

Abstract
The ability of mature organisms to stabilize phenotypes has enormous selective advantage across all phyla, but the mechanisms have been largely unexplored. Individuals with fibrodysplasia ossificans progressiva (FOP), a rare genetic disorder of progressive heterotopic ossification, undergo a pathological metamorphosis in which one normal tissue is transformed into another through a highly regulated process of tissue destruction and phenotype reassignment. This disabling metamorphosis is mediated by the FOP metamorphogene, which encodes a mutant bone morphogenetic protein (BMP) type I receptor that exhibits mild constitutive activity during development and severe episodic dysregulation postnatally. The discovery of the FOP metamorphogene reveals a highly conserved target for drug development and identifies a fundamental defect in the BMP signaling pathway that when triggered by injury and inflammation transforms one tissue into another.
Keywords: Fibrodysplasia ossificans progressiva, heterotopic ossification, bone morphogenetic protein (BMP) receptor, BMP signaling, metamorphosis, metamorphogene, ACVR1, activin-like kinase 2 (ALK2)
1. Introduction
1.1. FOP
FOP is a complex and disabling genetic disease characterized by congenital skeletal malformations and inflammation-induced extraskeletal bone formation through an endochondral process [1–3]. Heterotopic ossification begins in childhood, may occur without warning, or can be induced by trauma or various viral illnesses. Bone formation is episodic and progressive, leading to the formation of a heterotopic skeleton in well-defined spatial and temporal patterns [4–5]. Eventually, progressive ossification causes extraarticular ankylosis of nearly all axial and appendicular joints and permanent immobilization in a disabling “second skeleton” of heterotopic bone [1,2,4,6,7,8]. Misdiagnosis is common [9,10]. Death results most commonly by the fifth decade from complications of restrictive chest wall disease [11,12].
1.2. The BMP Signaling Pathway
A comprehensive understanding of the bone morphogenetic protein (BMP) signaling pathway is critical to understanding the pathophysiology of FOP. The bone morphogenetic proteins (BMPs) are a family of highly conserved extracellular signaling molecules that regulate progenitor cell fates [13–17].
BMPs act through complex biochemical signaling pathways and function in a wide variety of cells and tissues during embryonic development and postnatal life. BMPs signal by binding to and activating transmembrane heterotetrameric complexes of type I and type II BMP receptors. BMP signaling is mediated through three known type I receptors: BMPRIA/ALK3, BMPRIB/ALK6, and ACVR1/ALK2, and two type II receptors, BMPRII and ActRII. Until recently, BMPRIA/ALK3 and BMPRIB/ALK6 have been the overwhelming focus of most BMP type I receptor studies [13–16, 18–20].
A unique feature of all transforming growth factor-β/bone morphogenetic protein (TGF-β/BMP) type I receptors is a cytoplasmic juxtamembrane region rich in glycine and serine residues (GS domain). Following ligand binding, serines and threonines in this region are phosphorylated by the constitutively active BMP type II receptor, thus activating the BMP type I receptor to transmit signals through SMAD and mitogen-activated protein kinase (MAPK) signaling pathways to regulate nuclear transcription of BMP responsive target genes [13–19, 21–24].
The GS domain of all TGF-β/BMP type I receptors is a critical site for the activation of pathway-specific SMAD signaling proteins by constitutively active TGF-β/BMP type II receptors. Importantly, FKBP1A (also known as FKBP12), binds and stabilizes the inactive conformation of all TGF-β/BMP type I receptors including ACVR1/ALK2. When bound to the GS domain, FKBP1A prevents promiscuous or leaky activation of all type I receptors in the absence of ligand [25–28]. Importantly, FKBP1A also serves as a docking protein for SMAD/SMURF complexes that mediate ubiquitination, internalization, and degradation of all BMP type I receptors [29,30]. As a result, FKBP1A is predicted to regulate the steady-state concentration of all BMP type I receptors (ALKs 2, 3, 6) at the cell membrane.
1.3. Dysregulation of BMP Signaling in FOP Cells
Studies of the Drosophila decapentaplegic mutant, a dipteran model of dysregulated BMP signaling [31], as well as studies of the classic FOP phenotype, supported that the primary molecular pathology in FOP involved both embryonic patterning and postnatal response to injury. The association of these two developmentally associated processes strongly suggested that the bone morphogenetic protein (BMP) signaling pathway might be involved in the pathogenesis of FOP [31]. A series of discoveries in in vitro and in vivo systems provided compelling evidence of profound dysregulation of the BMP signaling pathway in FOP cells.
These findings included but were not limited to:
Failure to upregulate multiple BMP antagonists [36]
Failure to appropriately regulate the concentration of BMP in the extracellular space [37,38]
BMP expression in fibroproliferative cells of early FOP lesions [32]
Incomplete modulation of BMP signaling by cell-surface heparan sulfate proteoglycans [39,40]
Increased concentration of BMP type I receptors at the cell surface [38]
Failure to appropriately internalize and degrade BMP type I receptors [37,38,41]
Sustained BMP signaling in FOP cells in the absence and presence of BMP [38]
Dysregulated BMP-independent signaling through the SMAD pathway [42,43]
Dysregulated BMP-dependent signaling through both the SMAD and the p38 MAPK pathways [42,43]
Enhanced osteogenic differentiation of FOP connective tissue progenitor cells [42]
2. The FOP Metamorphogene
2.1 The Discovery of the FOP Metamorphogene
The FOP gene was mapped by genome-wide linkage analysis to chromosome 2q23-24, a region that includes the gene encoding Activin receptor A type I/activin-like kinase 2 (ACVR1/ALK2), a type I serine-threonine kinase receptor initially reported to mediate activin signaling but shown more recently to be a BMP type I receptor [44]. ACVR1/ALK2 is one of seven activin-like kinases (ALKs) in humans and one of the three classic BMP type I receptors encoded in the human genome [45]. Within the linkage interval, ACVR1/ALK2 was a prime candidate gene for FOP based on a number of studies supporting dysregulated BMP signaling in the pathogenesis of FOP.
A heterozygous missense mutation (c.617G >A; R206H) was identified in the glycine-serine (GS) activation domain of ACVR1/ALK2 in all classically affected FOP patients worldwide [44]. This single nucleotide mutation transforms a morphogen receptor gene into a metamorphogene and provides a permissive genetic background for the developmental and post-natal features of classic FOP (Figure 1). The mutant ACVR1/ALK2 protein activates BMP signaling in the absence of BMP and permits robust signaling in the presence of BMP [46]. Identification of the mutant transmembrane receptor (remarkably containing a single substituted amino acid residue) provides the basis for elucidating the molecular pathophysiology of dysregulated BMP signaling and resultant skeletal metamorphosis in FOP [47].
Figure 1. The FOP metamorphogene and its major phenotypes.

The FOP metamorphogene encodes a type I BMP receptor (ACVR1/ALK2; R206H) that dysregulates BMP signaling and accounts for all of the congenital and postnatal features of FOP including congenital malformations of the great toes, heterotopic endochondral ossification, and ectopic skeletogenesis in characteristic anatomic patterns.
Investigations of ACVR1/ALK2 function in embryonic development and in cell differentiation have been limited [48,49]. ACVR1/ALK2 is expressed in many tissues including skeletal muscle, blood vessels, and chondrocytes, providing support for its phenotypic effects in FOP including developmental joint malformations and postnatal heterotopic ossification consistent with the classic FOP phenotype [49]. Consistent with the classic FOP phenotype, constitutive activation of ACVR1/ALK2 induces alkaline phosphatase activity in C2C12 muscle satellite cells, upregulates BMP4, downregulates BMP antagonists, fails to appropriately regulate the concentration of BMP in the extra cellular space, dysregulates BMP signaling through the SMAD pathway, expands cartilage elements, induces ectopic chondrogenesis and stimulates joint fusions, findings similar to those seen in FOP [44,46,49–51].
Constitutively active ACVR1/ALK2 expression in embryonic chick limb buds induces expansion of chondrogenic anlage and leads to joint fusions, suggesting that promiscuous ACVR1/ALK2 signaling alters cell fate and induces undifferentiated mesenchyme to form cartilage and bone [46,49]. In the chick, however, heterotopic bone is seen at birth, whereas in humans with FOP, heterotopic bone is not seen at birth, and occurs later in childhood after an inflammatory trigger. These findings suggest that the mutant ACVR1/ALK2 receptor in FOP is only mildly constitutively active and responsive to receptor stimulation. Recent data support these findings [42,46]. Enhanced ACVR1/ALK2 activation in FOP is also supported by recent data showing increased BMP pathway-specific SMAD phosphorylation and expression of BMP transcriptional targets in FOP cells, as well as rescue of an ACVR1/ALK2 loss of function phenotype by mutant FOP ACVR1/ALK2 in zebrafish embryos [46].
Recent studies examined BMP signaling and osteogenic differentiation using connective tissue progenitor cells (SHED cells; stem cells from human exfoliated deciduous teeth) from discarded primary teeth of FOP patients and controls [42].
SHED cells from unaffected individuals transmitted BMP signals through both the SMAD and p38 mitogen-activated protein kinase (MAPK) limbs of the BMP signaling pathways and responded to BMP4 treatment by inducing BMP responsive genes. FOP SHED cells showed BMP-independent signaling as well as enhanced responsiveness to BMP stimulation. Further, FOP SHED cells showed more rapid differentiation to an osteogenic phenotype than control cells [42]. These data strongly support that both BMP-independent and BMP-dependent signaling occur as a result of mutant ACVR1/ALK2 activity in FOP cells. These data substantially extend our understanding of dysregulated BMP signaling in a progenitor cell population relevant to the pathogenesis of FOP [42].
2.2 A Novel Protein Encoded by the FOP Metamorphogene
Many abnormalities in the BMP signaling pathway were unveiled before the discovery of the FOP gene, and suggested that (BMPRIA/ALK3) may also be involved in the FOP disease process [38]. While still unresolved, these data are compatible with recent animal models of BMP signaling in which FOP features have been observed. In two classic model systems (Drosophila and Zebrafish), ACVR1/ALK2 homologues (Saxophone and Alk8), do not act independently, but rather in heteromeric complexes with other BMP type I receptors (Thickveins and Alk8), which are the Drosophila and Zebrafish orthologs of BMPRIA/ALK3 and human ACVR1/ALK2 respectively [52,53]. These data strongly suggest that the abnormal cellular metabolism of BMPRIA/ALK3 in FOP may arise from its association with ACVR1/ALK2 in heteromeric signaling complexes [52,53].
Protein homology modeling of the mutant FOP receptor predicts changes in both BMP-independent and BMP-dependent signaling in FOP cells [44,54]. Protein modeling of the FOP mutation predicts impaired FKBP1A (FKBP12) binding and/or activity with resultant BMP-independent as well as enhanced BMP-dependent signaling, coordinate features of BMP signal dysregulation that have recently been demonstrated in FOP cells [46,54]. Thus, it seems likely that the interaction of FKBP1A with the GS domain may be altered in FOP, leading in part to BMP-independent activity of ACVR1/ALK2. Recent preliminary data strongly support this hypothesis [46]. Further, loss of autoinhibition of ACVR1/ALK2 is predicted to change not only the intensity, but also the duration of BMP signaling (42,55), thus creating new repertoires of BMP activity not previously observed in nature.
2.3. Inflammation & the FOP Metamorphogene
The recurrent heterozygous ACVR1/ALK2 (R206H) mutation that causes FOP creates a novel form of the receptor that increases the risk of FOP to 100% for all affected individuals (44). Interestingly, while this mutation is necessary for FOP and may be directly responsible for the congenital malformations in the condition, it does not appear sufficient for the disabling, episodic, postnatal flare-ups of heterotopic ossification (56–59). Activation of FOP by the innate immune system appears to be an important trigger for post-natal flare-ups of the disease (60–64).
Presently five animal models reproduce various clinical and pathological features of FOP-like heterotopic ossification [61–67]. One in particular, an ACVR1/ALK2 mutant knock-in chimera, reproduces all of the phenotypic features of FOP and provides proof-of-concept that the canonical FOP metamorphogene causes all of the phenotypic features of classic FOP and with patterns and triggers similar to that in the human condition [65].
Four recent in vivo studies have examined the role of inflammation in triggering heterotopic ossification in a BMP-permissive environment [61–64]. In a unique FOP case study and associated murine bone marrow transplantation experiment, the innate immune system was required for disease flare-ups [62]. In a transgenic mouse model of BMP-induced heterotopic ossification, the activity of circulating monocytes and tissue macrophages was inhibited pharmacologically and genetically, and found to substantially abrogate inflammation-associated heterotopic ossification [61]. In a related animal study, local inflammation was sufficient to stimulate heterotopic ossification in a transgenic mouse model in which BMP4 was overexpressed at the neuromuscular junction [63]. In yet another study, the conditional activation of inflammatory pathways in a constitutively active ALK2 mouse model led to the activation of heterotopic ossification at sites of inflammation [64]. Together, these findings support that dysregulation of the BMP signaling pathway and an inflammatory microenvironment are both required for the formation of heterotopic ossification.
2.4. Developmental Effects of the FOP Metamorphogene
In addition to its effect on connective tissue metamorphosis, the FOP metamorphogene leads to a wide range of phenotype abnormalities during development and postnatal life including:
Benign skeletal neoplasms [69]
The congenital phenotypes caused by the FOP metamorphogene include an array of congenital malformations and skeletal anomalies including malformed great toes, fusion of the cervical facet joints, fusion of the costovertebral joints, proximal medial tibial osteochondromas, and short broad femoral necks [1,68–72]. Unlike post-natal heterotopic ossification, these anomalies all seem to occur without a histologically observable inflammatory component, suggesting that an inflammatory trigger may not be necessary for the congenital features of the disease.
Interestingly, identical twins with FOP have nearly identical congenital malformations, but have a divergent history of postnatal heterotopic ossification that more closely follows variable and individual postnatal inflammatory triggers such as trauma and viral illnesses [60]. Importantly, BMP signaling stimulates chondrocyte differentiation at multiple steps during embryonic cartilage development [73]. Profound over-activity of the BMP signaling pathway, as seen in Noggin Knockout mice in the absence of inflammatory triggers, leads to failure of formation of all diarthrodial joints [74]. Taken together, it is reasonable to hypothesize that the formation and maintenance of diarthrodial joints is sensitive to various gradients of BMP activity whereas postnatal heterotopic ossification is more dependent on inflammatory stimuli in the setting of dysregulated BMP signaling. A more complete analysis of the developmental effects of the FOP metamorphogene may be found in several recent articles and reviews [68–71].
2.5. Hypoxia & the FOP Metamorphogene
Hypoxia is predicted to increase BMP signaling from the mutant ACVR1/ALK2 receptor in both a pH-independent and a pH-dependent manner. Preliminary data support this hypothesis [75]. Importantly, generation of a hypoxic microenvironment triggered by BMP2 in skeletal muscle has recently been shown to be a critical step in the formation of heterotopic bone in a murine model [76]. To better understand the molecular constraints and physiological implications of an hypoxic microenvironment on the FOP mutation, detailed in silico modeling of wild type and mutant ACVR1/ALK2 were undertaken [54].
In silico modeling predicts that in the wild-type ACVR1/ALK2 model, the conserved arginine at codon 206 forms a salt bridge with an invariant aspartate residue at codon 269 [54]. In contrast, histidine at residue 206 (as occurs in FOP), would participate in a salt bridge with aspartate at residue 269 only at decreased intracellular pH and with extensive structural rearrangement predicting that substitution with histidine creates a pH-sensitive switch within the activation domain of the receptor [54]. For example, flare-ups of FOP might result from physiological changes such as trauma-associated hypoxia that lower intracellular pH and activate the switch. If so, perhaps, the intracellular pH of affected connective tissues could be modulated pharmacologically to suppress flare-ups of FOP or diminish the extent of heterotopic ossification [54].
Development of therapeutic approaches that abrogate hypoxic stress may serve to inhibit heterotopic bone formation in general, whether initiated by BMP ligand or by the aberrant ACVR1/ALK2 (R206H) receptor hypothesized to become hyperactive under conditions of hypoxia and/or intracellular acidosis (54). Such approaches may prove useful for a wide array of non-genetic forms of heterotopic ossification.
2.6. FOP Variants & Novel Mutations in the FOP Metamorphogene
All patients with classic clinical features of FOP (great toe malformations and progressive heterotopic ossification) carry the same heterozygous mutation (c.617G>A; R206H) in the glycine-serine residue (GS) activation domain of activin A type I receptor/activin-like kinase 2 (ACVR1/ALK2). However, among patients with FOP-like heterotopic ossification and/or toe malformations, we also identified patients with clinical features unusual for FOP. These atypical FOP patients form two classes: FOP-plus (classic defining features of FOP plus one or more atypical features) and FOP variants (major variations in one or both of the two classic defining features of FOP [68]. All patients in both classes have heterozygous ACVR1/ALK2 missense mutations in conserved amino acids in ACVR1/ALK2. While the canonical c.617G>A; R206H mutation was found in all cases of classic FOP and most cases of FOP-plus, novel ACVR1/ALK2 mutations occur in all FOP variants and in two cases of FOP-plus [68]. Protein structure homology modeling predicts that each of the amino acid substitutions seen in classic FOP or its variants activates the ACVR1/ALK2 protein to enhance receptor signaling [68]. We observed genotype-phenotype correlation between some ACVR1/ALK2 mutations and the age of onset of heterotopic ossification or on embryonic skeletal development.
3. Skeletal Metamorphosis in FOP
3.1 FOP & Skeletal Metamorphosis
The ability of mature organisms to stabilize tissue phenotypes has enormous selective advantage. However, the mechanisms by which tissues and organs retain their distinct phenotypic stability throughout adult life have been shrouded in mystery. FOP exemplifies a pathological process of metamorphosis, the promiscuous transformation of one normal tissue or organ into another - not through the transdifferentiation of one mature cell into another [77], but rather through a pathological process in which the normal structure and function of one tissue or organ is destroyed and replaced with that of another tissue or organ [47].
The histologic stages of skeletal metamorphosis in FOP have been well-described [47,66,78–80]. Skeletal metamorphosis in FOP begins with a catabolic phase in which a soft tissue injury triggers an inflammatory mononuclear cell infiltrate, and leads to widespread muscle cell injury and death. This catabolic phase is followed rapidly by an anabolic phase in which a highly angiogenic fibroproliferative lesion forms and matures through an endochondral process that culminates in the formation of a new heterotopic bone. As the process of metamorphosis continues and spreads to contiguous and adjacent sites, the new skeletal elements ramify to form a disabling second skeleton of heterotopic bone.
Inflammatory signals appear to be required for the initiation of skeletal regeneration and heterotopic ossification [61–64]. Cells of the innate immune system derived from hematopoietic precursors have been implicated in the skeletal metamorphosis of FOP [62]. However, the definitive contribution of hematopoietic cells to the pathogenesis of skeletal metamorphosis has, until recently, remained obscure. Examination of a patient with FOP who coincidentally developed intercurrent aplastic anemia demonstrated that bone marrow transplantation does not cure FOP, most likely because the hematopoietic cell population is not the target, or at least not the dominant target, of the intrinsic dysregulation of the BMP signaling pathway in FOP. However, following transplantation of bone marrow from a normal donor, immunosuppression of the immune system appeared to ameliorate activation of skeletal metamorphosis in the genetically susceptible chimeric host. Moreover, even a normal functioning innate immune system is apparently sufficient to trigger an FOP flare-up in the genetically susceptible chimera [62]. Thus, innate immune cells of hematopoietic origin may be required to trigger FOP lesions, but even normal cells of the innate immune system may be capable of doing this in an individual who harbors connective tissue progenitor cells that express the FOP metamorphogene [62].
Which connective tissue cells contribute to the fibroproliferative and chondrogenic mesenchymal anlagen in skeletal metamorphosis? Recent studies performed by two independent routes of investigation support that such cells arise from connective tissue progenitor cells that are, at least in part, of vascular origin [63,81]. The development of a knock-in mouse that replicates the identical mutation of classic FOP will facilitate the identification of the connective tissue progenitor cell(s) that is sensitive to dysregulated BMP signaling in FOP [65]. Such work is underway.
3.2. Principles of Skeletal Metamorphosis in FOP
A number of recent clinical and experimental observations, bone marrow transplantation studies, and progenitor lineage tracing experiments have elucidated a set of principles that guide our understanding of the process of metamorphosis in FOP and in animal models of the condition [61–64,82,83]. While basal activity of the mutant ACVR1/ALK2 receptor may be sufficient to drive the nearly invariant array of skeletal malformations seen in FOP patients, it appears insufficient to trigger episodic flare-ups of FOP that lead to progressive heterotopic ossification and cumulative disability. Data from FOP patients and from in vivo animal models of FOP strongly suggest that inflammatory signals, in response to soft tissue injury, mobilize resident connective tissue progenitor cells of vascular origin that contribute to every stage in the development of the heterotopic anlagen. These principles are summarized below and are depicted in Figure 2:
Figure 2. Working model of heterotopic ossification in FOP.
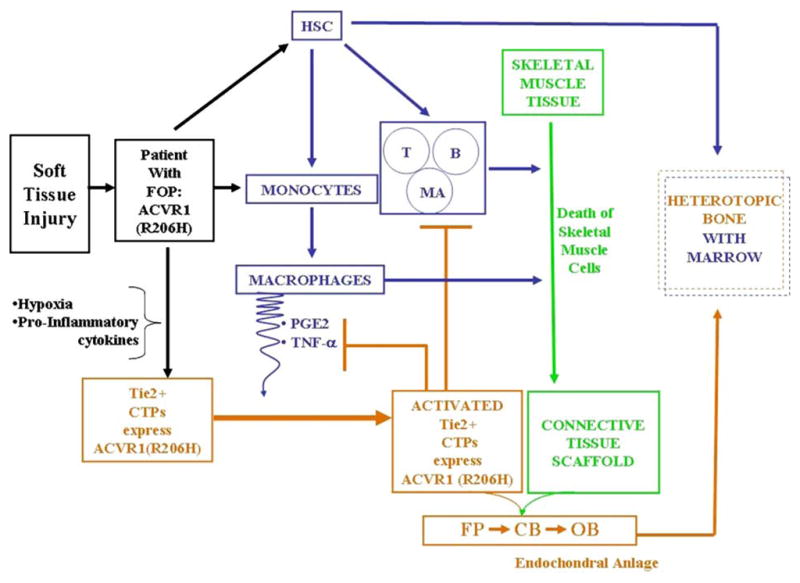
Injury to skeletal muscle and connective tissues leads to lymphocyte and monocyte invasion, macrophage and mast cell activation, tissue hypoxia, and upregulation of inflammatory cytokines and osteogenic factors that recruit vascular progenitor cells to form the heterotopic anlagen. Hypoxia and pro-inflammatory cytokines contribute to the Tie2+ angiogenic response. Hypoxia-related changes may further sensitize vascular progenitor cells in which the FOP metamorphogene is expressed. The inflammatory response to muscle injury, the ambient hypoxia, the presence of the promiscuously active FOP metamorphogene in vascular progenitor cells, and the cross-talk between cells of the innate and adaptive immune system stimulate the induction and propagation of an ectopic skeletal element. CTPs, connective tissue progenitor cells; HSC, hematopoietic stem cells; T, T-cells; B, B-cells; MA, mast cells; FP, fibroproliferative cells; CB, chondroblasts; OB, osteoblasts; PGE2, prostaglandin E2; TNFα, tumor necrosis factor-alpha; blue lines, hematopoietic-derived pathways; brown lines, CTP-derived pathways; blunt-end lines signify inhibitory pathways; arrows signify stimulatory pathways. (Modified from Lounev et al. J Bone Joint Surg Am 91: 652-663, 2009)
Inflammatory cells of hematopoietic origin trigger soft tissue metamorphosis to heterotopic bone [61,62].
Inflammatory signals, in response to soft tissue injury, and in part from inflammatory cells, are sufficient to induce the process of heterotopic ossification in a BMP-conducive environment [61–64].
Cells of the innate immune system, specifically of the monocyte/macrophage lineage, induce skeletal metamorphosis in a BMP conducive environment [61].
Cells of the adaptive immune system, specifically of the lymphocyte lineage, propagate the growth and expansion of metamorphic changes in a BMP conducive environment [61].
Immunosuppression ameliorates heterotopic ossification in a genetically susceptible host [62,64].
Bone marrow transplantation does not cure FOP and is ineffective in abrogating its progression, because even normal inflammatory cells are sufficient to trigger FOP in a genetically susceptible host [62].
Circulating osteoprogenitor cells of hematopoietic origin may seed areas of evolving endochondral ossification [82].
Inflammatory signals, in response to soft tissue injury, mobilize resident connective tissue progenitor cells of vascular origin that contribute to every stage in the development of the heterotopic anlagen [62,63,83].
Connective tissue progenitor cells of vascular origin transduce inflammatory signals in a BMP-conducive environment and contribute to every stage in the evolution of heterotopic anlagen [63].
Therapeutic regulation of progenitor cell population involved in FOP lesions holds promise for treatment of FOP and possibly other disorders of heterotopic ossification [63].
3.3. Prevention & Treatment of Skeletal Metamorphosis in FOP
The discovery of the FOP metamorphogene along with emerging insights into the pathophysiology of ACVR1/ALK2-mediated heterotopic ossification reveal at least four plausible approaches to the treatment and/or prevention of FOP, and possibly even more common forms of heterotopic ossification [2,45,84]. These approaches include:
Blocking the activity of the mutant FOP receptor
Blocking the inflammatory triggers
Blocking the responding connective tissue progenitor (CTP) cells
Altering the inductive and/or conducive microenvironments that promote heterotopic ossification.
Several recent articles provide a thorough review of this important topic [2,45,71,85]. Here, we will briefly highlight only one limited approach, that of blocking the mutant receptor using signal transduction inhibitors or siRNA.
3.3.1. Signal Transduction Inhibitor (STI) Strategy
Small molecule signal transduction inhibitors (STIs) are important molecular tools for investigating signal transduction pathways, and have the potential for development into powerful therapeutic agents [85–87]. Residues close to the ATP-binding site of ACVR1/ALK2 are being exploited to achieve selectivity, even among closely related receptor serine-threonine kinases such as BMPRIA/ALK3, BMPRIB/ALK6 and ACVR1/ALK2 [88]. Small soluble molecular inhibitors designed to specifically block ACVR1/ALK2 must have sufficient specificity, efficacy, tolerance to resistance, as well as acceptable safety profiles before they can be used in clinical trials of FOP.
Recently, Dorsomorphin, a small molecule STI was identified in a screen for compounds that perturb dorsoventral axis formation in zebrafish [89] Dorsomorphin and its derivatives inhibit all type I BMP receptors including ACVR1/ALK2, BMPR1A/ALK3, and BMPR1A/ALK6, and thus block BMP-mediated SMAD phosphorylation, target gene transcription, and osteogenic differentiation as shown in both in vitro and in vivo studies [46,51,64]. Additionally, Dorsomorphin blocks all BMP-specific SMAD signaling in FOP cells [46,51,72]. However, a safe and effective STI for FOP will likely inhibit ACVR1/ALK2 preferentially over ALK3 and ALK6. Also, while Dorsomorphin and its derivatives are able to inhibit SMAD activation, they have no appreciable affect on the ability of BMPs to activate p38 MAP kinases [89,90], a limb of the BMP signaling pathway that may plausibly play a role in the pathophysiology of FOP [43].
Compounds such as Dorsomorphin and its derivatives, as well as other more recently screened STIs, offer promise as pharmaceutical agents in the treatment of FOP and related disorders of heterotopic ossification [85]. Additional studies on BMP signaling will need to be evaluated in in vivo models of classic FOP to determine potential efficacy of this class of molecules in preventing inflammation-induced flare-ups of the condition.
3.3.2. Small Inhibitory RNA (siRNA) Strategy
Classic FOP results from a missense activating mutation in one of the two alleles of ACVR1/ALK2. Thus, strategies to specifically inhibit messenger RNA from the mutant allele while leaving messenger RNA from the normal allele unperturbed are desirable. Studies are underway to investigate the potential utility of a small inhibitory RNA (siRNA) strategy for treating FOP.
4. Concluding Remarks
FOP is a rare and illustrative genetic disorder that provides unique insight into the molecular, genetic and cellular principles of phenotype stability and metamorphosis. The single nucleotide heterozygous missense mutation that causes classic FOP in all affected individuals creates a metamorphogene that profoundly dysregulates BMP signaling in a highly conserved pathway involved in tissue development and repair. FOP is particularly instructive because it is caused by activating mutations in a BMP type I receptor that vastly alters not only body plan, but also tissue response to injury [91]. It is rare and perhaps unique that a single nucleotide mutation so vastly alters the identity of tissues and organ systems as a result of an inflammatory trigger and permits their promiscuous metamorphosis into other tissues and organs.
Identification of the molecular pathophysiology of the FOP metamorphogene reveals a critical and novel regulator of chondro-osseous differentiation that affects many physiological processes including morphogenesis, metamorphosis, and tissue maintenance and repair. The FOP metamorphogene and its encoded receptor are not only therapeutic targets for FOP, but also an important focal point for understanding a wide variety of developmental and repair processes under the control of BMP signaling, and for understanding the fundamental processes that regulate tissue stability in vertebrate tissue and organs.
As Thomas Maeder wrote, “FOP and its problems lie at the crossroads of several seemingly unrelated disciplines. Answers to questions that FOP poses will also address grander issues of how the body first creates its shape and then knows where to stop, how tissues decide to become what they are, and why they don’t turn into something else (Figure 2) [92].
As William Harvey, the discoverer of the circulatory system, noted, “Nature is nowhere accustomed more openly to display her secret mysteries than in cases where she shows traces of her workings apart from the beaten path; nor is there any better way to advance the proper practice of medicine than to give our minds to the discovery of the usual law of nature, by the careful investigation of cases of rarer forms of disease. For it has been found in all things, that what they contain of useful or of applicable (nature) is hardly perceived unless we are deprived of them or they become deranged in some way [87].”
Acknowledgments
This work was supported in part by the International Fibrodysplasia Ossificans Progressiva Association, the Center for Research in FOP and Related Disorders, the Ian Cali Endowment for FOP Research, the Whitney Weldon Endowment for FOP Research, the Isaac & Rose Nassau Professorship of Orthopaedic Molecular Medicine, and by grants from the Rita Allen Foundation, and the US National Institutes of Health (NIH R01-AR41916). Portions of this work were modified and adapted from the following references: 2, 45, 47, 71, 72, 85.
Biographies
 Frederick S. Kaplan, M.D. is The Isaac & Rose Nassau Professor of Orthopaedic Molecular Medicine, Chief of the Division of Orthopaedic Molecular Medicine, and Director of The Center for Research in FOP & Related Disorders at the University of Pennsylvania. He is an alumnus of The Johns Hopkins University (1972), and The Johns Hopkins University School of Medicine (1976), and was Chief Resident in Orthopaedic Surgery at The Hospital of The University of Pennsylvania and The Children’s Hospital of Philadelphia from 1980 to 1981.
Frederick S. Kaplan, M.D. is The Isaac & Rose Nassau Professor of Orthopaedic Molecular Medicine, Chief of the Division of Orthopaedic Molecular Medicine, and Director of The Center for Research in FOP & Related Disorders at the University of Pennsylvania. He is an alumnus of The Johns Hopkins University (1972), and The Johns Hopkins University School of Medicine (1976), and was Chief Resident in Orthopaedic Surgery at The Hospital of The University of Pennsylvania and The Children’s Hospital of Philadelphia from 1980 to 1981.
Kaplan was a Hartford Foundation Research Fellow in human genetics and molecular biology from 1989 to 1991 in the laboratory of his mentor and friend Dr. Michael Zasloff at The Children’s Hospital of Philadelphia. This experience led to his exploration of the mechanisms for heterotopic bone formation and skeletal metamorphosis in several disabling childhood diseases and to identifying the molecular pathology of fibrodysplasia ossificans progressiva (FOP). Kaplan also identified another genetic bone disorder of children, progressive osseous heteroplasia (POH) and co-discovered the genes that cause both FOP and POH. His lifelong research goals are to develop more effective treatments and a cure for FOP and POH.
Kaplan is the chief medical and scientific advisor of The International FOP Association, The Progressive Osseous Heteroplasia Association, and The Melorheostosis Association. He serves on the Board of Directors of The Paget Foundation.
Kaplan has received numerous international awards and honors. In July 2006, he was named one of 15 Great Americans by NEWSWEEK.
 Robert J. Pignolo, MD, PhD, is Assistant Professor in the Department of Medicine, Director of the Ralston-Penn Clinic for Osteoporosis and Related Bone Disorders, and the Ian Cali Clinical & Research Scholar in the Center for Research in FOP and Related Disorders at the University of Pennsylvania School of Medicine. Dr. Pignolo is a clinician-scientist with expertise in metabolic bone disease, gerontology, geriatric medicine, genetic and non-hereditary heterotopic ossification, as well as cell and molecular biology, especially related to cell proliferation, aging, and osteoblast differentiation. Dr. Pignolo’s research focuses on osteoblast differentiation in conditions that reflect either impaired or inappropriate osteogenesis. He studies age-related bone loss and disorders of extraskeletal ossification, seemingly disparate conditions linked by the common thread of dysregulated osteoblast differentiation.
Robert J. Pignolo, MD, PhD, is Assistant Professor in the Department of Medicine, Director of the Ralston-Penn Clinic for Osteoporosis and Related Bone Disorders, and the Ian Cali Clinical & Research Scholar in the Center for Research in FOP and Related Disorders at the University of Pennsylvania School of Medicine. Dr. Pignolo is a clinician-scientist with expertise in metabolic bone disease, gerontology, geriatric medicine, genetic and non-hereditary heterotopic ossification, as well as cell and molecular biology, especially related to cell proliferation, aging, and osteoblast differentiation. Dr. Pignolo’s research focuses on osteoblast differentiation in conditions that reflect either impaired or inappropriate osteogenesis. He studies age-related bone loss and disorders of extraskeletal ossification, seemingly disparate conditions linked by the common thread of dysregulated osteoblast differentiation.
 Eileen M. Shore, Ph.D. is an Associate Professor at the University of Pennsylvania School of Medicine in the Departments of Orthopaedic Surgery and Genetics, and the Co-Director of the Center for Research in FOP and Related Disorders. She is Scientific Advisor to the International Fibrodysplasia Ossificans Progressiva Association and the Progressive Osseous Heteroplasia Association. Dr. Shore is a long-standing and active member of the ASBMR and currently serves as President of the AIMM Board of Directors.
Eileen M. Shore, Ph.D. is an Associate Professor at the University of Pennsylvania School of Medicine in the Departments of Orthopaedic Surgery and Genetics, and the Co-Director of the Center for Research in FOP and Related Disorders. She is Scientific Advisor to the International Fibrodysplasia Ossificans Progressiva Association and the Progressive Osseous Heteroplasia Association. Dr. Shore is a long-standing and active member of the ASBMR and currently serves as President of the AIMM Board of Directors.
Dr. Shore attended the University of Notre Dame (B.S., Biology), Indiana University (M.A., Biology), and the University of Pennsylvania (Ph.D., Cell and Molecular Biology) with post-doctoral training at the Institute for Cancer Research, Fox Chase Cancer Center in Philadelphia.
Dr. Shore has applied her experience in molecular biology and genetics to her research interests in cell differentiation and development and their roles in human genetic disease. Her research focuses on fibrodysplasia ossificans progressiva (FOP) and progressive osseous heteroplasia (POH), two rare disorders characterized by de novo formation of bone. Dr. Shore’s work led to the discovery of the mutated genes in both conditions. She currently investigates the cellular targets and molecular pathways involved in induction of bone formation with the goal of developing treatments for these and other more common bone disorders.
Footnotes
Conflicts of Interest: The authors have no personal relationships with other people or organizations that could inappropriately influence or bias their work. The authors declare that there are no conflicts of interest in this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Frederick S. Kaplan, Departments of Orthopaedic Surgery and Medicine, and The Center for Research in FOP & Related Disorders, The University of Pennsylvania School of Medicine, Philadelphia, PA 19104, USA
Robert J. Pignolo, Department of Medicine, and The Center for Research in FOP & Related Disorders, The University of Pennsylvania School of Medicine, Philadelphia, PA 19104, USA
Eileen M. Shore, Departments of Orthopaedic Surgery and Genetics, and The Center for Research in FOP & Related Disorders, The University of Pennsylvania School of Medicine, Philadelphia, PA 19104, USA
References
- 1.Kaplan FS, Glaser DL, Shore EM, Deirmengian GK, Gupta R, Delai P, Morhart P, Smith R, Le Merrer M, Rogers JG, Connor JM, Kitterman JA. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone & Miner Metab. 2005;3:183–188. [Google Scholar]
- 2.Kaplan FS, LeMerrer M, Glaser DL, Pignolo RJ, Goldsby RE, Kitterman JA, Groppe J, Shore EM. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191–205. doi: 10.1016/j.berh.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shore EM, Feldman GJ, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clin Rev Bone & Miner Metab. 2005;3:201–204. [Google Scholar]
- 4.Cohen RB, Hahn GV, Tabas J, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. J Bone Joint Surg. 1993;75-A:215–219. doi: 10.2106/00004623-199302000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS. Age and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1994;301:243–248. [PubMed] [Google Scholar]
- 6.Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Joint Surg. 1982;64(1):76–83. doi: 10.1302/0301-620X.64B1.7068725. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan FS, Strear CM, Zasloff MA. Radiographic and scintigraphic features of modeling and remodeling in the heterotopic skeleton of patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1994;304:238–247. [PubMed] [Google Scholar]
- 8.Mahboubi S, Glaser DL, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva. Pediatr Radiol. 2001;31:307–314. doi: 10.1007/s002470100447. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan FS, Xu M, Glaser DL, Collins F, Connor M, Kitterman J, Sillence D, Zackai E, Ravitsky V, Zasloff M, Ganguly A, Shore EM. Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics. 2008;121:e1295–e1300. doi: 10.1542/peds.2007-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics. 2005;116:654–661. doi: 10.1542/peds.2005-0469. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan FS, Glaser DL. Thoracic insufficiency syndrome in patients with fibrodysplasia ossificans progressiva. Clin Rev Bone & Miner Metab. 2005;3:213–216. [Google Scholar]
- 12.Kussmaul WG, Esmail AN, Sagar Y, Ross J, Gregory S, Kaplan FS. Pulmonary and cardiac function in advanced fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1998;346:104–109. [PubMed] [Google Scholar]
- 13.Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- 14.Gazzerro E, Canalis E. Bone morphogenetic proteins and their antagonists. Rev Endocr Metab Disord. 2006;7:51–65. doi: 10.1007/s11154-006-9000-6. [DOI] [PubMed] [Google Scholar]
- 15.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 17.Urist MR. Bone formation by autoinduction. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- 18.Schmierer B, Hill CS. TGF-β-SMAD signal transduction: molecular specificity and functional flexibility. Nature Reviews Mol Cell Bio. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 19.Wagner TU. Bone morphogenetic protein signaling in stem cells – one signal many consequences. FEBS Journal. 2007;274:2968–2976. doi: 10.1111/j.1742-4658.2007.05839.x. [DOI] [PubMed] [Google Scholar]
- 20.Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. BMPR1A and BMPR1B have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA. 2005;102(14):5062–5067. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derynck R, Akhurst RJ. Differentiation plasticity regulated by the TGF-β family proteins in development and disease. Nature Cell Biology. 2007;9:1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- 22.Harradine KA, Akhurst RJ. Mutations of TGF-β signaling molecules in human disease. Annals Medicine. 2006;38:403–414. doi: 10.1080/07853890600919911. [DOI] [PubMed] [Google Scholar]
- 23.Hartung A, Sieber C, Knaus P. Yin and Yang in BMP signaling: impact on the pathology of diseases and potential for tissue regeneration. Signal Transduction. 2006;6:314–328. [Google Scholar]
- 24.Yoon BS, Lyons KM. Multiple functions of BMPs in chondrogenesis. J Cell Biochem. 2004;93(1):93–103. doi: 10.1002/jcb.20211. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y-G, Liu F, Massague J. Mechanism of TGF-β receptor inhibition by FKBP12. The EMBO J. 1997;16:3866–3876. doi: 10.1093/emboj/16.13.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huse M, Chen Y-G, Massague J, Kuriyan J. Crystal structure of the cytosplasmic domain of the type I TGF-β receptor in complex with FKBP12. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- 27.Huse M, Muir TW, Xu L, Chen YG, Kuriyan J, Massagué J. The TGF-β receptor activation process: an inhibitor-to-substrate binding switch. Mol Cell. 2001;8:671–682. doi: 10.1016/s1097-2765(01)00332-x. [DOI] [PubMed] [Google Scholar]
- 28.Wang T, Li B-Y, Danielson PD, Shah PC, Rockwell S, Lechleider RJ, Martin J, Manganaro T, Donahoe PK. The immunophilin FKBP12 functions as a common inhibitor of the TGF-β family type I receptors. Cell. 1996;86:435–444. doi: 10.1016/s0092-8674(00)80116-6. [DOI] [PubMed] [Google Scholar]
- 29.Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K. Smurf 1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276:12477–12480. doi: 10.1074/jbc.C100008200. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi T, Kurisaki A, Yamakawa N, Minakuchi K, Sugino H. FKBP12 functions as an adaptor of the Smad7-Smurf1 complex on activin type I receptor. J Mol Endocrinology. 2006;36:569–579. doi: 10.1677/jme.1.01966. [DOI] [PubMed] [Google Scholar]
- 31.Kaplan FS, Tabas JA, Zasloff MA. Fibrodysplasia ossificans progressiva: A clue from the fly? Calcif Tiss Int. 1990;47:117–125. doi: 10.1007/BF02555995. [DOI] [PubMed] [Google Scholar]
- 32.Gannon FH, Kaplan FS, Olmsted E, Finkel G, Zasloff MA, Shore EM. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Hum Pathol. 1997;28:339–343. doi: 10.1016/s0046-8177(97)90133-7. [DOI] [PubMed] [Google Scholar]
- 33.Olmsted EA, Kaplan FS, Shore EM. Bone morphogenetic protein 4 regulation in fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 2003;408:331–343. doi: 10.1097/00003086-200303000-00044. [DOI] [PubMed] [Google Scholar]
- 34.Roush W. Protein builds second skeleton. Science. 1996;273:1170. doi: 10.1126/science.273.5279.1170. [DOI] [PubMed] [Google Scholar]
- 35.Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, Kaplan FS. Over-expression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Engl J Med. 1996;335:555–561. doi: 10.1056/NEJM199608223350804. [DOI] [PubMed] [Google Scholar]
- 36.Ahn J, Serrano de la Peña L, Shore EM, Kaplan FS. Paresis of a bone morphogenetic protein-antagonist response in a genetic disorder of heterotopic skeletogenesis. J Bone Joint Surg. 2003;85-A:667–674. doi: 10.2106/00004623-200304000-00013. [DOI] [PubMed] [Google Scholar]
- 37.Kaplan FS, Fiori JL, Ahn J, Billings PC, Shore EM. Dysregulation of BMP4 receptor trafficking and signaling in fibrodysplasia ossificans progressiva. Clin Rev Bone & Miner Metab. 2005;3:217–223. [Google Scholar]
- 38.Serrano de la Peña L, Billings PC, Fiori JL, Ahn J, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J Bone Miner Res. 2005;20:1168–1176. doi: 10.1359/JBMR.050305. [DOI] [PubMed] [Google Scholar]
- 39.O’Connell MP, Billings PC, Fiori JL, Deirmengian G, Roach HI, Shore EM, Kaplan FS. HSPG modulation of BMP signaling in fibrodysplasia ossificans progressiva cells. J Cellular Biochem. 2007;102:1493–1503. doi: 10.1002/jcb.21370. [DOI] [PubMed] [Google Scholar]
- 40.Jiao X, Billings PC, O’Connell MP, Kaplan FS, Shore EM, Glaser DL. Heparan sulfate proteoglycans (HSPGs) modulate BMP2 osteogenic bioactivity in C2C12 cells. J Biol Chem. 2007;282 (2):1080–1086. doi: 10.1074/jbc.M513414200. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan FS, Fiori J, Serrano de la Peña L, Ahn J, Billings PC, Shore EM. Dysregulation of the BMP4 signaling pathway in fibrodysplasia ossificans progressiva. Ann NY Acad Sci. 2006;1068:54–65. doi: 10.1196/annals.1346.008. [DOI] [PubMed] [Google Scholar]
- 42.Billings PC, Fiori JL, Bentwood JL, O’Connell MP, Jiao X, Nussbaum B, Caron RJ, Shore EM, Kaplan FS. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva. J Bone Miner Res. 2008;23:305–313. doi: 10.1359/JBMR.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiori JL, Billings PC, Serrano de la Peña L, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP) J Bone Miner Res. 2006;21:902–909. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- 44.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho T-J, Choi IH, Connor JM, Delai P, Glaser DL, Le Merrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genetics. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 45.Kaplan FS, Glaser DL, Pignolo RJ, Shore EM. A new era of fibrodysplasia ossificans progressiva (FOP): a druggable target for the second skeleton. Exp Opin Biol Ther. 2004;7:705–712. doi: 10.1517/14712598.7.5.705. [DOI] [PubMed] [Google Scholar]
- 46.Shen Q, Little SC, Xu M, Ast C, Katagiri T, Mundlos S, Seemann P, Kaplan FS, Mullins MC, Shore EM. Fibrodysplasia ossificans progressiva ACVR1 (R206H) mutation activates BMP-independent chondrogenesis and ventralization of zebrafish embryos. J Clin Invest. 2009 doi: 10.1172/JCI37412. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaplan FS, Shore EM. Morphogen receptor genes and metamorphogenes: skeleton keys to metamorphosis. Ann NY Acad Sci. 2007;1116:113–133. doi: 10.1196/annals.1402.039. [DOI] [PubMed] [Google Scholar]
- 48.Mishina Y, Crombie R, Bradley A, Behringer RR. Multiple roles for activin-like kinase-2 signaling during mouse embryogenesis. Dev Biol. 1999;213(2):314–26. doi: 10.1006/dbio.1999.9378. [DOI] [PubMed] [Google Scholar]
- 49.Zhang D, Schwarz EM, Rosier RN, Zuscik MJ, Puzas JE, O’Keefe RJ. ALK2 functions as a BMP type I receptor and induces Indian Hedgehog in chondrocytes during skeletal development. J Bone Miner Res. 2003;18:1593–1604. doi: 10.1359/jbmr.2003.18.9.1593. [DOI] [PubMed] [Google Scholar]
- 50.Akiyama S, Katagiri T, Namiki M, Yamaji N, Yamamoto N, Miyama K, Shibuya H, Ueno N, Wozney JM, Suda T. Constitutively active BMP type I receptors transduce BMP2 signals without the ligand in C2C12 myoblasts. Exp Cell Res. 1997;235:362–369. doi: 10.1006/excr.1997.3680. [DOI] [PubMed] [Google Scholar]
- 51.Fukada T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem. 2009;284:7149–7156. doi: 10.1074/jbc.M801681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Little SC, Mullins MC. Bone morphogenetic protein heterodimers assemble heteromeric type I receptor complexes to pattern the dorsorventral axis. Nature Cell Biol. 2009;11:637–643. doi: 10.1038/ncb1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bangi E, Wharton K. Dual function of the Drosophila Alk1/Alk2 ortholog Saxophone shapes the BMP activity gradient in the wing imaginal disc. Development. 2006;133(17):3295–3303. doi: 10.1242/dev.02513. [DOI] [PubMed] [Google Scholar]
- 54.Groppe JC, Shore EM, Kaplan FS. Functional modeling of the ACVR1 (R206H) mutation in FOP. Clin Orthop Rel Res. 2007;462:87–92. doi: 10.1097/BLO.0b013e318126c049. [DOI] [PubMed] [Google Scholar]
- 55.Fuentealba LC, Eivers E, Ikeda A, Hurtado C, Kuroda H, Pera EM, De Robertis EM. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/SMAD1 signal. Cell. 2007;131:980–993. doi: 10.1016/j.cell.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaplan FS, Shore EM, Gupta R, Billings PC, Glaser DL, Pignolo RJ, Graf D, Kamoun M. Immunological features of fibrodysplasia ossificans progressiva and the dysregulated BMP4 Pathway. Clin Rev Bone & Miner Metab. 2005;3:189–193. [Google Scholar]
- 57.Lanchoney TF, Cohen RB, Rocke DM, Zasloff MA, Kaplan FS. Permanent heterotopic ossification at the injection site after diphtheria-tetanus-pertussis immunizations in children who have fibrodysplasia ossificans progressiva. J Pediatrics. 1995;126:762–764. doi: 10.1016/s0022-3476(95)70408-6. [DOI] [PubMed] [Google Scholar]
- 58.Luchetti W, Cohen RB, Hahn GV, Rocke DM, Helpin M, Zasloff M, Kaplan FS. Severe restriction in jaw movement after routine injection of local anesthetic in patients who have fibrodysplasia ossificans progressiva. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;81:21–25. doi: 10.1016/s1079-2104(96)80141-7. [DOI] [PubMed] [Google Scholar]
- 59.Scarlett RF, Rocke DM, Kantanie S, Patel JB, Shore EM, Kaplan FS. Influenza-like viral illnesses and flare-ups of fibrodysplasia ossificans progressiva (FOP) Clin Orthop Rel Res. 2004;423:275–279. doi: 10.1097/01.blo.0000129557.38803.26. [DOI] [PubMed] [Google Scholar]
- 60.Hebela N, Shore EM, Kaplan FS. Three pairs of monozygotic twins with fibrodysplasia ossificans progressiva: the role of environment in the progression of heterotopic ossification. Clin Rev Bone & Miner Metab. 2005;3:205–208. [Google Scholar]
- 61.Kan L, Liu Y, McGuire TL, Berger DMP, Atwatramani RB, Dymecki SM, Kessler JA. Dysregulation of local stem/progenitor cells as a common cellular mechanism for heterotopic ossification. Stem Cells. 2009;27:150–156. doi: 10.1634/stemcells.2008-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaplan FS, Glaser DL, Shore EM, Pignolo RJ, Xu M, Zhang Y, Senitzer D, Forman SJ, Emerson SG. Hematopoietic stem-cell contribution to ectopic skeletogenesis. J Bone Joint Surg Am. 2007;89:347–357. doi: 10.2106/JBJS.F.00472. [DOI] [PubMed] [Google Scholar]
- 63.Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment ADA, Shore EM, Glaser EM, Goldhamer DJ, Kaplan FS. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surgery Am. 2009;91:652–663. doi: 10.2106/JBJS.H.01177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD. BMP type I receptor inhibition prevents ectopic ossification in mouse model of fibrodysplasia ossificans progressiva (FOP) Nature Medicine. 2008;14:1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chakkalakal SA, Zhang D, Raabe T, Richa J, Hankenson K, Kaplan FS, Shore EM. ACVR1 knock-in mouse model for fibrodysplasia ossificans progressiva. J Bone Miner Res. 2008;23(suppl 1) [Google Scholar]
- 66.Glaser DL, Economides AN, Wang L, Liu X, Kimble RD, Fandl JP, Wilson JM, Stahl N, Kaplan FS, Shore EM. In vivo somatic cell gene transfer of an engineered noggin mutein prevents BMP4-induced heterotopic ossification. J Bone Joint Surg. 2003;85-A:2332–2342. doi: 10.2106/00004623-200312000-00010. [DOI] [PubMed] [Google Scholar]
- 67.Kan L, Hu M, Gomes WA, Kessler JA. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am J Pathol. 2004;165:1107–1115. doi: 10.1016/S0002-9440(10)63372-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaplan FS, Xu M, Seemann P, Connor M, Glaser DL, Carroll L, Delai P, Fastnacht-Urban E, Forman SJ, Gillessen-Kaesbach G, Hoover-Fong J, Köster B, Pauli RM, Reardon W, Zaidi S-A, Zasloff M, Morhart R, Mundlos S, Groppe J, Shore EM. Classic and atypical FOP phenotypes caused by mutations in the BMP type I receptor ACVR1. Human Mutation. 2009;30:379–390. doi: 10.1002/humu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deirmengian GK, Hebela NM, O’Connell M, Glaser DL, Shore EM, Kaplan FS, Deirmengian GK. Proximal tibial osteochondromas in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am. 2008;90:366–374. doi: 10.2106/JBJS.G.00774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schaffer AA, Kaplan FS, Tracy MR, O’Brien ML, Dormans JP, Shore EM, Harland RM, Kusumi K. Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome. Spine. 2005;30:1379–1385. doi: 10.1097/01.brs.0000166619.22832.2c. [DOI] [PubMed] [Google Scholar]
- 71.Kaplan FS, Groppe J, Seemann P, Pignolo RJ, Shore EM. Fibrodysplasia ossificans progressiva: developmental implications of a novel metamorphogene. In: Bronner, Farah-Carson, Roach, editors. Chapter 14 in Bone Development. Vol. 6. 2009. Topics in Bone Biology. in press. [Google Scholar]
- 72.Kaplan FS, Shen Q, Lounev, Seemann P, Groppe J, Katagiri T, Pignolo RJ, Shore EM. Skeletal metamorphosis in fibrodysplasia ossificans progressiva (FOP) J Bone Miner Metab. 2008;26:521–530. doi: 10.1007/s00774-008-0879-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kobayashi T, Lyons KM, McMahon AP, Kronenberg HM. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. PNAS. 2005;102:18023–18027. doi: 10.1073/pnas.0503617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- 75.Wang H, Shore EM, Kaplan FS, Groppe J, Pignolo RJ. Hypoxia promotes ligand-independent activation of the ACVR1 (R206H) mutant receptor in C2C12 cells. J Bone Miner Res. 2008;23:s433. [Google Scholar]
- 76.Olmsted-Davis E, Gannon FH, Ozen M, Ittmann MM, Gugala Z, Hipp JA, Moran KM, Fouletier-Dilling CM, Schumara-Martin S, Lindsey RW, Heggeness MH, Brenner MK, Davis AR. Hypoxic adipocytes pattern early heterotopic bone formation. Am J Pathol. 2007;170:620–632. doi: 10.2353/ajpath.2007.060692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cobaleda C, Jochum W, Busslinger M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature. 2007;449:473–477. doi: 10.1038/nature06159. [DOI] [PubMed] [Google Scholar]
- 78.Gannon FH, Valentine BA, Shore EM, Zasloff MA, Kaplan FS. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1998;346:19–25. [PubMed] [Google Scholar]
- 79.Kaplan FS, Zasloff MA. The histopathology of fibrodysplasia ossificans progressiva: an endochondral process. J Bone Joint Surg. 1993;75-A:220–230. doi: 10.2106/00004623-199302000-00009. [DOI] [PubMed] [Google Scholar]
- 80.Pignolo RJ, Suda RK, Kaplan FS. The fibrodysplasia ossificans progressiva lesion. Clin Rev Bone & Miner Metab. 2005;3:195–200. [Google Scholar]
- 81.Hegyi L, Gannon FH, Glaser DL, Shore EM, Kaplan FS, Shanahan CM. Stromal cells of fibrodysplasia ossificans progressiva lesions express smooth muscle lineage markers and the osteogenic transcription factor Runx2/Cbfa-1: clues to a vascular origin of heterotopic ossification. J Pathol. 2003;201:141–148. doi: 10.1002/path.1413. [DOI] [PubMed] [Google Scholar]
- 82.Suda RK, Billings PC, Egan KP, Kim JH, McCarrick-Walmsley R, Glaser DL, Porter DL, Shore EM, Pignolo RJ. Circulating osteogenic precursor cells in heterotopic bone formation. Stem Cells. 2009 doi: 10.1002/stem.150. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gannon FH, Glaser D, Caron R, Thompson LD, Shore EM, Kaplan FS. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum Pathol. 2001;32:842–848. doi: 10.1053/hupa.2001.26464. [DOI] [PubMed] [Google Scholar]
- 84.Kaplan FS, Glaser DL, Hebela N, Shore EM. Heterotopic ossification. J Am Acad Orthop Surg. 2004;12:116–125. doi: 10.5435/00124635-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 85.Kaplan F, Groppe J, Shore EM. When one skeleton is enough: approaches and strategies for the treatment of fibrodysplasia ossificans progressiva. Drug Discovery Today: Therapeutic Strategies. 2009 doi: 10.1016/j.ddstr.2008.11.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fishman MC, Porter JA. A new grammar for drug discovery. Nature. 2005;437:491–493. doi: 10.1038/437491a. [DOI] [PubMed] [Google Scholar]
- 87.Kaplan FS. The key to the closet is the key to the kingdom: a common lesson of rare diseases. Orphan Disease Update. 2006;24(3):1–9. [Google Scholar]
- 88.Cuny GD, Yu PB, Laha JK, Xing X, Liu JF, Lai CS, Deng DY, Sachidanandan C, Bloch KD, Peterson RT. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorganic Medicinal Chemistry Letters. 2008;18:4388–4392. doi: 10.1016/j.bmcl.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Block KD, Peterson RT. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nature Chem Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anderson GJ, Darshan D. Small-molecule dissection of BMP signaling. Nature Chem Biol. 2008;4:15–16. doi: 10.1038/nchembio0108-15. [DOI] [PubMed] [Google Scholar]
- 91.Ingber DE, Levin M. What lies at the interface of regenerative medicine and developmental biology? Development. 2007;134:2541–2547. doi: 10.1242/dev.003707. [DOI] [PubMed] [Google Scholar]
- 92.Maeder T. A few hundred people turned to bone. Atlantic Monthly. 1988;281:81–89. [Google Scholar]