

Summary
Background
Fibrillar amyloid-β (Aβ) is thought to begin accumulating in the brain many years before the onset of clinical impairment in patients with Alzheimer’s disease. By assessing the accumulation of Aβ in people at risk of genetic forms of Alzheimer’s disease, we can identify how early preclinical changes start in individuals certain to develop dementia later in life. We sought to characterise the age-related accumulation of Aβ deposition in presenilin 1 (PSEN1) E280A mutation carriers across the spectrum of preclinical disease.
Methods
Between Aug 1 and Dec 6, 2011, members of the familial Alzheimer’s disease Colombian kindred aged 18–60 years were recruited from the Alzheimer’s Prevention Initiative’s registry at the University of Antioquia, Medellín, Colombia. Cross-sectional assessment using florbetapir PET was done in symptomatic mutation carriers with mild cognitive impairment or mild dementia, asymptomatic carriers, and asymptomatic non-carriers. These assessments were done at the Banner Alzheimer’s Institute in Phoenix, AZ, USA. A cortical grey matter mask consisting of six predefined regions. was used to measure mean cortical florbetapir PET binding. Cortical-to-pontine standard-uptake value ratios were used to characterise the cross-sectional accumulation of fibrillar Aβ deposition in carriers and non-carriers with regression analysis and to estimate the trajectories of fibrillar Aβ deposition.
Findings
We enrolled a cohort of 11 symptomatic individuals, 19 presymptomatic mutation carriers, and 20 asymptomatic non-carriers, ranging in age from 20 to 56 years. There was greater florbetapir binding in asymptomatic PSEN1 E280A mutation carriers than in age matched non-carriers. Fibrillar Aβ began to accumulate in PSEN 1E280A mutation carriers at a mean age of 28·2 years (95% CI 27·3–33·4), about 16 years and 21 years before the predicted median ages at mild cognitive impairment and dementia onset, respectively. 18F florbetapir binding rose steeply over the next 9·4 years and plateaued at a mean age of 37·6 years (95% CI 35·3–40·2), about 6 and 11 years before the expected respective median ages at mild cognitive impairment and dementia onset. Prominent florbetapir binding was seen in the anterior and posterior cingulate, precuneus, and parietotemporal and frontal grey matter, as well as in the basal ganglia. Binding in the basal ganglia was not seen earlier or more prominently than in other regions.
Interpretation
These findings contribute to the understanding of preclinical familial Alzheimer’s disease and help set the stage for assessment of amyloid-modifying treatments in the prevention of familial Alzheimer’s disease.
Funding
Avid Radiopharmaceuticals, Banner Alzheimer’s Foundation, Nomis Foundation, Anonymous Foundation, Forget Me Not Initiative, Colciencias, National Institute on Aging, and the State of Arizona.
Introduction
Fibrillar amyloid-β (Aβ) deposition is a cardinal neuropathological feature of Alzheimer’s disease.1 Findings from histopathological, PET, and CSF studies show that fibrillar Aβ is present in a substantial number of cognitively normal old adults and suggest that it might reach neuropathologically diagnostic concentrations at least 10 years before the onset of dementia.2–6 These findings have led investigators to propose biomarker models to characterise the preclinical stages of Alzheimer’s disease.7
Longitudinal studies using amyloid PET imaging may enable researchers to identify the extent to which cortical fibrillar Aβ deposition in asymptomatic individuals predicts subsequent clinical decline. Recent failures of anti-amyloid therapies in patients with Alzheimer’s disease have highlighted the need for disease-modifying treatments to be used before the onset of clinical symptoms, when the neuropathological features of Alzheimer’s disease are already extensive.8 Research is ongoing to assess biomarkers of brain function and pathological abnormalities in cognitively normal people at genetic risk to characterise the changes associated with their predisposition to Alzheimer’s disease. These studies include the comparison of people with and without the apolipoprotein E (APOE) ε4 allele,4,9–11 a major susceptibility gene for late-onset Alzheimer’s disease, and the comparison of carriers and non-carriers of uncommon mutations that cause autosomal dominant early-onset Alzheimer’s disease.2,12–14
As recently shown by the Dominantly Inherited Alzheimer’s Network (DIAN),2 in a study of a heterogeneous cohort of presymptomatic mutation carriers of various autosomal dominant Alzheimer’s disease mutations, the investigation of genetically driven forms of Alzheimer’s disease can provide insights into pre-symptomatic biomarker features associated with predictable future clinical outcomes. Cross-sectional studies of autosomal dominant Alzheimer’s disease can be undertaken to analyse an individual’s biomarker profile in comparison with clinical outcomes in affected family members, such as their age at onset of clinical symptoms, and enable a better understanding of the predictive characteristics of the biomarker under study, that could hypothetically emulate longitudinal outcomes.
Because different autosomal dominant mutations in the presenilin 1 (PSEN1), PSEN2, and amyloid precursor protein (APP) genes vary in clinical presentation and pathological features,15,16 assessing presymptomatic biomarkers in a single-mutation kindred would be useful to reduce this heterogeneity. The PSEN1 E280A (Glu280Ala) mutation kindred comprises an extended family living in the region of Antioquia, Colombia, with about 5000 members, of whom about 1500 carry this mutation, which causes Alzheimer’s disease.17 Findings from 15 years of observational studies from 1995 to 2010 in 449 PSEN1 E280A mutation carriers showed a predictable clinical course with a median age of 44 years (95% CI 43–45) at onset of mild cognitive impairment (MCI) and 49 years (49–50) at onset of dementia.12
In a previous study,18 our group assessed young PSEN1 E280A mutation carriers and non-carriers, and found significantly higher CSF and plasma Aβ1–42 concentrations in mutation carriers, consistent with Aβ1–42 overproduction, as well as functional and structural MRI abnormalities comparable to those reported in patients with sporadic late-onset Alzheimer’s disease. We subsequently sought to characterise the accumulation of cortical Aβ deposition in mutation carriers across a broad age range, that is, across the spectrum of stages of preclinical disease. We undertook florbetapir (18F) amyloid PET analyses in presymptomatic PSEN1 E280A mutation carriers, non-carriers, and symptomatic patients from this single-mutation autosomal dominant Alzheimer’s disease cohort.
Methods
Study design and participants
Between Aug 1 and Dec 6, 2011, members of the PSEN1 E280A mutation Colombian kindred were recruited from the Alzheimer’s Prevention Initiative (API) registry. The API registry was developed in 2010 to establish a database of all living family members who were known through ongoing observational studies since 1995,12 in preparation for planned biomarker and preclinical treatment trials. This registry was searched for individuals potentially meeting the enrolment criteria for the present study, with potential volunteers contacted by one of the study staff (MGG) by phone.
Participants had to be 18–60 years old to be included in the study. Asymptomatic participants had to show no cognitive impairment on a standard cognitive battery, including a clinical diagnostic rating scale (CDR) score of 0 and a Folstein mini-mental state examination (MMSE) score of 28 or greater. Symptomatic mutation carriers were required to have a CDR score of 0·5 or greater with mild dementia due to Alzheimer’s disease according to the National Institute of Aging–Alzheimer’s Association (NIA–AA) criteria19 and an MMSE score of at least 18, or MCI due to Alzheimer’s disease according to NIA–AA criteria.20 To facilitate a desired age distribution among cognitively normal participants, individuals were enrolled into 18–34 years and 35–60 years age groups; within each of these age groups, asymptomatic mutation carriers and non-carriers were matched for sex, age, and educational level.
Participants or their legal representatives, or both, provided written informed consent before enrolment and understood that they would not receive information about their PSEN1 or APOE genotypes, which were analysed as previously described.12,21 Participants were studied under guidelines approved by local institutional review boards. Ethics approval was obtained from the University of Antioquia Ethics Committee for procedures undertaken in Colombia and the Western Institutional Review Board for procedures undertaken in the USA. All data were acquired by investigators who were masked to the participants’ genetic status.
Procedures
Between Sept 26 and Dec 6, 2011, all clinical measures were undertaken at the University of Antioquia (Medellín, Colombia), and amyloid PET scanning was done at the Banner Alzheimer’s Institute (Phoenix, AZ, USA). Neurocognitive testing included the MMSE, CDR, and a Spanish version of the Consortium to Establish a Registry for Alzheimer’s Disease battery, which has been adapted to this Colombian population.12 Additional testing consisted of the Yesavage geriatric depression scale22 and the functional assessment staging test, which were done during screening and at baseline, before imaging.23
Volumetric and safety screening MRI scans (3D T1, T2 fluid-attenuated inversion recovery, and long echo time gradient-echo imaging) were done on a Siemens 1.5T Avanto scanner at the Hospital Pablo Tobón Uribe (Medellín, Colombia) after cognitive screening but before amyloid imaging. Finally, participants and University of Antioquia staff travelled to the Banner Alzheimer’s Institute in Phoenix (AZ, USA) for the participants’ florbetapir PET scans. These scans were done on a Siemens Biograph 16 HiRez PET/CT scanner after intravenous injection of about 10 mCi of florbetapir (18F), a 50 min radiotracer uptake period, a 10 min emission scan, and a CT scan for correction of radiation attenuation. Images were reconstructed using an iterative algorithm, measured attenuation–correction, and a 5 mm full-width-at-half-maximum Gaussian filter.
An automated algorithm (Statistical Parametric Mapping version 8 [SPM8], Wellcome Trust Centre for Neuroimaging, London, UK) was used to deform each PET image into the coordinates of a standard brain atlas to visually inspect individual images in a standardised fashion, assess cortical and reference regions of interest (ROIs), and create statistical brain maps. Mean cortical florbetapir standard uptake value ratios (SUVRs) were computed in each participant using six cortical grey matter ROIs.5,24,25 The pons, rather than the cerebellum, was used as a reference region for SUVR calculations because increased cerebellar Aβ concentrations have been reported in this PSEN1 E280A cohort and in other cohorts of individuals with dominant mutations that cause Alzheimer’s disease.6 Also, the pons has previously been validated for use in ageing and Alzheimer’s disease as a reference region in amyloid PET.26,27 We also computed a basal ganglia SUVR with automatic ana tomical labelled caudate, putamen, and globus pallidus ROIs11 because of reports of unusually high striatal Aβ deposition in people with some but not all autosomal dominant mutations that cause Alzheimer’s disease.13,28
Statistical analysis
The enrolment goal was for a convenience sample of 40 asymptomatic individuals, 50% with the PSEN1 E280A mutation, and ten symptomatic family members. This goal was based on projected availability of eligible participants and finite study resources that would allow for an adequate sample size to show statistically significant group differences.
To characterise the spatial pattern of fibrillar Aβ deposition in presymptomatic PSEN1 E280A mutation carriers in contrast to that of non-carriers, we did a two-sample voxel-wise t test between these two groups. We used SPM8 to smooth the spatially standardised images using a 12 mm full-width-at-half-maximum Gaussian filter and to compute a statistical brain map of greater cortical-to-pontine SUVRs. Regression modelling was used to characterise the age-related accumulation in mean cortical and ROI SUVRs in mutation carrier and non-carrier groups. To identify the model that best defined the relation between age and SUVRs, we compared goodness of fit between linear, multiple piecewise linear, and curvilinear models. For the curvilinear regression model in mutation carriers, we assumed the relation between SUVR and age would be sigmoidal, as previously suggested,4,29 such that there would be no detectable Aβ at the youngest ages, followed by an age-related increase in concentrations, and a plateau in concentrations at older ages and symptomatic stages of Alzheimer’s disease. The goodness of fit of every model was assessed using R2 testing. The extra sum of squares f test was used to compare the goodness of fit between the simple linear and piecewise linear models. Linear regression models were compared with the sigmoidal model using Akaike’s information criterion (AIC), which is based on information theory, because the two models are not nested. We did this model comparison with the GraphPad Prism software package (GraphPad Software, La Jolla, CA, USA). This method measures how well the data support each model, taking into account both the goodness of fit (sum of squares) and the number of parameters in the model. The results are expressed as the probability that each model is correct, with the probabilities summing to 100%.
The sigmoidal model is represented by the following equation:
Unknown parameters α1–α4 were estimated using SUVR data and independent non-linear curve fitting. To identify the two age inflexion points of the sigmoidal curves, we defined the absence of the SUVR change (flatness) as a slope less than or equal to 0·02 SUVR per year. We did a Monte-Carlo simulation procedure with 5000 iterations to assess the variations of these age inflexion points. Finally, we used an automated algorithm30 in SPM831 in post-hoc analyses to identify the extent to which mean cortical SUVR measurements and the statistical brain maps were affected by the combined effects of brain atrophy and partial-volume averaging.
Role of the funding source
This study was designed and undertaken by the study investigators, who had unrestricted use rights to the data. All authors had full access to all the data in the study. At the time of the study, florbetapir was approved under a US Food and Drug Administration investigational new drug application through Avid Radiopharmaceuticals and did not yet have approval for clinical use. For this reason, data collection was monitored for compliance by Avid Radiopharmaceuticals. Avid Radiopharmaceuticals also provided consultation for florbetapir PET acquisition and analysis. Avid Radiopharmaceuticals had no other role in the study design, data collection, data analysis, data interpretation, or writing of the report. Other funding sources had no access to data or role in the study. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Our study cohort consisted of 11 symptomatic individuals, 19 presymptomatic mutation carriers, and 20 asymptomatic non-carriers, ranging in age from 20 to 56 years. Table 1 lists participants’ characteristics. The symptomatic group was older than the presymptomatic carriers and non-carriers, and scored worse on all cognitive test measures. Symptomatic participants scored worse on all neuropsychological tests (p ≤ 0·00011; table 1). There were no differences between non-carriers and presymptomatic carriers on any of the neuro psychological tests undertaken. No differences were noted between the three groups in proportions of APOEε4 gene carriers: APOEε4 negative (2/3, 3/3), APOEε4 heterozygotes (2/4, 3/4), and homozygotes (4/4).
Table 1.
Demographics, APOE allele status, and key cognitive scores of study participants
| Non-carriers (n=20) | Presymptomatic carriers (n=19) | p value vs non-carriers | Symptomatic carriers (n=11) | p value vs non-carriers | p value vs presymptomatic | |
|---|---|---|---|---|---|---|
| Age (years) | 33·9 (8·7, 20–50) | 32·6 (8·2, 20–43) | 0·66 | 47·5 (4·6, 41–56) | <0·0001 | <0·0001 |
|
| ||||||
| Sex | 0·90 | 0·66 | 0·59 | |||
| Men | 7 | 7 | 3 | ·· | ·· | |
| Women | 13 | 12 | 8 | ·· | ·· | |
|
| ||||||
| Education (years) | 11·2 (3·3) | 12·3 (2·8) | 0·29 | 8·8 (3·5) | 0·070 | 0·0068 |
|
| ||||||
| Apolipoprotein E εallele | 0·36 | 0·66 | 1·00 | |||
| 2/3 | 1 | 1 | 1 | ·· | ·· | |
| 2/4 | 1 | 3 | 0 | ·· | ·· | |
| 3/3 | 13 | 10 | 8 | ·· | ·· | |
| 3/4 | 4 | 5 | 2 | ·· | ·· | |
| 4/4 | 1 | 0 | 0 | ·· | ·· | |
|
| ||||||
| Clinical diagnostic rating sum of boxes | 0·05 (0·1) | 0·026 (0·1) | 0·59 | 3·9 (2·3) | <0·0001 | <0·0001 |
|
| ||||||
| Mini-mental state examination | 29·8 (0·5) | 29·8 (0·4) | 0·78 | 23·1 (3·5) | <0·0001 | <0·0001 |
|
| ||||||
| CERAD word list delayed recall | 19·4 (3·1) | 20·5 (3·0) | 0·28 | 6·4 (3·1) | <0·0001 | <0·0001 |
|
| ||||||
| Trail making A | 48·3 (18·9) | 39·6 (11·3) | 0·093 | 143·5 (94·5) | <0·0001 | <0·0001 |
|
| ||||||
| Category fluency, animals | 19·4 (2·8) | 21·3 (5·5) | 0·19 | 10·6 (4·4) | <0·0001 | <0·0001 |
|
| ||||||
| Verbal fluency | 33·4 (6·6) | 35·9 (12·1) | 0·43 | 17·1 (8·7) | <0·0001 | 0·00011 |
Data are mean (SD, range), number, or mean (SD). APOE ratio tests were compared between three APOE groups: 2/3 and 3/3, 2/4 and 3/4, and 4/4. CERAD=Consortium to Establish a Registry for Alzheimer’s Disease.
Florbetapir binding was evident in all symptomatic and asymptomatic mutation carriers over age 30 years; there was no apparent binding in the grey matter in carriers under age 30 years or in any of the non-carriers (figure 1). Non-specific white matter binding, which was probably unrelated to fibrillar Aβ,24 was seen in all images in all participants. Voxel-wise comparison showed greater florbetapir binding in asymptomatic PSEN1 E280A mutation carriers than in age-matched non-carriers (figure 2). The cerebral pattern of fibrillar Aβ deposition resembled that found in people clinically affected by or at risk for late-onset Alzheimer’s disease. This includes preferential florbetapir binding in posterior cingulate, precuneus, parietotemporal, frontal, and basal ganglia regions.
Figure 1. Florbetapir (18F) binding in members of the Colombian kindred by age group and clinical diagnosis.
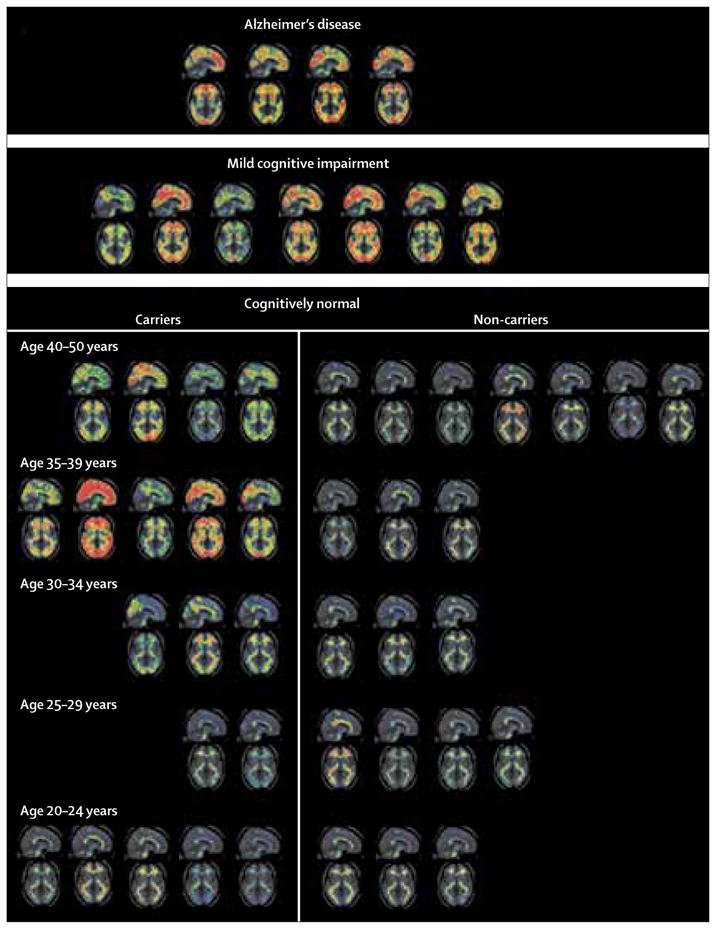
Sagittal and axial standard uptake value ratio maps are presented in standardised atlas space. Cortical amyloid-β is evident in all patients with mild cognitive impairment and dementia and in all PSEN1 E280A mutation carriers aged 30 years or older. There is variability of visible tracer binding within each age subdivision and within the mild cognitive impairment and dementia groups. Typical patterns of non-specific white matter binding occurred in carriers under age 30 years and in all non-carriers. Participants’ data are displayed in age groupings to help protect individual anonymity.
Figure 2. Comparison of florbetapir (18F) levels in asymptomatic participants and non-carriers.
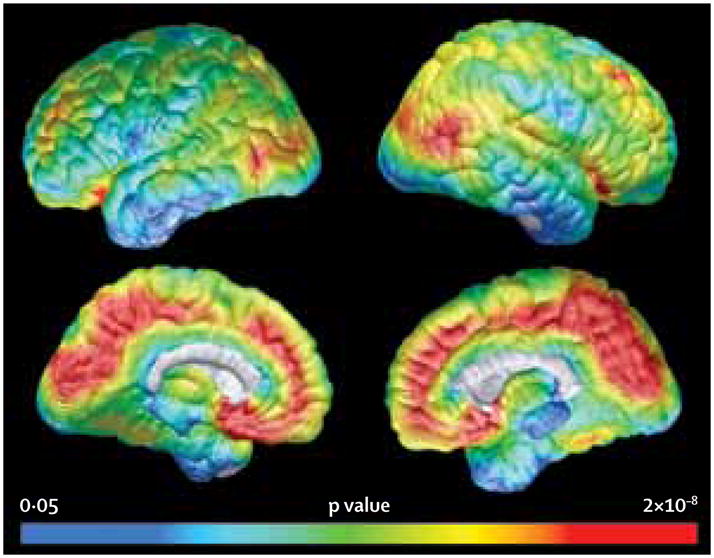
Statistical map showing significantly greater cortical-to-pontine florbetapir standard uptake value ratios in 19 asymptomatic mutation carriers compared with 20 non-carriers. The pattern of cortical amyloid-β deposition is similar to that reported in patients with late-onset Alzheimer’s disease. See Online for appendix
The relation between age and mean cortical SUVRs fitted well to both a simple linear regression model (R=0·74, p<0·0001) and a sigmoidal shaped curve (R2 =0·72, p<0·0001; appendix). However, the AIC comparison of these two models, accounting for the comparable goodness of fit to the data and the number of estimated parameters in the models, suggested that the sigmoidal model was more accurate for mean cortical SUVR and all individual ROIs tested (appendix). The probability of the sigmoidal model properly representing the data was 98·4% versus a 1·6% probability that the simple linear regression model was correct. Additional support of this non-linear model came from assessment with piecewise linear regression modelling. There were significant slope inflection points on a three-piece linear model, with an initial flat slope followed by an upward inflexion in mean cortical SUVR at age 28·5 years (95% CI 20·7–36·2) and plateauing at age 37·8 years (33·6–41·9; appendix). Although similar to the sigmoidal model, the three-piece linear model was inferior (probability of accuracy 9·6%) to the sigmoidal model (90·4%) on AIC comparison (appendix).
Based on the preferred sigmoidal model, we estimated that fibrillar Aβ began to accumulate in PSEN 1 E280A mutation carriers at a mean age of 28·2 years (95% CI 27·3–33·4); it then rose steeply over the next 9·4 years and began to plateau at 37·6 years (95% CI 35·3–40·2; table 2; video). Mean cortical SUVRs exceeded the highest level that occurred in the non-carrier group, an SUVR of 0·87, at 31 years of age in the PSEN1 E280A mutation carriers (figure 3). On average, the non-carrier group had mean cortical SUVRs of 0·78 (SD 0·05), which was similar to the levels reported in pre-symptomatic carriers below age 30 years (figure 3). The SUVRs increase was not associated with age in non-carriers (r=0·12, p=0·62).
Table 2.
Estimated ages at fibrillar amyloid-β onset and plateau in PSEN1 E280A mutation carriers
| Age at onset (years) | Age at plateau (years) | R2 (model goodness of fit) | Absolute sum of squares | |
|---|---|---|---|---|
| Mean cortical | 28·2 (27·3–33·4) | 37·6 (35·3–40·2) | 0·72 | 0·23 |
| Precuneus | 26·5 (26·0–27·6) | 36·6 (35·2–37·4) | 0·77 | 0·43 |
| Basal ganglia | 26·6 (25·6–27·7) | 34·6 (32·8–36·3) | 0·71 | 0·17 |
| Frontal | 27·0 (26·2–28·9) | 36·2 (35·1–39·3) | 0·77 | 0·17 |
| Anterior cingulate | 27·3 (26·3–30·7) | 37·4 (36·3–41·3) | 0·79 | 0·35 |
| Parietal | 27·9 (26·7–36·2) | 35·0 (33·7–41·3) | 0·55 | 0·44 |
| Temporal | 28·8 (27·5–33·1) | 37·1 (36·0–41·3) | 0·70 | 0·23 |
| Posterior cingulate | 29·0 (26·0–32·1) | 38·2 (36·2–41·3) | 0·70 | 0·37 |
Data are mean (95% CI).
Figure 3. Mean cortical standard uptake value ratios at different ages.

The individual values of non-carriers are shown as artificially clustered at 5-year intervals to help preserve anonymity of carriers versus non-carriers related to specific ages. All datapoints over age 40 years are clustered at 42·5 years on the x axis because of limited non-carrier datapoints above age 40 years. SUVR=standard uptake value ratio.
When SUVR versus age sigmoidal regression curves for mean cortical, component ROIs, and the basal ganglia ROI were compared, mean ages at which amyloid began to increase ranged from 26 to 29 years and plateaued around 35–38 years (table 2, figure 4). 95% CIs overlapped for all measures between all regions. In a post-hoc analysis of carriers versus non-carriers aged 40 years or older, when plateau SUVR levels in ROIs were ranked, the highest levels occurred in the anterior cingulate and the precuneus regions, with posterior cingulate and frontal ROIs showing mid-range differences in florbetapir binding levels between carriers and non-carriers, and mean cortical, basal ganglia, temporal, and lateral parietal levels in the lower half of the SUVR difference range (p=0·0008; figure 4). Basal ganglia binding was not seen earlier or more prominently than in other regions (table 2, figure 4). Age-related SUVR findings in PSEN1 E280A mutation carriers remained significant after controlling for the combined effects of brain atrophy and partial volume averaging in each of our ROI and voxel-based analyses (data not shown).
Figure 4. Regional standard uptake value ratio differences according to participants’ age.

Sigmoidal regression curves relating regional florbetapir PET SUVRs to age in PSEN1 E280A mutation carriers compared with non-carriers. Lines represent the differences between mutation carrier and non-carrier regression lines, accounting for variability in non-specific florbetapir binding between different regions. The greatest maximal plateau differences between carriers and non-carriers occurred in the anterior cingulate and precuneus regions (p=0·0008). SUVR=standard uptake value ratio.
Discussion
In this study, we characterised the pattern of fibrillar amyloid deposition and estimated its temporal relation with clinical onset in PSEN1 E280A mutation carriers and non-carriers from the largest known kindred with autosomal dominant Alzheimer’s disease. The cerebral pattern of fibrillar Aβ accumulation in the late preclinical and clinical stages of Alzheimer’s disease was similar to that reported in individuals with late-onset Alzheimer’s disease,5 including striatal uptake that was not out of proportion with that in other cortical regions. Fibrillar Aβ, as measured by 18F-PET, may begin to accumulate about 16 years before the median age at MCI onset and 21 years before the median age at dementia onset in this kindred. Accumulation exceeded the highest level noted in non-carriers at age 31 years in the PSEN1 E280A carriers, 13 years and 18 years before expected MCI and dementia onset, respectively. Fibrillar Aβ concentrations may increase steeply over about 9 years, and then begin to plateau about 6 years before the median age at MCI onset and 11 years before the median age at dementia onset. This pattern of cortical Aβ progression is consistent with hypothetical models proposed in late-onset Alzheimer’s disease.7
In this study, we capitalised on data from a large, clinically and neuropathologically well characterised kindred, which allowed us to explore the association between fibrillar Aβ deposition with the estimated ages at MCI and dementia onset.12 Other biomarker studies in autosomal dominant Alzheimer’s disease have either been limited to small cohorts or assessed a heterogeneous cohort of autosomal dominant Alzheimer’s disease mutation carriers from several different families (panel).2,13,14,28 The largest of these studies included 88 mutation carriers and 40 non-carriers from 51 different genetic kindreds, including various APP, PSEN1, and PSEN2 mutations.2 The study reported 11C-Pittsburgh compound B PET increases about 15 years before the mean expected age of symptom onset of 45·7 years (SD 6·8). These data are consistent with our findings, although there are several differences in study design. Since different mutations for autosomal dominant Alzheimer’s disease can produce differences in clinical presentation and pathology,15,16 the study of a single mutation kindred would be expected to reduce variability in clinical and pathological outcome measures such as age at clinical onset and patterns of amyloid distribution. Also, unlike previous cohort studies, we used an 18F amyloid PET tracer, which might detect different binding patterns than the more commonly used 11C-Pittsburgh compound B in these populations. Also, the DIAN study used a semi-structured interview and available historical data from a first-degree relative of each participant to estimate their age of expected symptomatic onset, whereas in our study we report median age at which participants were expected to meet formal diagnostic criteria for MCI (44 years, 95% CI 43–45) and Alzheimer’s disease (49 years, 49–50) estimated from a cohort of 449 single-mutation carriers.12 Because in the PSEN1 E280A mutation kindred age is predictive of clinical onset, cross-sectional assessments across a wide age range in this well-defined cohort are perhaps analogous to what might be expected from the assessment of longitudinal trajectories of biomarker change. Our findings are also similar to proposed trajectories of asymptomatic cortical amyloidosis in cross-sectional studies of ageing and late-onset Alzheimer’s disease, with amyloid buildup occurring 15–20 years before typical symptomatic disease onset.4,25 However, unlike cross-sectional studies of asymptomatic cortical amyl oidosis in healthy elderly individuals, all PSEN1 E280A mutation carriers are destined to develop Alzheimer’s disease. For these reasons, this study offered a unique opportunity to assess biomarker progression in Alzheimer’s disease without needing decades of longitudinal data to assess true data-driven trajectories of change. Our group previously assessed fluid and imaging biomarkers of Alzheimer’s disease in a younger cohort (mean age 22 years, range 18–26 years) from this kindred.18 We showed that, at this earlier age range, differences occurred in functional and structural MRI findings, suggesting that, unlike in some hypothetical models of late-onset Alzheimer’s disease7 and findings from the recent DIAN study,2 structural and functional imaging changes might be detectable before cortical amyloid deposition. However, although eight participants from the previous study18 also participated in this subsequent amyloid PET study, with investigators remaining masked to mutation carrier status, the previous study of younger individuals cannot be directly compared with the amyloid data presented here. In the DIAN study,2 hippocampal volume was reduced in carriers compared with non-carriers 15 years before expected symptom onset. One reason why we were able to identify structural brain changes at an earlier age might be because of potential heterogeneity of pathological abnormalities in the DIAN study, or their use of a less sensitive measure of hippocampal volume than the voxel-based findings reported in the study by Reiman and colleagues.18
Another important difference between our findings and those from other amyloid PET studies is absence of early prominent and disproportionate binding to the striatum in this PSEN1 E280A cohort (figure 1 and figure 4). Why some autosomal dominant mutation carriers might have more or less striatal fibrillar plaque burden than others is not clear. However, the presence of striatal plaques post mortem has been well documented in sporadic Alzheimer’s disease, occurring in both the striatum and the thalamus in nearly all sporadic Alzheimer’s disease cases.34–37 The presence of fibrillar deposition in the striatum has been reported on amyloid PET in both patients with sporadic late-onset38,39 and autosomal dominant Alzheimer’s disease.13,14,28,32,33 Although fibrillar amyloid might be present, neuritic plaques in the striatum are less common than in cortical grey matter,36,38 and why some patients with autosomal dominant Alzheimer’s disease have neurotoxicity related to the presence of fibrillar amyloid in the striatum leading to clinical symptoms associated with striatal pathology is not clear.32 Further studies that directly compare carriers of different mutations and evaluate the molecular properties of subsequent amyloid by-products are necessary.
Although the data presented here may support development of prevention trial protocols in this cohort, what is less clear is how our findings could inform the design of trials in patients with late-onset Alzheimer’s disease. Although this study included a relatively large number of mutation carriers and non-carriers, limitations in sample size introduce uncertainties in the extent to which our data fit the prevailing sigmoidal model and the estimated ages at which fibrillar Aβ accumulation begins and plateaus. Also, the estimated ages of onset of disease symptoms presented here are representative of a larger historical cohort and have no absolute predictive value for the individuals in this study. Larger studies are needed to confirm our findings. Also, florbetapir PET might underestimate the extent of Aβ deposition in autosomal dominant Alzheimer’s disease because of low affinity for diffuse plaques,40 as opposed to fibrillar plaques, or because of the possible presence of fibrillar Aβ in the reference region. Lastly, although we argue that this population is informative for understanding longitudinal change associated with disease progression, this cannot be proven without longitudinal follow-up.
Supplementary Material
Panel: Research in context.
Systematic review
We searched PubMed for the terms “amyloid imaging” AND “autosomal dominant Alzheimer”. The search was repeated with the terms “amyloid PET” replacing “amyloid imaging” and “early onset”, “genetic”, and “familial” replacing “autosomal dominant”. As of Aug 27, 2012, we found reports of nine studies with amyloid PET in patients with autosomal dominant Alzheimer’s disease that had sample sizes greater than one. The largest single-mutation study assessed six individuals with a common genetic variant.14 Findings from these studies showed increased amyloid concentrations on PET in mutation carriers at various stages of disease, most of whom had Alzheimer’s disease-like patterns, but some who also had early and prominent increased striatal binding.13,14,28,32,33 No longitudinal studies have been reported, with only one cross-sectional study assessing amyloid PET in relation to age at expected onset of symptomatic disease.2 In a study of 88 mutation carriers, 51 different mutation pedigrees were combined, with a mean age of increased amyloid concentrations estimated at about 15 years before the typical age of clinical disease onset.
Interpretation
In this cross-sectional assessment of florbetapir PET in a kindred with a single common autosomal dominant mutation who were destined to develop Alzheimer’s disease, we report that cortical amyloid began to increase about 16 years before the expected age of onset of mild cognitive impairment and 21 years before dementia, followed by a later plateauing of cortical deposition just before the estimated age of symptom onset. Longitudinal studies are needed to better characterise the natural history of amyloid deposition. The findings from this study provide insight into the pathological progression of disease and could inform the design of presymptomatic treatment trials in this population and potentially in other forms of Alzheimer’s disease.
Acknowledgments
This study was funded by Avid Radiopharmaceuticals, Banner Alzheimer’s Foundation, Nomis Foundation, Anonymous Foundation, Forget Me Not Initiative, Colciencias (1115-408-20512, 1115-408-20543P30), National Institute on Aging (R01 AG031581, P30 AG19610), and State of Arizona. Avid Radiopharmaceuticals provided funding for travel of participants to the USA, and PET scanning, and provided the florbetapir tracer. We thank Daniel Skovronsky, Mark Mintun, and Michael Pontecorvo for their technical guidance and support; Sergio Alvarez, Andres Arbelaez, Natalia Montes, Feliza Restrepo, and their colleagues from the Hospital Pablo Tobon Uribe and the University of Antioquia, and Dan Bandy, Sandy Goodwin, Nicole Richter, Hillary Protas, Stephanie Parks, Xiaofen Liu, Candy Monarrez, and Lazaro Martinez from the Banner Alzheimer’s Institute for roles in data management and collection; Paul Thompson and his colleagues for the software used to generate the cortical surface maps in figures 2 and 3; and our research participants and other members of the PSEN1 E280A mutation kindred for their invaluable dedication and inspiration.
Footnotes
For more on the API see http://endalznow.org/
Contributors
ASF, YTQ, LJJ, FL, and EMR conceived and designed the study. ASF, FL, and EMR supervised the study. ASF, LJJ, MGG, CML, SM, NA-B, MG, GG, and FL acquired data. ASF, KC, YTQ, NA, AR, PT, WL, RAR, MJH, KSK, and EMR analysed and interpreted the data. KC, NA, AR, PT, WL, and HM did the statistical analyses. ASF, KC, YTQ, LJJ, MGG, CML, JBSL, NA, AR, PT, WL, HM, LL, SM, NA-B, MG, GG, RAR, MJH, KSK, PNT, FL, and EMR wrote the manuscript. ASF, KC, NA, AR, PT, and WL prepared the figures and tables. ASF, PNT, and EMR obtained funding. ASF, KC, FL, and EMR revised the manuscript.
Conflicts of interest
ASF served as a consultant to Eli Lilly and Avid Radiopharmaceuticals and received grant funding from Avid Radiopharmaceuticals. The Banner Alzheimer’s Foundation has received compensation for undertaking educational programs for Avid Radiopharmaceuticals and Eli Lilly staff, which ASF contributed time towards. KSK serves as a scientific adviser to Amgen, Genentech, iPierian, and Noscira; serves on the board of Minerva Biotechnologies; and has had research contracts with the NIA and the California Institute for Regenerative Medicine. PNT has received consulting fees from Abbott Laboratories, AC Immune, Adamas, Allergan, Avanir, Boehringer-Ingelheim, Chase Pharmaceuticals, Chiesi, Eisai, Elan, Medavante, Merz, Neuroptix, Novartis, Otsuka, Sanofi-Aventis, Schering-Plough, and Worldwide Clinical Trials; consulting fees and research support from AstraZeneca, Avid Radiopharmaceuticals, Bristol-Myers Squibb, Genentech, GlaxoSmithKline, Janssen, Eli Lilly, Medivation, Merck and Company, Pfizer, Roche, Toyama, and Wyeth Laboratories; research support from Baxter Healthcare, Functional Neuromodulation, GE Healthcare, and Targacept; and other research support from NIA, National Institute of Mental Health, Alzheimer’s Association, and Arizona Department of Health Services. PNT also has stock options in Medavante and Adamas and holds a patent for biomarkers of Alzheimer’s disease. EMR has received research funding from Avid Radiopharmaceuticals and holds a paid consultation role for Eli Lilly. KC, YTQ, LJJ, MGG, CML, JBSL, NA, AR, PT, WL, HM, LL, SM, NA-B, MG, GG, RAR, MJH, and FL declare that they have no conflicts of interest.
References
- 1.Braak H, Braak E. Staging of Alzheimer-related cortical destruction. Int Psychogeriatr. 1997;9 (suppl 1):257–61. [PubMed] [Google Scholar]
- 2.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris JC, Price AL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci. 2001;17:101–18. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- 4.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–83. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Fleisher AS, Chen K, Liu X, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol. 2011;68:1404–11. doi: 10.1001/archneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 6.Duque-Castano A, Roldan MI, Arango-Viana JC, Arcos-Burgos M, Cubillo H, Lopera F. Neuropathological findings in early-onset Alzheimer’s disease (E280a-PS1 mutation) Rev Neurol. 1999;29:1–6. [Spanish] [PubMed] [Google Scholar]
- 7.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reiman EM, Langbaum JB, Tariot PN. Alzheimer’s prevention initiative: a proposal to evaluate presymptomatic treatments as quickly as possible. Biomark Med. 2010;4:3–14. doi: 10.2217/bmm.09.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reiman EM, Caselli RJ, Yun LS, et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–58. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 10.Fleisher AS, Houston WS, Eyler LT, et al. Identification of Alzheimer disease risk by functional magnetic resonance imaging. Arch Neurol. 2005;62:1881–88. doi: 10.1001/archneur.62.12.1881. [DOI] [PubMed] [Google Scholar]
- 11.Reiman EM, Chen K, Liu X, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106:6820–25. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acosta-Baena N, Sepulveda-Falla D, Lopera-Gomez CM, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. Lancet Neurol. 2011;10:213–20. doi: 10.1016/S1474-4422(10)70323-9. [DOI] [PubMed] [Google Scholar]
- 13.Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27:6174–84. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholl M, Almkvist O, Axelman K, et al. Glucose metabolism and PIB binding in carriers of a His163Tyr presenilin 1 mutation. Neurobiol Aging. 2011;32:1388–99. doi: 10.1016/j.neurobiolaging.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 15.Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115:1449–57. doi: 10.1172/JCI24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Lopera F, Ardilla A, Martinez A, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997;277:793–99. [PubMed] [Google Scholar]
- 18.Reiman EM, Quiroz YT, Fleisher AS, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012 doi: 10.1016/S1474-4422(12)70228-4. published online Nov 6. http://dx.doi.org/10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed]
- 19.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–69. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–79. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 22.Sheikh JI, Yesavage JA. Geriatric depression scale (GDS): recent evidence and development of a shorter version. In: Brink TL, editor. Clinical gerontology: a guide to assessment and intervention. New York: The Haworth Press; 1986. pp. 165–73. [Google Scholar]
- 23.Reisberg B. Functional assessment staging (FAST) Psychopharmacol Bull. 1988;24:653–59. [PubMed] [Google Scholar]
- 24.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011;305:275–83. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleisher AS, Chen K, Liu X, et al. APOE4 and age effects on florbetapir PET in healthy aging and Alzheimer disease. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.04.017. published online May 28. [DOI] [PubMed] [Google Scholar]
- 26.Villain N, Chetelat G, Grassiot B, et al. Regional dynamics of amyloid-beta deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: a voxelwise PiB-PET longitudinal study. Brain. 2012;135:2126–39. doi: 10.1093/brain/aws125. [DOI] [PubMed] [Google Scholar]
- 27.Edison P, Hinz R, Ramlackhansingh A, et al. Can target-to-pons ratio be used as a reliable method for the analysis of [11C]PiB brain scans? Neuroimage. 2012;60:1716–23. doi: 10.1016/j.neuroimage.2012.01.099. [DOI] [PubMed] [Google Scholar]
- 28.Villemagne VL, Ataka S, Mizuno T, et al. High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol. 2009;66:1537–44. doi: 10.1001/archneurol.2009.285. [DOI] [PubMed] [Google Scholar]
- 29.Sojkova J, Zhou Y, An Y, et al. Longitudinal patterns of beta-amyloid deposition in nondemented older adults. Arch Neurol. 2011;68:644–49. doi: 10.1001/archneurol.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meltzer CC, Cantwell MN, Greer PJ, et al. Does cerebral blood flow decline in healthy aging? A PET study with partial-volume correction. J Nucl Med. 2000;41:1842–48. [PubMed] [Google Scholar]
- 31.Tohka J, Zijdenbos A, Evans A. Fast and robust parameter estimation for statistical partial volume models in brain MRI. Neuroimage. 2004;23:84–97. doi: 10.1016/j.neuroimage.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 32.Koivunen J, Verkkoniemi A, Aalto S, et al. PET amyloid ligand [11C] PIB uptake shows predominantly striatal increase in variant Alzheimer’s disease. Brain. 2008;131:1845–53. doi: 10.1093/brain/awn107. [DOI] [PubMed] [Google Scholar]
- 33.Remes AM, Laru L, Tuominen H, et al. Carbon 11-labeled pittsburgh compound B positron emission tomographic amyloid imaging in patients with APP locus duplication. Arch Neurol. 2008;65:540–44. doi: 10.1001/archneur.65.4.540. [DOI] [PubMed] [Google Scholar]
- 34.Braak H, Braak E. Alzheimer’s disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol. 1990;49:215–24. [PubMed] [Google Scholar]
- 35.Suenaga T, Hirano A, Llena JF, Yen SH, Dickson DW. Modified Bielschowsky stain and immunohistochemical studies on striatal plaques in Alzheimer’s disease. Acta Neuropathol. 1990;80:280–86. doi: 10.1007/BF00294646. [DOI] [PubMed] [Google Scholar]
- 36.Brilliant MJ, Elble RJ, Ghobrial M, Struble RG. The distribution of amyloid beta protein deposition in the corpus striatum of patients with Alzheimer’s disease. Neuropathol Appl Neurobiol. 1997;23:322–25. [PubMed] [Google Scholar]
- 37.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 38.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 39.Price JC, Klunk WE, Lopresti BJ, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh compound-B. J Cereb Blood Flow Metab. 2005;25:1528–47. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- 40.Choi SR, Golding G, Zhuang Z, et al. Preclinical properties of 18F-AV-45: a PET agent for Abeta plaques in the brain. J Nucl Med. 2009;50:1887–94. doi: 10.2967/jnumed.109.065284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.