

Abstract
Background:
The aim of this study was to describe the high-resolution CT (HRCT) scan features that characterize familial interstitial pneumonia (FIP).
Methods:
FIP was defined by the presence of two or more cases of probable or definite idiopathic interstitial pneumonia (IIP) in individuals related within three degrees. The cases were collected consecutively from three centers. We identified 371 individuals with potential FIP from 289 families, including 340 individuals who had HRCT scans. Two chest radiologists independently reviewed the HRCT scans, scoring the extent and distribution of HRCT scan findings, and assessed the overall radiologic diagnosis.
Results:
HRCT scan abnormalities suggestive of IIP were present in 85% (289 of 340 subjects). The most frequent findings were reticular pattern (n = 238, 82%) and ground-glass opacity (GGO) associated with reticular abnormality (n = 231, 80%). Other changes included GGO in 116 (40%), honeycombing in 92 (32%), and micronodules in 65 (22%). In the 289 cases with evidence of IIP, the findings were diffusely distributed in the craniocaudal plane in 186 (64%), and the lower lung zones were predominantly involved in 89 (31%). In the axial plane, 194 (67%) had a subpleural distribution; 88 (30%) were diffuse. The imaging pattern was classified as definite or probable usual interstitial pneumonia (UIP) in only 62 subjects (22%) and definite or probable nonspecific interstitial pneumonia (NSIP) in 35 subjects (12%). In 160 subjects (55%), the imaging findings did not conform to previously described UIP or NSIP patterns.
Conclusions:
Reticulation and a mixed GGO/reticular pattern are the most common HRCT scan findings in FIP. The parenchymal abnormalities are most often diffuse in the craniocaudal dimension and have a predominantly peripheral distribution in the axial dimension. Although a radiologic UIP pattern is not uncommon, most cases do not conform to typical UIP or NSIP patterns.
Idiopathic pulmonary fibrosis (IPF) is a progressive and fatal interstitial lung disease (ILD). The pathogenesis is unknown, and to date there are no effective therapies. Since the earliest descriptions of IPF, familial clusters of seemingly similar cases have been identified.1 Marshall et al2 estimated that familial cases accounted for 0.5% to 2.2% of all patients with IPF. It has been suggested more recently that this estimate might actually prove to be 10-fold higher, because 19% of lung transplant patients with IPF reported a family history of lung fibrosis.3 We evaluated the clinical characteristics of 309 affected patients in 111 families and found that the clinical features of familial IPF were similar to those of sporadic IPF.4 Either by high-resolution CT (HRCT) scan or histopathologic pattern, usual interstitial pneumonia (UIP) was by far the most common pattern, being present in 80% of individuals, followed by nonspecific interstitial pneumonia (NSIP) (6%), and organizing pneumonia (OP) (1%). Twelve percent of cases did not conform to a recognized radiologic phenotype. Genetic analysis suggested autosomal-dominant inheritance with reduced penetrance. These results were confirmed by other investigators.5 The clinical and histologic similarities between sporadic and familial IPF support the hypothesis that there may be, in part, a genetic basis for IPF. Furthermore, the gene variants associated with familial interstitial pneumonia (FIP) (TERT, TERC, SP-C, SP-A2, and MUC5B) are also associated with IPF,6 suggesting that the familial and sporadic forms of this disease share a similar cause.
To our knowledge, no studies have reported the detailed radiologic features of a large FIP cohort, and, therefore, there is not a complete understanding of the radiologic similarities and differences between sporadic interstitial pneumonia and FIP. The purpose of this study was to describe the HRCT scan findings of FIP.
Materials and Methods
Subjects
We studied family members enrolled in a large, ongoing, multicenter study of FIP.4,7 The study was approved by the National Jewish Health institutional review board (approval number HS 1441A). A certificate of confidentiality was obtained from the National Institutes of Health (Bethesda, Maryland), and all patients gave informed consent.
FIP was defined by the presence of two or more cases of probable or definite idiopathic interstitial pneumonia (IIP) in individuals related within three degrees.4,6 From a total of 722 individuals, 371 individuals with known or suspected FIP from 289 families underwent HRCT scanning. Thirty-one of these were excluded because CT image quality was inadequate (n = 24), an acute process obscured the underlying lung disease on CT scan (n = 4), or there was another potential cause of their ILD (n = 3). The resultant cohort of 340 patients (male [female] = 176 [164]; median age, 71 years) was used for data analysis. Among the final 340 subjects, there were 38 subsets with two family members, eight with three family members, and one subset each containing four, five, and six family members. Of the 223 individuals, 147 (66%) were current or former smokers, similar to the prevalence reported for sporadic interstitial pneumonia.
HRCT Images and Review
HRCT scans of all patients were obtained at end inspiration in the supine position using a variety of scanners and without IV contrast material. The protocols consisted of 1- to 2-mm collimation sections reconstructed with a high-spatial-frequency algorithm at 1- or 2-cm intervals. All images were viewed on a workstation, at window settings appropriate for viewing lung parenchyma (window level, −600 or −700 Hounsfield units [HU]; window width, 1,500 or 1,750 HU) and mediastinum (window level, 40-50 HU; window width, 250-400 HU).
Two chest radiologists (with 15 and 7 years of CT scan interpretation experience), who were blinded, independently evaluated the CT images. The CT scans in each case were scored for the presence of IIP (0, no evidence of IIP; 1, abnormality equivocal for IIP; 2, abnormality suspicious for IIP). HRCT images were also assessed for the presence of specific parenchymal abnormalities, including reticulation, honeycombing, ground-glass opacity (GGO) associated with reticulation, GGO,8 micronodules, consolidation, and mosaic attenuation. The extent of these abnormalities and the overall extent of lung involvement were determined for each lung using a five-point scale: 0, no involvement; 1, 1% to 25% involvement; 2, 26% to 50% involvement; 3, 51% to 75% involvement; and 4, 76% to 100% involvement. A fibrotic score, which was estimated to the nearest 5% of parenchymal involvement of reticulation, GGO related to reticulation, and honeycombing were also assigned based on the percentage of lung parenchyma involved. The predominant craniocaudal distribution of ILD-related parenchymal abnormalities was scored as predominance in the upper, middle, or lower lungs, or diffuse. Transverse distribution of ILD-related parenchymal abnormalities was also recorded as either subpleural, peribronchovascular, or diffuse.
After scoring the CT scan features, both observers reached the most likely diagnosis based on criteria established by the American Thoracic Society and the European Respiratory Society relating to the radiographic classification of patients with ILD.9 The degree of probability of each CT scan diagnosis was also categorized: definite (≥ 90%) when all the criteria in the American Thoracic Society and European Respiratory Society classification were met, probable (≥ 50%), and possible (< 50%). In the case of probable or possible diagnoses, more than one diagnosis could be selected. Other CT scan diagnoses, including hypersensitivity pneumonitis and sarcoidosis, were also considered when the CT scan features were typical for these disorders. Inconsistencies between the individual reviewers were resolved by consensus, using a third reviewer.
Surgical lung biopsy specimens were available in 37 subjects, and the HRCT scan diagnosis was compared with the histologic finding established by the consensus of two pulmonary pathologists. The relatively small number of surgical tissues available for comparison reflects the treating physician’s opinion of the need for this procedure and/or the patient’s refusal when the biopsy was recommended.
Statistical Analysis
Interobserver agreement between the two radiologists for ILD-related CT scan findings was determined through intraclass correlation coefficients10 for continuous variables and the κ value for categorical variables. κ Agreement scores were determined by the following agreement levels: 0-0.2, poor; 0.21-0.4, fair; 0.41-0.6, moderate; 0.61-0.80, substantial; 0.81-1.0, excellent.11 We compared demographic, clinical, and radiologic features among FIP subtypes using the Kruskal-Wallis test or Fisher exact test. A P value < .05 was considered indicative of a significant difference. When multiple comparisons were performed, the cutoff level of α error was reduced to 0.05/number of tests (Bonferroni correction). Statistical analysis was conducted by using SPSS software (SPSS for Windows, version 15.0, 2006; IBM).
Results
Interobserver agreement for the presence or absence of interstitial abnormality on the CT scans was excellent (κ = 0.95). Among the 340 patients, 40 (12%) did not have evidence of interstitial pneumonia and 11 (3%) were equivocal. Infiltrative lung disease was identified in 289 of 340 patients (85%). Infiltrative lung disease was thought to be definitely present in 278 (96%) and probable in 11 (4%). Of these, the most frequent findings were reticulation and GGO associated with reticulation, both occurring in > 80% of cases (Table 1).These abnormalities involved < 25% of the lung parenchyma in 99% and 94% of individuals, respectively. Honeycombing was identified in 92 patients (32%). Interobserver agreement for most of the ILD-related CT scan findings was very good, whereas the interobserver agreement for pure reticulation, honeycombing, micronodules, and mosaic attenuation was good (Table 1).
Table 1.
—HRCT Scan Findings
| Finding | No. (%) | Agreement (95% CI) |
| Reticulation | 238 (82) | 0.60 (0.41-0.78) |
| GGO associated with reticulation | 231 (80) | 0.89 (0.81-0.96) |
| GGO | 116 (40) | 0.73 (0.59-0.86) |
| Honeycombing | 92 (32) | 0.68 (0.54-0.83) |
| Micronodules | 65 (22) | 0.62 (0.45-0.79) |
| Consolidation | 29 (10) | 0.83 (0.68-0.97) |
| Mosaic attenuation | 32 (11) | 0.63 (0.44-0.83) |
| Craniocaudal direction | 0.84 (0.75-0.93) | |
| Diffuse distribution | 186 (64) | |
| Lower predominance | 89 (31) | |
| Upper predominance | 14 (5) | |
| Middle predominance | 0 (0) | |
| Axial direction | 0.85 (0.78-0.93) | |
| Diffuse distribution | 88 (30) | |
| Subpleural predominance | 194 (67) | |
| Peribronchovascular predominance | 7 (3) | |
| Fibrotic score | 25 ± 18a | 0.81 (0.62-0.90) |
Based on the 289 subjects with evidence of infiltrative lung disease. Agreement was determined by κ value, except for distribution (weighted κ value) and fibrotic score (intraclass correlation coefficients). HRCT = high-resolution CT; GGO = ground-glass opacity.
Mean percentage of involvement ± SD.
Regarding the regional distribution of abnormalities, 186 (64%) had diffuse distribution in the craniocaudal plane (Fig 1), and the lower lung zones were predominantly involved in only 31% (Table 1). In the transverse plane, subpleural predominance was present in 67%. The median score for extent of fibrosis was 1, corresponding to 1% to 25% involvement.
Figure 1.

High-resolution CT (HRCT) images of interstitial abnormalities of nontypical CT scan pattern show bilateral patchy areas of ground-glass opacity superimposed on fine reticulation, distributed diffusely in the craniocaudal and axial planes. A, B, and C, Transverse thin-section CT scans. D, Coronal scan.
The first two reviewers disagreed on 10 cases, and resolution by a third reviewer was needed. A definite or probable CT scan diagnosis was possible in only 129 patients (45%) (Table 2). In the remaining cases, the imaging features, although clearly abnormal, did not permit a specific diagnosis of UIP, NSIP, or other IIP. Definite or probable UIP was determined in only 22%, and definite or probable NSIP in 12%. In 6% of cases, the pattern met the criteria for definite or probable HP (Figs 2, 3). In six cases, the diagnosis of cryptogenic OP was made because of the predominant finding of consolidation. In four cases, the diagnosis of sarcoidosis was made because of perilymphatic nodules, sometimes associated with architectural distortion and mediastinal and/or hilar lymphadenopathy.12 Features of DIP were found in two cases and consisted of extensive GGO with basal and peripheral predominance, with or without cysts.13 The diagnosis of respiratory bronchiolitis was made in two cases because of mild centrilobular nodularity and areas of GGO.13 In the remaining 55%, imaging findings did not conform to established HRCT scan features of IIP (Fig 1).
Table 2.
—Radiologic Subtypes of Infiltrative Lung Disease Among Cases of Familial Interstitial Pneumonia
| Definite or Probable | No. (% of 289) |
| UIP | 62 (22) |
| NSIP | 35 (12) |
| Other diagnoses | 32 (11) |
| HP | 18 (6) |
| COP | 6 (2) |
| Sarcoidosis | 4 (1) |
| DIP | 2 (1) |
| RB | 2 (1) |
| Total | 129 (45) |
COP = cryptogenic organizing pneumonia; DIP = desquamative interstitial pneumonia; HP = hypersensitivity pneumonitis; NSIP = nonspecific interstitial pneumonia; RB = respiratory bronchiolitis; UIP = usual interstitial pneumonia.
Figure 2.

Representative abnormal HRCT image of definite usual interstitial pneumonia pattern. Transverse thin-section CT scan of basal segments of lower lobes shows reticulation and honeycombing. See Figure 1 legend for expansion of the abbreviation.
Figure 3.
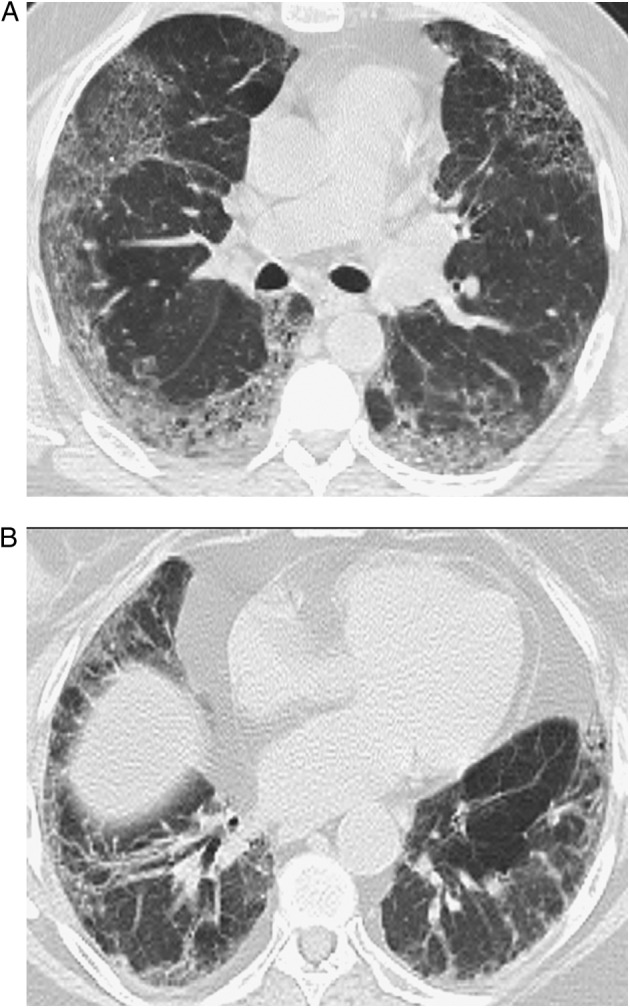
A, B, Representative abnormal HRCT images of probable nonspecific interstitial pneumonia pattern. Transverse thin-section CT scans of basal segments of lower lobes show peripheral predominant ground-glass opacity with mild reticulation. See Figure 1 legend for expansion of the abbreviation.
Details of demographic, clinical, and radiologic appearances of the individuals with an HRCT scan diagnosis of UIP, NSIP, HP, and nontypical CT scan patterns are shown in Table 3. Subjects with probable or definite UIP were more likely to be male, were older, had relatively lower diffusion capacity of the lung for carbon monoxide, and had higher fibrotic scores than did those with the nontypical pattern.
Table 3.
—Prevalence of Clinical and Radiologic Features of Individuals With Typical Patterns of NSIP, UIP, HP, and Nontypical CT Scan Pattern
| Consensus CT Scan Diagnosis |
|||||
| Clinical and Radiologic Features | Probable or Definite UIP (n = 62) | Probable or Definite NSIP (n = 35) | Probable or Definite HP (n = 18) | Nontypical CT Scan Pattern (n = 160) | P Value |
| Sex, male | 46 (74) | 19 (54) | 7 (39) | 73 (46) | .003a |
| Age, y | 69.5 ± 8.4 | 69.2 ± 10.2 | 62.1 ± 17.7 | 64.1 ± 12.2 | .002b |
| PFTs (n = 239) | |||||
| FVC, % predicted | 69.7 ± 13.7 | 73.3 ± 15.2 | 65.8 ± 17.2 | 69.0 ± 17.2 | .844 |
| Dlco, % predicted | 44.5 ± 14.8 | 50.8 ± 19.2 | 49.1 ± 14.4 | 55.9 ± 21.6 | .011c |
| Grade of dyspnea (n = 236)d | .471 | ||||
| 0 | 11 (24) | 9 (29) | 3 (27) | 44 (32) | |
| 1 | 8 (17) | 4 (13) | 1 (9) | 24 (18) | |
| 2 | 3 (7) | 5 (16) | 0 (0) | 14 (10) | |
| 3 | 1 (2) | 2 (6) | 0 (0) | 7 (5) | |
| 4 | 23 (50) | 11 (26) | 7 (63) | 47 (34) | |
| CT scan findings | |||||
| Reticulation | 55 (89) | 35 (100) | 14 (78) | 129 (81) | .971 |
| GGO associated with reticulation | 58 (94) | 34 (97) | 17 (94) | 122 (76) | .135 |
| GGO | 15 (24) | 7 (20) | 12 (67) | 82 (51) | .172 |
| Honeycombing | 52 (84) | 4 (11) | 5 (28) | 31 (19) | .361 |
| Micronodules | 6 (10) | 6 (17) | 8 (44) | 41 (26) | .226 |
| Consolidation | 3 (5) | 2 (6) | 2 (11) | 17 (11) | .909 |
| Mosaic attenuation | 1 (2) | 3 (9) | 9 (50) | 17 (11) | .404 |
| Craniocaudal direction | .028e | ||||
| Diffuse distribution | 36 (58) | 19 (54) | 18 (100) | 103 (64) | |
| Lower predominance | 26 (42) | 16 (46) | 0 (0) | 44 (28) | |
| Upper predominance | 0 (0) | 0 (0) | 0 (0) | 13 (8) | |
| Middle predominance | 0 (0) | 0 (0) | 0 (0) | 0 (0) | |
| Axial direction | < .001f | ||||
| Diffuse distribution | 7 (11) | 6 (17) | 13 (72) | 62 (39) | |
| Subpleural predominance | 55 (89) | 29 (83) | 5 (28) | 102 (64) | |
| Peribronchovascular predominance | 0 (0) | 0 (0) | 0 (0) | 2 (1) | |
| Fibrotic score | 37 ± 16 | 29 ± 12 | 34 ± 22 | 27 ± 19 | .002g |
Data are presented as mean ± SD or No. (%), unless indicated otherwise. Fourteen subjects with a consensus CT scan diagnosis of either COP, sarcoidosis, or RB are not included in this table, so the total included in the table is 275. Post hoc test with Bonferroni. Dlco = diffusion capacity of the lung for carbon monoxide; PFT = pulmonary function test. See Table 1 and 2 legends for expansion of other abbreviations.
UIP vs nontypical (P = .001).
UIP vs nontypical (P = .029).
UIP vs nontypical (P = .006).
Based on American Thoracic Society score.
HP vs nontypical (P = .017).
UIP vs HP (P < .001), UIP vs nontypical (P = .01), NSIP vs HP (P < .001), HP vs nontypical (P = .009).
HP vs nontypical (P = .017).
Surgical lung biopsy specimens were available in 37 cases (Table 4). A probable or definite CT scan diagnosis of UIP was concordant with histologic diagnosis in five of seven cases (71%) with histology. However, the typical features of UIP histology were present in one case of a CT scan diagnosis of HP and one with a CT scan diagnosis of NSIP. Moreover, in 12 cases in which a specific CT scan diagnosis could not be established, histologic UIP was present.
Table 4.
—Relationship Between Radiologic and Histologic Diagnoses in 37 Cases
| Probable or Definite Histologic Diagnosis |
|||||
| Probable or Definite Radiologic Diagnosis | UIP | NSIP | HP | RB-ILD | No Specific Diagnosis |
| UIP | 5 | 1 | … | … | 1 |
| NSIP | 1 | 1 | … | … | 1 |
| COP | … | … | 1 | … | … |
| HP | 1 | … | … | … | 1 |
| DIP | … | … | … | … | 1 |
| No specific diagnosis | 12 | 2 | … | 2 | 7 |
ILD = interstitial lung disease. See Table 2 for expansion of other abbreviations.
Discussion
Although both genetic and environmental factors have been implicated, the cause of IPF is unknown. Treatments that target immune suppression have been unsuccessful.14 To better understand the pathophysiology of the disease and to explore possible new treatment targets, several studies have focused on genetic variations in IPF.7,15,16
This report identifies differences between the CT scan appearances seen in FIP and previous descriptions of sporadic IPF with the underlying lesion of UIP. Lower lobe predominance was identified in only 31% of study subjects, compared with 70% in sporadic IPF.12,17‐19 Subpleural predominance was present in 67% in this study compared with 80% to 95% in sporadic cases. Honeycombing was present in 32% of the familial population, compared with 71% previously reported in sporadic IPF cases.20 Moreover, we were unable to establish a UIP or NSIP radiologic diagnosis based on HRCT scan features in 192 of 289 study subjects (66%). A partial explanation for this unexpected finding could be the selection of a population with earlier disease, as suggested by the relatively young age, higher diffusion capacity of the lung for carbon monoxide, and higher fibrotic score in the group with nontypical CT scan findings. Additionally, there is a well-described overlap of HRCT scan appearances of UIP and NSIP.21 The number of patients without a classifiable HRCT scan, as well as the number of non-IPF HRCT scans, is consistent with the heterogeneity of FIP as previously reported.4 However, the differences in craniocaudal and axial distributions may reflect a difference in the phenotypic manifestation of some cases of FIP compared with non-FIP. It seems unlikely that lead-time bias could account for the more diffuse craniocaudal distribution of abnormality identified in the FIP population, because even early cases of IIP usually show lower lung predominance.
Two studies have described the CT scan appearances in FIP. Lee et al5 evaluated the HRCT scan findings of 17 patients with FIP. Bilateral irregular linear opacities were seen in all cases. A subpleural predominance of linear opacities was present in 94% of patients. Honeycombing was observed in 29% of patients. These authors concluded that the CT scan findings were consistent with IPF. Notably, in contrast to the current study, these authors reported that the CT scan findings of pulmonary fibrosis in FIP were distributed in the basilar regions of the lung. However, the authors did not report the specific prevalence of basal lung distribution. In a study of nine individuals with FIP, Nishiyama et al22 found that all had GGO, intralobular reticular opacities, and irregular thickening of the interlobular septa; seven (78%) had small foci of consolidation; and three (33%) had honeycombing. The abnormalities were primarily in the lower lung zones in six subjects and the upper lung zones in two patients, and one had no zonal predominance. The authors commented that the prevalence of lower lung zone predominance of FIP in this study was lower than in sporadic IIP. However, the small number of cases in this study limits the generalizability of the findings.
The current study suggests that FIP should be considered in patients with diffuse or predominantly upper lung distribution of fibrosis in the craniocaudal plane, and this finding should prompt a detailed family history. If FIP is suspected, patients and their physicians may consider early screening in the family and further genetic evaluation for specific genetic polymorphisms in genes encoding the telomerase,15 surfactant proteins, and gel-forming mucins.6,23,24
As for the correlation with histologic interpretation, our findings showed substantial discrepancy between CT scan diagnosis and histologic diagnosis, although only a small number of cases were available for the histologic interpretation. This discordance between histologic and CT scan diagnosis may be due to selection of radiologically atypical cases for surgical biopsy, as suggested by a study,25 which showed that 34 of 55 biopsy-proven IPF cases (62%) were assigned alternative diagnoses on HRCT scan. In these atypical IPF cases, the first-choice diagnoses were NSIP (53%), HP (12%), sarcoidosis (9%), and OP (3%); in eight of 34 cases (23%), no single diagnosis was favored by more than one observer. Similarly, in a study of 98 patients, Flaherty et al21 showed that 26 of 73 patients (35%) with sporadic IPF and surgically proven UIP had an HRCT scan appearance more compatible with NSIP.
Our study was limited inherently by its retrospective design and potentially by selection bias in the identification of families. However, the large number of cases derived from three centers suggests that the study population provides a broad representation of FIP. Additionally, as discussed earlier, the screening method may have led to selection of a relatively early, and less radiologically specific, phase of FIP.
In conclusion, the parenchymal abnormalities of FIP are most often diffuse in the craniocaudal dimension and have a predominantly peripheral distribution in the axial dimension. Although a radiologic UIP pattern is most common, most cases do not conform to typical UIP or NSIP patterns.
Acknowledgments
Author contributions: Dr Lynch is guarantor of the manuscript and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Dr Lee: contributed to the study design, CT scan reading, statistical analyses, and was the primary author of the manuscript.
Dr Seo: contributed to the CT scan readings and was the secondary author of the manuscript.
Dr Steele: contributed to the confirmation of the diagnosis of familial pulmonary fibrosis in a large proportion of the cohort and editing of the manuscript.
Dr Schwarz: contributed to the clinical review of cases and editing of the manuscript.
Dr Brown: contributed to the clinical review of cases and editing of the manuscript.
Dr Loyd: contributed to the confirmation of the diagnosis of familial pulmonary fibrosis in a large proportion of cohort and editing of the manuscript.
Ms Talbert: contributed to results interpretation and drafting of the manuscript.
Dr Schwartz: contributed to the establishment of the familial pulmonary fibrosis cohort as the project reader and drafting of the manuscript.
Dr Lynch: contributed to the establishment of the radiology project, design of the radiology study, performance of the consensus reads, and editing of the manuscript.
Financial/nonfinancial disclosures: The authors have reported to CHEST the following conflicts of interest: Dr Lynch has served as a consultant to several trials in IPF, sponsored by Gilead; Actelion Pharmaceuticals Ltd; Johnson & Johnson Services, Inc; and InterMune. He is also a consultant for Perceptive Informatics, Inc and receives research support from Siemens AG. The remaining authors have reported that no potential conflicts of interest exist with any companies/organizations whose products or services may be discussed in this article.
Role of sponsors: The sponsors had no role in the design of the study, the collection and analysis of the data, or in the preparation of the manuscript.
Abbreviations
- FIP
familial interstitial pneumonia
- GGO
ground-glass opacity
- HRCT
high-resolution CT
- HU
Hounsfield unit
- IIP
idiopathic interstitial pneumonia
- ILD
interstitial lung disease
- IPF
idiopathic pulmonary fibrosis
- NSIP
nonspecific interstitial pneumonia
- OP
organizing pneumonia
- UIP
usual interstitial pneumonia
Footnotes
Dr Lee is currently at the Department of Radiology and Center for Imaging Science, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, South Korea.
Dr Seo is currently at the Department of Radiology and Research Institute of Radiology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, South Korea.
Funding/Support: This study was supported by grants from the National Heart, Lung and Blood Institute [RO1-HL095393 and RO1-HL097163], both to Dr Schwartz.
Reproduction of this article is prohibited without written permission from the American College of Chest Physicians. See online for more details.
References
- 1.Allam JS, Limper AH. Idiopathic pulmonary fibrosis: is it a familial disease?. Curr Opin Pulm Med. 2006;12(5):312-317 [DOI] [PubMed] [Google Scholar]
- 2.Marshall RP, Puddicombe A, Cookson WO, Laurent GJ. Adult familial cryptogenic fibrosing alveolitis in the United Kingdom. Thorax. 2000;55(2):143-146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loyd JE. Pulmonary fibrosis in families. Am J Respir Cell Mol Biol. 2003;29(suppl 3):S47-S50 [PubMed] [Google Scholar]
- 4.Steele MP, Speer MC, Loyd JE, et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005;172(9):1146-1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee HL, Ryu JH, Wittmer MH, et al. Familial idiopathic pulmonary fibrosis: clinical features and outcome. Chest. 2005;127(6):2034-2041 [DOI] [PubMed] [Google Scholar]
- 6.Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503-1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang IV, Burch LH, Steele MP, et al. Gene expression profiling of familial and sporadic interstitial pneumonia. Am J Respir Crit Care Med. 2007;175(1):45-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246(3):697-722 [DOI] [PubMed] [Google Scholar]
- 9.Travis W, King T, Bateman E, et al. ; American Thoracic Society ; European Respiratory Society American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165(2):277-304 [DOI] [PubMed] [Google Scholar]
- 10.Winer-Muram HT, Jennings SG, Meyer CA, et al. Effect of varying CT section width on volumetric measurement of lung tumors and application of compensatory equations. Radiology. 2003;229(1):184-194 [DOI] [PubMed] [Google Scholar]
- 11.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159-174 [PubMed] [Google Scholar]
- 12.Lynch JP, III, Ma YL, Koss MN, White ES. Pulmonary sarcoidosis. Semin Respir Crit Care Med. 2007;28(1):53-74 [DOI] [PubMed] [Google Scholar]
- 13.Lynch DA, Travis WD, Müller NL, et al. Idiopathic interstitial pneumonias: CT features. Radiology. 2005;236(1):10-21 [DOI] [PubMed] [Google Scholar]
- 14.Lawson WE, Loyd JE. The genetic approach in pulmonary fibrosis: can it provide clues to this complex disease?. Proc Am Thorac Soc. 2006;3(4):345-349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317-1326 [DOI] [PubMed] [Google Scholar]
- 16.Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(7):729-737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Souza CA, Müller NL, Lee KS, Johkoh T, Mitsuhiro H, Chong S. Idiopathic interstitial pneumonias: prevalence of mediastinal lymph node enlargement in 206 patients. AJR Am J Roentgenol. 2006;186(4):995-999 [DOI] [PubMed] [Google Scholar]
- 18.Ichikado K, Suga M, Müller NL, et al. Acute interstitial pneumonia: comparison of high-resolution computed tomography findings between survivors and nonsurvivors. Am J Respir Crit Care Med. 2002;165(11):1551-1556 [DOI] [PubMed] [Google Scholar]
- 19.Tomiyama N, Müller NL, Johkoh T, et al. Acute respiratory distress syndrome and acute interstitial pneumonia: comparison of thin-section CT findings. J Comput Assist Tomogr. 2001;25(1):28-33 [DOI] [PubMed] [Google Scholar]
- 20.Johkoh T, Müller NL, Cartier Y, et al. Idiopathic interstitial pneumonias: diagnostic accuracy of thin-section CT in 129 patients. Radiology. 1999;211(2):555-560 [DOI] [PubMed] [Google Scholar]
- 21.Flaherty KR, Thwaite EL, Kazerooni EA, et al. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax. 2003;58(2):143-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishiyama O, Taniguchi H, Kondoh Y, et al. Familial idiopathic pulmonary fibrosis: serial high-resolution computed tomography findings in 9 patients. J Comput Assist Tomogr. 2004;28(4):443-448 [DOI] [PubMed] [Google Scholar]
- 23.Amin RS, Wert SE, Baughman RP, et al. Surfactant protein deficiency in familial interstitial lung disease. J Pediatr. 2001;139(1):85-92 [DOI] [PubMed] [Google Scholar]
- 24.Newman B, Kuhn JP, Kramer SS, Carcillo JA. Congenital surfactant protein B deficiency—emphasis on imaging. Pediatr Radiol. 2001;31(5):327-331 [DOI] [PubMed] [Google Scholar]
- 25.Sverzellati N, Wells AU, Tomassetti S, et al. Biopsy-proved idiopathic pulmonary fibrosis: spectrum of nondiagnostic thin-section CT diagnoses. Radiology. 2010;254(3):957-964 [DOI] [PubMed] [Google Scholar]