AD. Evolution of transcription networks—lessons from yeasts. Curr Biol. 2010;20:R746–R753. doi: 10.1016/j.cub.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod D, Charlton J, Mullins J, Bird AP. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994;8:2282–2292. doi: 10.1101/gad.8.19.2282. [DOI] [PubMed] [Google Scholar]
- Maglott DR, Katz KS, Sicotte H, Pruitt KD. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco E, Garcia-Nieto R, Gago F. Assessment by molecular dynamics simulations of the structural determinants of DNA-binding specificity for transcription factor Sp1. J Mol Biol. 2003;328:9–32. doi: 10.1016/s0022-2836(03)00243-2. [DOI] [PubMed] [Google Scholar]
- Nagy KA. Field metabolic rate and body size. J Exp Biol. 2005;208:1621–1625. doi: 10.1242/jeb.01553. [DOI] [PubMed] [Google Scholar]
- Northcutt RG. Paleontology. Evolving large and complex brains. Science. 2011;332:926–927. doi: 10.1126/science.1206915. [DOI] [PubMed] [Google Scholar]
- Nutiu R, et al. Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument. Nat Biotechnol. 2011;29:659–664. doi: 10.1038/nbt.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom DT, et al. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet. 2007;39:730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka S, et al. NMR structure of transcription factor Sp1 DNA binding domain. Biochemistry. 2004;43:16027–16035. doi: 10.1021/bi048438p. [DOI] [PubMed] [Google Scholar]
- Otto W, et al. Measuring transcription factor-binding site turnover: a maximum likelihood approach using phylogenies. Genome Biol Evol. 2009;1:85–98. doi: 10.1093/gbe/evp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabo CO, Sauer RT. Transcription factors: structural families and principles of DNA recognition. Annu Rev Biochem. 1992;61:1053–1095. doi: 10.1146/annurev.bi.61.070192.005201. [DOI] [PubMed] [Google Scholar]
- Pedersen AG, Baldi P, Chauvin Y, Brunak S. The biology of eukaryotic promoter prediction—a review. Comput Chem. 1999;23:191–207. doi: 10.1016/s0097-8485(99)00015-7. [DOI] [PubMed] [Google Scholar]
- Peter IS, Davidson EH. Evolution of gene regulatory networks controlling body plan development. Cell. 2011;144:970–985. doi: 10.1016/j.cell.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipsen S, Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock DD, et al. Bayesian analysis of high-throughput quantitative measurement of protein-DNA interactions. PLoS One. 2011;6:e26105. doi: 10.1371/journal.pone.0026105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud'homme B, Gompel N, Carroll SB. Emerging principles of regulatory evolution. Proc Natl Acad Sci U S A. 2007;104(Suppl 1):8605–8612. doi: 10.1073/pnas.0700488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Katz KS, Sicotte H, Maglott DR. Introducing RefSeq and LocusLink: curated human genome resources at the NCBI. Trends Genet. 2000;16:44–47. doi: 10.1016/s0168-9525(99)01882-x. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Maglott DR. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 2001;29:137–140. doi: 10.1093/nar/29.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Nielsen K. Animal physiology: adaptation and environment. Cambridge (UK), New York: Cambridge University Press; 1997. [Google Scholar]
- Stern DL. Evolutionary developmental biology and the problem of variation. Evolution. 2000;54:1079–1091. doi: 10.1111/j.0014-3820.2000.tb00544.x. [DOI] [PubMed] [Google Scholar]
- Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- Vinogradov AE. Bendable genes of warm-blooded vertebrates. Mol Biol Evol. 2001;18:2195–2200. doi: 10.1093/oxfordjournals.molbev.a003766. [DOI] [PubMed] [Google Scholar]
- Wagner GP, Otto W, Lynch V, Stadler PF. A stochastic model for the evolution of transcription factor binding site abundance. J Theor Biol. 2007;247:544–553. doi: 10.1016/j.jtbi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Wierstra I. Sp1: emerging roles—beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun. 2008;372:1–13. doi: 10.1016/j.bbrc.2008.03.074. [DOI] [PubMed] [Google Scholar]
- Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- Wray GA, et al. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 2003;20:1377–1419. doi: 10.1093/molbev/msg140. [DOI] [PubMed] [Google Scholar]
- Yokoyama KD, Ohler U, Wray GA. Measuring spatial preferences at fine-scale resolution identifies known and novel cis-regulatory element candidates and functional motif-pair relationships. Nucleic Acids Res. 2009;37:e92. doi: 10.1093/nar/gkp423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama KD, Thorne JL, Wray GA. Coordinated genome-wide modifications within proximal promoter cis-regulatory elements during vertebrate evolution. Genome Biol Evol. 2010;3:66–74. doi: 10.1093/gbe/evq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Meng A. Sp1-like transcription factors are regulators of embryonic development in vertebrates. Dev Growth Differ. 2005;47:201–211. doi: 10.1111/j.1440-169X.2005.00797.x. [DOI] [PubMed] [Google Scholar]