

Abstract
Recent findings shed light on the coordination of two fundamental, yet mechanistically opposing, processes in the early mammalian embryo. During the oocyte-to-embryo transition and early preimplantation development nuclear reprogramming occurs. This resetting of the epigenome in maternal and paternal pronuclei to a ground state is the essential step ensuring totipotency in the zygote, the first embryonic stage. Radical, global DNA demethylation, which occurs actively in the paternal and passively in the maternal genome, is a prominent feature of nuclear reprogramming; yet, this process poses a danger to a subset of methylated sequences that must be preserved for their germline to soma inheritance. Genomic imprinting and its importance were demonstrated three decades ago by a series of experiments generating non-viable mammalian uniparental embryos. Indeed, imprinted loci, gene clusters with parent-of-origin specific gene expression patterns, must retain their differential methylation status acquired during gametogenesis throughout embryogenesis and in adult tissues. It is just recently that the molecular players that protect/maintain imprinting marks during reprogramming in preimplantation embryos have been identified, in particular, an epigenetic modifier complex formed by ZFP57 and TRIM28/KAP1. The interaction of these and other molecules with the newly formed embryonic chromatin and imprinted genes is discussed and highlighted herein.
Keywords: Kap1, Trim28, Zfp57, 5-methyl-cytosine, embryogenesis, epigenetics, imprinting, reprogramming
Genomic Imprinting
Mammalian parthenotes are not viable,1 suggesting that differential and essential traits are contributed to the newly formed embryo by both egg and sperm. By generating embryos from two female pronuclei (biparental gynogenetic embryo) or from two male pronuclei (biparental androgenetic embryos) using pronuclear manipulation in mouse zygotes, the requirement of the “genomic contribution” from both the mother and the father to the next generation was demonstrated.2 Nearly three decades later the true nature of this parental imprint is still elusive; however, it is well established that asymmetric chromatin and cytosine methylation on DNA at a number of genomic loci results in parent-of-origin specific gene expression in the progeny. Thus, gynogenetic and androgenetic mammalian embryos differ from each other and from normal embryos in their expression of essential developmental genes, explaining their incompatibility with life.3,4
Imprinted genes are usually arranged in clusters, with each cluster containing a local imprinting control region (ICR) (Fig. 1A). Different, independently evolved mechanisms ensure allele-specific expression at over 80 different imprinted regions, involving insulator proteins and microRNAs, yet each of them ultimately depends on differential DNA methylation at ICRs. This methylation, established at specific loci in the maternal or the paternal germ line is conserved throughout development and is generally maintained in adult tissues.5-7
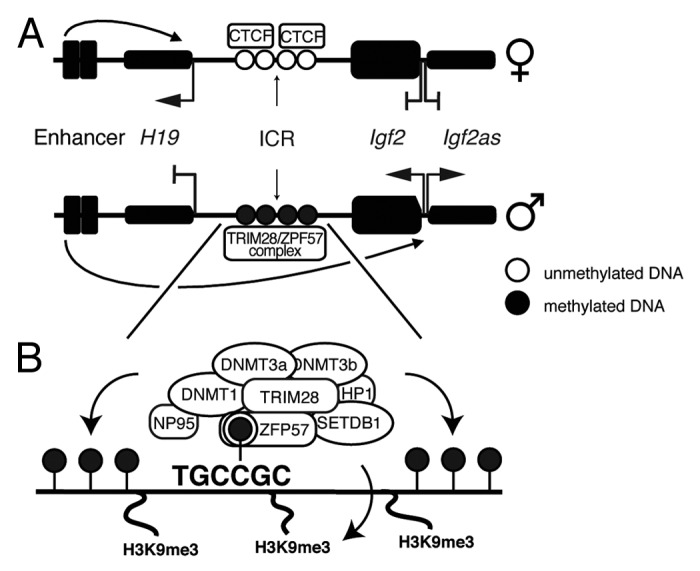
Figure 1. Schematic representation of the imprinted H19 locus and TRIM28/ZFP57 complex binding to the ICR. (A) Gene expression from the H19 imprinted locus is controlled by an enhancer, whose activity is directed to the H19 promoter by binding the insulator CTCF to the unmethylated ICR on the maternal allele. Methylation of the ICR on the paternal allele prevents CTCF binding, thus directing the enhancer activity to the Igf2/Igf2as promoters. (B) The ICR of the H19 locus contains a ZFP57 binding site. ZFP57 binds DNA in a sequence- and methylation-dependent manner, attracting TRIM28 and several chromatin and DNA modifying components.
Over the decades, genomic imprinting has served as an epigenetic paradigm, and many mechanistic aspects of imprinting control, establishment, maintenance and mitotic inheritance relate to general epigenetic principles (for a review see ref. 8). In particular, DNA methylation is a prominent and globally applied mechanism and has crucial roles not only in imprinting but also in X-chromosome inactivation, retrotransposon repression, chromosome structure and gene silencing.9-11 However, epigenetic marks on imprinted loci, due to their heritability from generation to generation, exhibit unique characteristics, especially in regard to epigenetic reprogramming (for reviews see refs. 7 and 12).
Epigenetic Reprogramming
All cells in an individual organism (with few exceptions) carry identical genetic information. Accordingly, functional specialization of cells during development is the outcome of differential transcriptional programs, not different genetic information. These programs are governed by the transcription/translation machinery, which in turn is guided and controlled by epigenetic (i.e., chemical) modifications of both DNA and chromatin.13,14 The cell’s epigenome, which does not affect the genetic code, is itself heritable at each mitotic cell division.15 These epigenetic states and their consequent transcriptional programs engendered in each cell are acquired throughout development as each cell specializes, concomitantly sacrificing its developmental potential.
To initiate a new life cycle, the epigenome must revert to a blank slate state. In vitro, this can be achieved by ectopic expression of transcription factors in somatic cells, forcing reprogramming to an “induced pluripotency” state.16 Epigenetic reprogramming can also occur pathologically in somatic cells in vivo but, importantly, it is the key process in primordial germ cell (PGC) determination and the oocyte-to-embryo transition (OET) (reviewed in ref. 17).
In both female and male PGCs DNA demethylation of genic and intergenic regions is completed by embryonic day E13.5.18,19 This process is absolute and includes reactivation of the second X-chromosome in females and full demethylation of all imprinted gene clusters.20 The demethylation in early PGCs is essential, as new all-paternal or all-maternal ICR methylation patterns are, and must be, established in sperm and oocytes, respectively (Fig. 2). This reacquisition of methylation is DNA methyltransferase 3A (DNMT3A)-dependent and occurs in males during late fetal development and begins in females postnatally in growing oocytes.21-23 The activity of DNMT3A relies on the enzymatically inactive regulatory factor DNMT3L, which enables binding and methylation of DNA in the first place. The loss of DNMT3L equally results in loss of maternal and paternal imprints. In male germ cells, DNMT3L is also required to repress retrotransposons, potentially linking genomic imprinting and silencing of repetitive elements. This observation may help to answer how DNMT3A is specifically targeted to the ICRs in the first place.24,25

Figure 2. Global and ICR-specific DNA methylation dynamics in germ cells and embryonic development. Maternal and paternal genomes are methylated during germ cell specification, including allele-specific methylation of imprinted genes by DNMT3A. Epigenetic reprogramming ensues after fertilization of the oocyte by sperm. The paternal genome becomes actively demethylated in the zygote and the maternal genome undergoes passive demethylation until the early blastocyst stage. STELLA and potentially TRIM28/ZFP57 protect ICRs from active demethylation in preimplantation embryos. ICR methylation is most likely maintained by TRIM28/ZFP57-guided DNMT1 activity.
ICRs are not the only remethylated regions in maturing germ cells. The average DNA methylation level in sperm is a remarkable (89.4%), reflective of its terminal differentiation and the dense packaging of genetic information in these highly specialized cells.26 Methylation of the maternal genome in oocytes is less dense (40%),26 yet still remarkably high compared with blastocysts (21%) and readily detectable by immunofluorescent analysis of 5-methyl-cytosine (5-mC).27-29 Similar results have been reported recently from genome-wide sperm, other oocyte and embryo methylome studies.30,31
In contrast to the exceptionally restricted potential of germ cells, the zygote is the only truly totipotent entity in mammals. This dramatic turnabout of potency is initiated after fertilization when immediate, drastic epigenetic reprogramming ensues to produce the totipotent state, which is necessary for further embryonic development.10 The enormity of this step to the genome is most apparent when the dynamic of DNA demethylation is examined. Within 7 to 8 h of fertilization, before DNA replication initiates, the paternal pronucleus loses most of its 5-mC marks. Conversely to this active demethylation, the maternally contributed genome undergoes replication-dependent, passive demethylation over several cell divisions (Fig. 2).27-29,32 Either way, in contrast to the epigenetic reprogramming in PGCs, reprogramming in preimplantation embryos is more selective, conserving DNA methylation at ICRs and thus allowing the inheritance of the parental imprints by the new embryo (Fig. 2).12 Although a control mechanism permitting the ‘demethylation machinery’ to distinguish imprinted from non-imprinted regions and even paternal from maternal genomes was postulated early on, it is only recently that the pieces to this puzzling process have fallen into place.
STELLA
The first genetic evidence for a molecule with such a sentry function came from analysis of embryos derived from a maternal knockout of the developmental pluripotency-associated 3 protein (DPPA3), also known as PGC7 or STELLA.33-35 Zygotes lacking maternal contribution of STELLA do not differentiate the maternal from paternal genome, actively demethylating both as a result.34 These mutants arrest before blastocyst formation. Whether active demethylation vs. the normal passive demethylation of the maternal pronucleus is the cause of this effect is still unclear, as some paternally and maternally methylated imprinted regions and retrotransposable elements are also errantly demethylated. A recent study has not resolved this question; yet, it has shown that an underlying chromatin modification, H3K9me2, specifically recruits STELLA to the maternal genome and to the subset of paternal ICRs, preventing active DNA demethylation by TET (ten-eleven translocation) enzymes at these loci.34,36,37
Remarkably, STELLA is also expressed in primordial germ cells, detectable first in embryos at E7.5 and remaining expressed throughout PGC specification. Its presence in PGCs, however, prevents neither active nor passive DNA demethylation of ICRs.33
TRIM28 and ZFP57
The loss of STELLA, while only affecting a subset of the imprinted regions, globally affects the maternal genome. Conversely, a chromatin-modifier complex has recently been shown to confer reprogramming resistance to imprinted regions in a global, but much more specific manner.38-40 TRIM28 (also known as KAP1 or TIF1b) is the central scaffolding component of this heterochromatin-inducing complex containing multiple chromatin-remodeling factors. Along with other components, it binds the H3K9me3-catalyzing histone methyltransferase SETDB1 (or ESET), the nucleosome remodeling and histone deacetylation (NuRD) complex as well as heterochromatin protein 1 (HP1).41-43 Recently, this list of interaction partners has been extended to include, at least in embryonic stem cells (ESCs), the maintenance and de novo DNA methyltransferases DNMT1, DNMT3A and DNMT3B.40,44 The DNA binding specificity of the complex is conveyed by interaction of TRIM28 with various, tissue-specifically expressed Krüppel-associated box-containing zinc-finger proteins (KRAB-ZFPs), such as ZFP57.38
ZFP57 was the first component of the complex to be linked to imprinting control. Ablating maternal-zygotic expression of ZFP57 in mouse embryos causes misexpression of imprinted genes and loss of methylation at both paternally and maternally methylated ICRs.38 Also in humans loss-of-function mutations in Zfp57 cause hypomethylation at several ICRs, ultimately resulting in transient neo-natal diabetes.45 The effect on imprint maintenance in mice is less pronounced and more variable in zygotic mutants and the loss of maternal ZFP57 alone can be fully rescued by zygotic expression of the paternal allele.38
Conversely, the deletion of maternal Trim28 alone is lethal, though the phenotypes of affected individuals are variable.39 DNA methylation analysis of post-implantation maternal Trim28 mutants also revealed an effect on several maternally and paternally methylated ICRs in genetically identical individuals.
ICR Recognition
Despite the fact that ICRs are not all affected to the same extent in mutant Trim28 or Zfp57 embryos, the binding of TRIM28 and ZFP57 and the presence of H3K9me3, product of the histone methyltransferase SETDB1, were detected at all imprinted loci tested.39 This global, yet highly specific, binding was confirmed by genome-wide DNA binding analysis in ESCs, showing that 91 loci, including all known ICRs, are bound by the complex in a fully ZFP57-dependent manner.40 Predictably, deletion of Zfp57 in ESCs also affects DNA methylation, with five tested ICRs (Snrpn, KvDMR, Rasgrf1, Peg3 and Gnas) being fully demethylated. In contrast, deletion of Trim28 in ESCs results in a rapid loss of pluripotency and growth arrest, preventing comprehensive analysis of DNA-demethylation dynamics in these cells.40
Interestingly, in maternal Trim28 mutant embryos with hypomethylated ICRs, TRIM28 and ZFP57 binding and accumulation of H3K9me3 was lost and never recovered, even when TRIM28 from the paternal allele was expressed (unpublished observation).39 This observation provided the first in vivo indication that DNA-binding of ZFP57 is 5-mC-dependent. Once DNA methylation at the ICR is lost it cannot be restored. In vitro, this irrevocability has been illustrated by forced expression of wild-type ZFP57 in a Zfp57 null embryonic stem cell line, which does not restore DNA methylation at ICRs. Using an inducible deletion/expression system it was moreover demonstrated, that wild-type ZFP57, but not a TRIM28 interaction-deficient ZFP57 protein, can prevent loss of DNA methylation, further cementing the requirement for TRIM28 in this process.44
Analysis of the binding sites discovered by ChIP-seq in ESCs revealed a hexanucleotide consensus ZFP57-binding sequence (TGCCGC), which was highly conserved in 81 of 91 bound loci.40 Further, in vitro gel-shift assays confirmed preferential binding of the ZFP57 zinc-finger domain to the methylated oligonucleotide.
In a Nutshell
Genomic imprinting is regulated by DNA methylation at control regions, which must be preserved throughout an individual’s life span even in the face of epigenetic reprogramming at early embryonic stages. The zinc-finger protein ZFP57 binds both maternally and paternally methylated ICRs in a sequence- and methylation-specific manner. Through the interaction of ZFP57 with TRIM28, the chromatin- and DNA-modifying factors, which are all maternally expressed, are attracted to the methylated ICRs. This complex remains bound to ICRs throughout preimplantation development, preserving the 5-mCs in an environment poised to reprogram and demethylate genomes (Figs. 1 and 2).
Genetic evidence from various knockout studies established the components required and, to some extent, their inter-relationships in this process. However, open questions remain especially regarding the actual mechanism employed by the TRIM28/ZFP57 complex to preserve DNA methylation.
Protection, Maintenance or Both?
In maternal Trim28 mutants demethylation of the ICRs is not complete. In fact, the most affected locus (H19) is completely demethylated in only 50% of the analyzed embryos. Other loci are less frequently affected and some appear to be unaffected. This could result from paternal TRIM28 rescue of the not yet demethylated ICRs that still retain bound ZFP57. Theoretically, after zygotic gene activation ZFP57 could engage newly synthesized TRIM28 expressed from the paternal allele to attract the whole modifier complex and recover normal imprinting. It is therefore likely that the rate of demethylation and of TRIM28 transcription from the paternal genome dictates the severity and variability of the mutant phenotype.39 Testing this hypothesis would require ICR methylation analysis of individual, maternal-zygotic Trim28 mutant embryos, which to date is prevented by technical limitations.
The rate of DNA demethylation in the embryo depends on whether it occurs actively or passively. It is tempting to consider that binding of the ZFP57/TRIM28 complex to ICRs could prevent active DNA demethylation, similar to STELLA function. However, combined bisulphite restriction analysis (COBRA) showed little effect on the H19 ICR in 2-cell stage embryos, and loss of methylation only became apparent at the 4- to 8-cell stages, suggesting DNA demethylation happens slowly. Thus, active DNA demethylation through base excision repair (BER) as observed in primordial germ cells and suggested to occur in zygotes46 is unlikely, suggestive of a passive mechanism. On the other hand, active DNA demethylation by TET enzymes produces the intermediate 5-hmC, which can persist in preimplantation embryos until it is passively diluted by DNA replication.37,47,48
Since bisulfite conversion-based detection of DNA methylation does not allow distinction between 5-mC and 5-hmC, i.e., active from passive demethylation,49 it is necessary to analyze ICRs in 2-cell stage embryos by other methods capable of such a distinction. However, the classical methods (i.e., MeDIP/hMeDIP or immunofluorescent detection) are not applicable to the small amounts of genomic material available from preimplantation embryos or do not allow the required resolution. Two related, recently presented methods, however, permit the quantitative mapping of 5-hmC and data from preimplantation embryos will undoubtedly be available soon.50,51 Whether or not active DNA demethylation could affect imprinting maintenance at all will depend on whether ZFP57 is also able to bind 5-hmC in its consensus sequence, which remains to be shown.
The direct interaction of TRIM28 with several DNMTs in ESCs argues in favor of a maintenance mechanism preventing the passive, replication-dependent dilution of 5-mC. In particular, the interaction of TRIM28 with DNMT1 and NP95, both previously shown to be involved in maintenance of imprints, is an intriguing finding.40,44,52,53 Branded the “maintenance DNA methyltransferase,” DNMT1 recognizes hemimethylated DNA, propagating methylation on the newly synthesized, “naked” strand in mitotic cells.54,55 The new evidence suggests ZFP57-guided targeting of DNMT1 to ICRs. The absence of ZFP57, or the linker TRIM28, during cell division would therefore prevent such maintenance and cause the observed dilution of methylated ICRs.38,39 In a Trim28 maternal-mutant embryo only two cells would carry one hemimethylated allele at any stage before zygotic rescue (Fig. 3B). With 91 loci bound by the complex, randomly distributed among the blastomeres, permutations of methylation defects could be vast, leading to a wide range of phenotypes. In this scenario, ZFP57 recognizes hemimethylation on either DNA strand in combination with its hexameric (non-palindromic) binding site, a hypothesis that remains to be tested (Fig. 3A).

Figure 3. Possible DNA methylation maintenance scenario in normal and Trim28 maternal mutant embryos. (A) ZFP57-dependent recognition of ICRs and TRIM28-mediated recruitment of DNMT1 during/after replication allows methylation of the newly synthesized strand. Note that this mechanism requires ZFP57-recognition of hemimethylated DNA on either strand in combination with the non-palindromic hexanucleotide ZFP57-binding sequence (bold). (B) In the absence of TRIM28, DNMT1 is not recruited to ICRs, resulting in hemimethylated alleles in the 2-cell stage embryo. Further DNA replication, in absence of TRIM28, produces two hemimethylated and two unmethylated alleles (highlighted) to be inherited at the 4-cell stage. (For simplicity, only the methylated parental allele and only one daughter cell are depicted.)
The question as to whether the TRIM28/ZFP57 complex inhibits active or passive demethylation cannot be conclusively answered at this time. Additional experiments, such as conditional deletion of Zfp57 in ESCs and timed, comparative MeDIP/hMeDIP experiments could, however, provide considerable insights. Possibly, a combination of both active and passive demethylation contribute to the loss of imprinting in maternal Trim28 mutants, especially since the paternally methylated H19 locus is most strongly affected. This combination of both demethylation mechanisms further enhances the unpredictability of the phenotypic and molecular outcome for individual blastomeres, and even more so for the embryos, fitting our in vivo observations.39
The implication that the ZFP57/TRIM28/DNMT1 complex could maintain imprints in a regulated way in preimplantation embryos, rather than simply providing mere protection from demethylation, raises the question as to whether this mechanism could also affect later developmental stages and adult tissues. Zygotic Trim28 mutants die at gastrulation, failing to induce mesoderm differentiation and showing extreme upregulation of IAP retrotransposons.56,57 However, a comprehensive analysis of the phenotype and its cause was not performed, and DNA methylation at ICRs has not been analyzed. Intriguingly, conditional deletion of Trim28 in the hippocampus of mice, a region also expressing ZFP57,58 led to disregulation of some imprinted loci,59 suggesting a similar function in adult tissues, which requires further investigation.
Yet, preimplantation embryos and ESC are well known for their labile epigenetic states. The requirement for parental genome activation followed directly by the preparation of blastomeres for disparate cell fates in the first lineage segregations demands great epigenetic plasticity. This plasticity is recognizable in co-expression of both demethylating (TETs) and methylating (DNMTs) enzymes, and in the presence of bivalent chromatin marks.60-62 Such an environment might just require specific, specialized targeting of a maintenance complex to ICRs to ensure epigenetic integrity in these volatile cells. For instance, recent careful reexamination of methylation patterns of imprinted loci during preimplantation development has uncovered an unexpected loss of 5-mC, especially at the periphery of ICRs.63 The new evidence, however, shows that as long as the specific 5-mC is maintained in the ZFP57 binding site, such demethylation “noise” could be inconsequential, as these neighboring methyl marks can be recovered by attracting de novo DNA methyltransferases to the ICRs.40
Concluding Remarks
Taken together, the recent discoveries allow a remarkably complete insight of how imprints are conserved during epigenetic reprogramming in preimplantation embryos. Yet, major discrepancies between the different genetic models, such as in the viability of maternal Zfp57 mutants vs. lethality of maternal Trim28 mutants and the non-overlapping effects on specific ICRs (e.g., H19 ICR) in vivo, require further investigation.38,39 These differences might in part originate from independent functions beyond imprinting control (mainly for TRIM28),41,56,59 the timing of gene-specific zygotic activation and the just-in-time availability of newly translated proteins to the complex, but they could also derive from partial functional redundancy of zinc finger proteins (there are over 400 KRAB-ZFPs encoded in the mouse genome).64 Especially, the intriguing loss of methylation at the H19 DMR in maternal Trim28 but not Zfp57 mutants could be based on such redundancy. It would therefore be interesting to see if TRIM28 remains bound to the H19 DMR in the absence of ZFP57. Unfortunately, due to the labile methylation state of the H19 locus in ESCs in general, this analysis was previously uninformative,40,44 and must therefore be performed in vivo on Zfp57-null embryonic tissues.
Now that the major molecules involved in the process are identified and with the knowledge that maintenance of a single CpG island in a ZFP57 binding site ensures the maintenance of imprinting, the experimental paradigm shifts. Future assays must address DNA methylation at these specific sites and reexamination of existing data might be required.
Finally, evidence from maternal Zfp57 mutants suggests a role for ZFP57 in establishing methylation at the Snrpn ICR in the maternal germ line.38 Even though this finding was not confirmed in maternal Trim28 mutants (unpublished observation), it opens the exciting prospect that profound analysis of ZFP57 function and its binding partners might shed light onto the other major, still unanswered, question of imprinting: How is DNA methylation established at ICRs in the first place?
Acknowledgments
I want to specially acknowledge Barbara B. Knowles and Davor Solter for insightful discussions, their support and input for this article. I further thank Heike Wollmann and Adam Hirst for critically reading and improving the manuscript. My work is supported by the Agency for Science, Technology and Research (A*STAR), Singapore.
Glossary
Abbreviations:
- ESC
embryonic stem cell
- ICR
imprinting control region
- OET
oocyte-to-embryo transition
- PGC
primordial germ cell
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/21337
References
- 1.Kaufman MH, Barton SC, Surani MA. Normal postimplantation development of mouse parthenogenetic embryos to the forelimb bud stage. Nature. 1977;265:53–5. doi: 10.1038/265053a0. [DOI] [PubMed] [Google Scholar]
- 2.McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell. 1984;37:179–83. doi: 10.1016/0092-8674(84)90313-1. [DOI] [PubMed] [Google Scholar]
- 3.Barton SC, Surani MA, Norris ML. Role of paternal and maternal genomes in mouse development. Nature. 1984;311:374–6. doi: 10.1038/311374a0. [DOI] [PubMed] [Google Scholar]
- 4.Surani MA, Barton SC, Norris ML. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 1984;308:548–50. doi: 10.1038/308548a0. [DOI] [PubMed] [Google Scholar]
- 5.Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum Mol Genet. 2005;14(Spec No 1):R47–58. doi: 10.1093/hmg/ddi114. [DOI] [PubMed] [Google Scholar]
- 6.Ferguson-Smith AC, Surani MA. Imprinting and the epigenetic asymmetry between parental genomes. Science. 2001;293:1086–9. doi: 10.1126/science.1064020. [DOI] [PubMed] [Google Scholar]
- 7.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet. 2011;12:565–75. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- 9.Brockdorff N. Chromosome silencing mechanisms in X-chromosome inactivation: unknown unknowns. Development. 2011;138:5057–65. doi: 10.1242/dev.065276. [DOI] [PubMed] [Google Scholar]
- 10.Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nat Rev Mol Cell Biol. 2009;10:526–37. doi: 10.1038/nrm2727. [DOI] [PubMed] [Google Scholar]
- 11.Probst AV, Almouzni G. Heterochromatin establishment in the context of genome-wide epigenetic reprogramming. Trends Genet. 2011;27:177–85. doi: 10.1016/j.tig.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–93. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 13.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 14.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–6. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–81. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 17.Surani MA, Hayashi K, Hajkova P. Genetic and epigenetic regulators of pluripotency. Cell. 2007;128:747–62. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 18.Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, et al. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/S0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Inoue K, Ono R, Ogonuki N, Kohda T, Kaneko-Ishino T, et al. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development. 2002;129:1807–17. doi: 10.1242/dev.129.8.1807. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi K, Surani MA. Self-renewing epiblast stem cells exhibit continual delineation of germ cells with epigenetic reprogramming in vitro. Development. 2009;136:3549–56. doi: 10.1242/dev.037747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–3. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 22.Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452:877–81. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucifero D, Mann MR, Bartolomei MS, Trasler JM. Gene-specific timing and epigenetic memory in oocyte imprinting. Hum Mol Genet. 2004;13:839–49. doi: 10.1093/hmg/ddh104. [DOI] [PubMed] [Google Scholar]
- 24.Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248–51. doi: 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–9. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–2. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 28.Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, et al. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10:475–8. doi: 10.1016/S0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- 29.Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol. 2002;241:172–82. doi: 10.1006/dbio.2001.0501. [DOI] [PubMed] [Google Scholar]
- 30.Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, et al. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet. 2011;43:811–4. doi: 10.1038/ng.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–44. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wossidlo M, Arand J, Sebastiano V, Lepikhov K, Boiani M, Reinhardt R, et al. Dynamic link of DNA demethylation, DNA strand breaks and repair in mouse zygotes. EMBO J. 2010;29:1877–88. doi: 10.1038/emboj.2010.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bortvin A, Goodheart M, Liao M, Page DC. Dppa3 / Pgc7 / stella is a maternal factor and is not required for germ cell specification in mice. BMC Dev Biol. 2004;4:2. doi: 10.1186/1471-213X-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamura T, Arai Y, Umehara H, Masuhara M, Kimura T, Taniguchi H, et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol. 2007;9:64–71. doi: 10.1038/ncb1519. [DOI] [PubMed] [Google Scholar]
- 35.Payer B, Saitou M, Barton SC, Thresher R, Dixon JP, Zahn D, et al. Stella is a maternal effect gene required for normal early development in mice. Curr Biol. 2003;13:2110–7. doi: 10.1016/j.cub.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, Matoba S, et al. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–9. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- 37.Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–10. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, et al. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15:547–57. doi: 10.1016/j.devcel.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science. 2012;335:1499–502. doi: 10.1126/science.1216154. [DOI] [PubMed] [Google Scholar]
- 40.Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44:361–72. doi: 10.1016/j.molcel.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iyengar S, Farnham PJ. KAP1 protein: an enigmatic master regulator of the genome. J Biol Chem. 2011;286:26267–76. doi: 10.1074/jbc.R111.252569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–32. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schultz DC, Friedman JR, Rauscher FJ., 3rd Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001;15:428–43. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J Biol Chem. 2012;287:2107–18. doi: 10.1074/jbc.M111.322644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–51. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- 46.Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, et al. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- 48.Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. doi: 10.1126/science.1212483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hayatsu H, Shiragami M. Reaction of bisulfite with the 5-hydroxymethyl group in pyrimidines and in phage DNAs. Biochemistry. 1979;18:632–7. doi: 10.1021/bi00571a013. [DOI] [PubMed] [Google Scholar]
- 50.Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the Mammalian genome. Cell. 2012;149:1368–80. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–7. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 52.Hirasawa R, Chiba H, Kaneda M, Tajima S, Li E, Jaenisch R, et al. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008;22:1607–16. doi: 10.1101/gad.1667008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–12. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 54.Bestor TH. Activation of mammalian DNA methyltransferase by cleavage of a Zn binding regulatory domain. EMBO J. 1992;11:2611–7. doi: 10.1002/j.1460-2075.1992.tb05326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. doi: 10.1016/0092-8674(92)90611-F. [DOI] [PubMed] [Google Scholar]
- 56.Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010;463:237–40. doi: 10.1038/nature08674. [DOI] [PubMed] [Google Scholar]
- 57.Cammas F, Mark M, Dollé P, Dierich A, Chambon P, Losson R. Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development. 2000;127:2955–63. doi: 10.1242/dev.127.13.2955. [DOI] [PubMed] [Google Scholar]
- 58.Alonso MB, Zoidl G, Taveggia C, Bosse F, Zoidl C, Rahman M, et al. Identification and characterization of ZFP-57, a novel zinc finger transcription factor in the mammalian peripheral nervous system. J Biol Chem. 2004;279:25653–64. doi: 10.1074/jbc.M400415200. [DOI] [PubMed] [Google Scholar]
- 59.Jakobsson J, Cordero MI, Bisaz R, Groner AC, Busskamp V, Bensadoun JC, et al. KAP1-mediated epigenetic repression in the forebrain modulates behavioral vulnerability to stress. Neuron. 2008;60:818–31. doi: 10.1016/j.neuron.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 60.Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 61.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 63.Tomizawa S, Kobayashi H, Watanabe T, Andrews S, Hata K, Kelsey G, et al. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development. 2011;138:811–20. doi: 10.1242/dev.061416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Looman C, Abrink M, Mark C, Hellman L. KRAB zinc finger proteins: an analysis of the molecular mechanisms governing their increase in numbers and complexity during evolution. Mol Biol Evol. 2002;19:2118–30. doi: 10.1093/oxfordjournals.molbev.a004037. [DOI] [PubMed] [Google Scholar]