

Abstract
In the title compound, C13H16N4O2, the pyrazole ring forms a dihedral angle of 50.61 (6)° with the 3-nitro-phenyl ring. The plane of the nitro group is twisted by 6.8 (7)° out of the plane of the phenyl ring. In the crystal, the molecules are linked by N—H⋯N and N—H⋯O hydrogen bonds, forming sheets in the bc plane. In addition, a weak C—H⋯N interaction is observed.
Related literature
For background to pyrazole-based ligands, see; Ahmed et al. (2005 ▶); Abonia et al. (2002 ▶, 2004 ▶, 2010 ▶); Guerrero et al. (2009 ▶); Quiroga et al. (2008 ▶); Schutznerová, et al. (2012 ▶). For structure of an isomer of the title compound, see: Low et al. (2004 ▶).
Experimental
Crystal data
C13H16N4O2
M r = 260.30
Monoclinic,
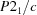
a = 11.9421 (14) Å
b = 9.6419 (11) Å
c = 11.7694 (13) Å
β = 93.504 (2)°
V = 1352.6 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.09 mm−1
T = 298 K
0.46 × 0.36 × 0.32 mm
Data collection
Bruker SMART APEX CCD area-detector diffractometer
14529 measured reflections
2486 independent reflections
2036 reflections with I > 2σ(I)
R int = 0.044
Refinement
R[F 2 > 2σ(F 2)] = 0.040
wR(F 2) = 0.115
S = 1.05
2486 reflections
181 parameters
2 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.17 e Å−3
Δρmin = −0.17 e Å−3
Data collection: SMART (Bruker, 1999 ▶); cell refinement: SAINT (Bruker, 1999 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812042791/bt6847sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812042791/bt6847Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N4—H4A⋯N2i | 0.90 (1) | 2.23 (1) | 3.1195 (17) | 172 (2) |
| N4—H4B⋯O1ii | 0.90 (1) | 2.39 (1) | 3.241 (2) | 160 (2) |
| C14—H14⋯N4iii | 0.93 | 2.54 | 3.403 (2) | 155 |
Symmetry codes: (i) 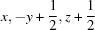 ; (ii)
; (ii) 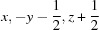 ; (iii)
; (iii) 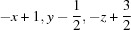 .
.
Acknowledgments
FCC and RAG thanks the Universidad del Valle and the Universidad del Quindío for financial support to project 542. ACO thanks the DGAPA–UNAM for financial support (PAPIIT IN203209). SHO thanks the Consejo Superior de Investigaciones Científicas (CSIC) of Spain for the award of a licence for the use of the Cambridge Structural Database.
supplementary crystallographic information
Comment
The recent past has evidenced an ever-increasing interest in pyrazole based ligands. The interest in such compounds is due, first of all, to their variety of coordination complexes with a great number of metal ions and, second, to their ability to provide an extensive variety of coordination geometries and significant structural nuclearity when introducing different kinds of heteroatoms (Ahmed et al. 2005; Schutznerová et al. 2012). The past few years have seen considerable rise in interest in the design of various pyrazole-based ligands for particular metal binding site (Guerrero et al. 2009).
As a part of our current research work focused on the development of new bioactive heterocyclic compounds and continuing with the use of pyrazolic Schiff bases (Quiroga et al., 2008; Abonia et al. 2002, 2004, 2010), in the synthesis of pyrazolopyrimidines, we want to describe the compound 5-amino-3-tert-butyl-1-(3-nitro-phenyl)-1H-pyrazole (I), which is a structural isomer of a related compound previously reported by Low (Low et al., 2004).
The structure of the title compound is shown in Figure 1. The compound consists of a ring pyrazole substituted by 3-nitro-phenyl ring bonded to N1, amino group in C5 and tertbutyl group in C3. The pyrazole and phenyl rings are not coplanar, they are forming a dihedral angle of 50.61 (6)°. The nitro group is rotated around C14—N3 bond by 6.8 (3)°. These angle values are larger than those described for the isomeric compound 5-amino-3-tertbutyl-1-(4-nitro-phenyl)-1H-pyrazole (Low et al., 2004). In the crystal, the molecules are linked by N—H–N and N—H–O intermolecular hydrogen bonds forming sheets in the bc plane. In additon, a weak intermolecular C-H···N interaction is observed (Figure 2, Table 1).
Experimental
To a solution of conc hydrochloric acid (3.8 ml) in water (33 ml), 3-nitrophenylhydrazine (1.5001 g, 9.87 mmol) and 4,4-dimethyl-3-oxopentanenitrile (1.8502 g, 14.80 mmol) were added. The mixture was heated at 70 °C for 1 h. Then, conc hydrochloric acid (3.8 ml) was added and the mixture was heated for 1 h more. After cooling, crushed ice was added and neutralized with conc ammonium hydroxide. The resulting solid was filtered under reduced pressure, washed with cold water (3 X 5 ml) and dried at ambient temperature affording the title compound (I) as a yellow solid [yield 1.744 g, 68%, m.p. 375 K]. MS (70 eV) m/z (%): 260 (55), 245 (100), 218 (88), 190 (73). Anal. Calc. for C13H16N4O2; C 59.99; H 6.20; N 21.52%, found C 60.36; H 6.42; N 21.88%. Crystals of the title compound suitable for single-crystal X-ray diffraction were grown by slow diffusion of pentane into a CH2Cl2 solution of the title compound.
Refinement
The positional parameters of the amino H atom were refined with a distance restraint of 0.90 (1)Å while those of the other H atoms were calculated geometrically (C—H = 0.93–0.98 Å). All H atoms were refined with Uiso(H) = 1.2Ueq of the parent atom.
Figures
Fig. 1.

Structure of (I), with the numbering scheme. The displacement ellipsoids are drawn to 40% of probability.
Fig. 2.

The crystal packing of (I), only the H atoms involved in intermolecular interaction were drawn.
Crystal data
| C13H16N4O2 | F(000) = 552 |
| Mr = 260.30 | Dx = 1.278 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 7587 reflections |
| a = 11.9421 (14) Å | θ = 2.7–25.3° |
| b = 9.6419 (11) Å | µ = 0.09 mm−1 |
| c = 11.7694 (13) Å | T = 298 K |
| β = 93.504 (2)° | Prism, orange |
| V = 1352.6 (3) Å3 | 0.46 × 0.36 × 0.32 mm |
| Z = 4 |
Data collection
| Bruker SMART APEX CCD area-detector diffractometer | 2036 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.044 |
| Graphite monochromator | θmax = 25.4°, θmin = 2.7° |
| Detector resolution: 0.83 pixels mm-1 | h = −14→14 |
| ω scans | k = −11→11 |
| 14529 measured reflections | l = −14→14 |
| 2486 independent reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.040 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.115 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0644P)2 + 0.1233P] where P = (Fo2 + 2Fc2)/3 |
| 2486 reflections | (Δ/σ)max < 0.001 |
| 181 parameters | Δρmax = 0.17 e Å−3 |
| 2 restraints | Δρmin = −0.17 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the those in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.61664 (14) | −0.30475 (15) | 0.45577 (16) | 0.1090 (6) | |
| O2 | 0.76468 (15) | −0.18160 (16) | 0.45837 (14) | 0.1006 (5) | |
| N1 | 0.71780 (9) | 0.17992 (11) | 0.75434 (9) | 0.0403 (3) | |
| N2 | 0.78018 (9) | 0.26705 (11) | 0.68907 (9) | 0.0426 (3) | |
| N3 | 0.67228 (15) | −0.20551 (15) | 0.48900 (13) | 0.0686 (4) | |
| N4 | 0.68987 (11) | 0.12825 (14) | 0.94996 (10) | 0.0516 (3) | |
| H4A | 0.7163 (13) | 0.1491 (17) | 1.0213 (9) | 0.062* | |
| H4B | 0.6699 (13) | 0.0411 (11) | 0.9321 (14) | 0.062* | |
| C3 | 0.84041 (11) | 0.34268 (13) | 0.76494 (11) | 0.0401 (3) | |
| C4 | 0.81984 (11) | 0.30418 (14) | 0.87650 (11) | 0.0433 (3) | |
| H4 | 0.8523 | 0.3421 | 0.9433 | 0.052* | |
| C5 | 0.74255 (11) | 0.19985 (13) | 0.86760 (11) | 0.0400 (3) | |
| C6 | 0.91855 (12) | 0.45492 (15) | 0.72699 (13) | 0.0488 (4) | |
| C7 | 0.99208 (17) | 0.3989 (2) | 0.63582 (17) | 0.0768 (6) | |
| H7A | 1.0374 | 0.3241 | 0.6669 | 0.115* | |
| H7B | 1.0398 | 0.4716 | 0.6110 | 0.115* | |
| H7C | 0.9453 | 0.3657 | 0.5723 | 0.115* | |
| C8 | 0.85048 (16) | 0.57837 (16) | 0.67990 (16) | 0.0698 (5) | |
| H8A | 0.8051 | 0.5500 | 0.6138 | 0.105* | |
| H8B | 0.9005 | 0.6509 | 0.6594 | 0.105* | |
| H8C | 0.8030 | 0.6119 | 0.7368 | 0.105* | |
| C9 | 0.99367 (15) | 0.50386 (18) | 0.82913 (16) | 0.0657 (5) | |
| H9A | 0.9483 | 0.5454 | 0.8845 | 0.098* | |
| H9B | 1.0464 | 0.5709 | 0.8044 | 0.098* | |
| H9C | 1.0334 | 0.4260 | 0.8626 | 0.098* | |
| C11 | 0.64947 (11) | 0.07495 (13) | 0.70143 (11) | 0.0413 (3) | |
| C12 | 0.69297 (12) | −0.00946 (14) | 0.62034 (12) | 0.0458 (4) | |
| H12 | 0.7658 | 0.0027 | 0.5985 | 0.055* | |
| C13 | 0.62517 (13) | −0.11245 (14) | 0.57271 (12) | 0.0490 (4) | |
| C14 | 0.51684 (14) | −0.13283 (15) | 0.60126 (14) | 0.0555 (4) | |
| H14 | 0.4727 | −0.2023 | 0.5668 | 0.067* | |
| C15 | 0.47534 (14) | −0.04795 (17) | 0.68205 (15) | 0.0598 (4) | |
| H15 | 0.4022 | −0.0603 | 0.7032 | 0.072* | |
| C16 | 0.54076 (12) | 0.05548 (16) | 0.73219 (13) | 0.0535 (4) | |
| H16 | 0.5117 | 0.1124 | 0.7869 | 0.064* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.1062 (12) | 0.0705 (9) | 0.1473 (15) | −0.0008 (8) | −0.0181 (10) | −0.0588 (10) |
| O2 | 0.1094 (13) | 0.0912 (11) | 0.1050 (12) | −0.0042 (9) | 0.0366 (10) | −0.0347 (9) |
| N1 | 0.0438 (6) | 0.0374 (6) | 0.0390 (6) | −0.0066 (5) | −0.0035 (5) | 0.0012 (5) |
| N2 | 0.0464 (7) | 0.0400 (6) | 0.0408 (6) | −0.0063 (5) | −0.0029 (5) | 0.0030 (5) |
| N3 | 0.0837 (11) | 0.0507 (8) | 0.0697 (9) | 0.0057 (8) | −0.0079 (8) | −0.0137 (7) |
| N4 | 0.0619 (8) | 0.0505 (7) | 0.0418 (7) | −0.0070 (6) | −0.0016 (6) | 0.0068 (6) |
| C3 | 0.0411 (7) | 0.0346 (7) | 0.0435 (7) | 0.0009 (6) | −0.0056 (6) | 0.0008 (6) |
| C4 | 0.0487 (8) | 0.0402 (7) | 0.0398 (7) | −0.0025 (6) | −0.0089 (6) | −0.0019 (6) |
| C5 | 0.0429 (7) | 0.0370 (7) | 0.0393 (7) | 0.0041 (6) | −0.0037 (6) | 0.0025 (5) |
| C6 | 0.0518 (8) | 0.0404 (7) | 0.0530 (8) | −0.0076 (6) | −0.0062 (7) | 0.0043 (6) |
| C7 | 0.0860 (13) | 0.0648 (11) | 0.0827 (13) | −0.0206 (10) | 0.0292 (11) | 0.0050 (10) |
| C8 | 0.0834 (12) | 0.0428 (9) | 0.0797 (12) | −0.0123 (8) | −0.0248 (10) | 0.0123 (8) |
| C9 | 0.0610 (10) | 0.0575 (10) | 0.0756 (11) | −0.0208 (8) | −0.0191 (9) | 0.0098 (8) |
| C11 | 0.0442 (8) | 0.0361 (7) | 0.0426 (7) | −0.0044 (6) | −0.0060 (6) | 0.0030 (6) |
| C12 | 0.0460 (8) | 0.0427 (8) | 0.0478 (8) | −0.0014 (6) | −0.0044 (6) | 0.0011 (6) |
| C13 | 0.0606 (9) | 0.0365 (7) | 0.0484 (8) | 0.0008 (7) | −0.0084 (7) | −0.0012 (6) |
| C14 | 0.0612 (10) | 0.0420 (8) | 0.0611 (9) | −0.0129 (7) | −0.0132 (8) | 0.0031 (7) |
| C15 | 0.0495 (9) | 0.0606 (10) | 0.0690 (10) | −0.0154 (8) | −0.0001 (8) | −0.0018 (8) |
| C16 | 0.0494 (9) | 0.0525 (9) | 0.0585 (9) | −0.0054 (7) | 0.0020 (7) | −0.0050 (7) |
Geometric parameters (Å, º)
| O1—N3 | 1.2159 (19) | C7—H7B | 0.9600 |
| O2—N3 | 1.204 (2) | C7—H7C | 0.9600 |
| N1—C5 | 1.3613 (17) | C8—H8A | 0.9600 |
| N1—N2 | 1.3858 (15) | C8—H8B | 0.9600 |
| N1—C11 | 1.4198 (16) | C8—H8C | 0.9600 |
| N2—C3 | 1.3297 (17) | C9—H9A | 0.9600 |
| N3—C13 | 1.470 (2) | C9—H9B | 0.9600 |
| N4—C5 | 1.3735 (18) | C9—H9C | 0.9600 |
| N4—H4A | 0.901 (9) | C11—C12 | 1.380 (2) |
| N4—H4B | 0.895 (9) | C11—C16 | 1.382 (2) |
| C3—C4 | 1.4005 (19) | C12—C13 | 1.378 (2) |
| C3—C6 | 1.5140 (19) | C12—H12 | 0.9300 |
| C4—C5 | 1.3650 (19) | C13—C14 | 1.370 (2) |
| C4—H4 | 0.9300 | C14—C15 | 1.370 (2) |
| C6—C8 | 1.526 (2) | C14—H14 | 0.9300 |
| C6—C7 | 1.526 (2) | C15—C16 | 1.377 (2) |
| C6—C9 | 1.530 (2) | C15—H15 | 0.9300 |
| C7—H7A | 0.9600 | C16—H16 | 0.9300 |
| C5—N1—N2 | 111.43 (10) | C6—C8—H8A | 109.5 |
| C5—N1—C11 | 127.92 (11) | C6—C8—H8B | 109.5 |
| N2—N1—C11 | 120.20 (10) | H8A—C8—H8B | 109.5 |
| C3—N2—N1 | 104.32 (10) | C6—C8—H8C | 109.5 |
| O2—N3—O1 | 123.17 (17) | H8A—C8—H8C | 109.5 |
| O2—N3—C13 | 118.64 (15) | H8B—C8—H8C | 109.5 |
| O1—N3—C13 | 118.17 (17) | C6—C9—H9A | 109.5 |
| C5—N4—H4A | 113.3 (11) | C6—C9—H9B | 109.5 |
| C5—N4—H4B | 115.7 (11) | H9A—C9—H9B | 109.5 |
| H4A—N4—H4B | 120.1 (16) | C6—C9—H9C | 109.5 |
| N2—C3—C4 | 111.44 (12) | H9A—C9—H9C | 109.5 |
| N2—C3—C6 | 120.78 (12) | H9B—C9—H9C | 109.5 |
| C4—C3—C6 | 127.78 (12) | C12—C11—C16 | 120.04 (13) |
| C5—C4—C3 | 106.26 (12) | C12—C11—N1 | 119.55 (12) |
| C5—C4—H4 | 126.9 | C16—C11—N1 | 120.40 (13) |
| C3—C4—H4 | 126.9 | C13—C12—C11 | 118.00 (13) |
| N1—C5—C4 | 106.53 (11) | C13—C12—H12 | 121.0 |
| N1—C5—N4 | 122.60 (12) | C11—C12—H12 | 121.0 |
| C4—C5—N4 | 130.77 (13) | C14—C13—C12 | 122.92 (14) |
| C3—C6—C8 | 109.90 (12) | C14—C13—N3 | 118.85 (14) |
| C3—C6—C7 | 110.23 (12) | C12—C13—N3 | 118.23 (14) |
| C8—C6—C7 | 109.71 (14) | C15—C14—C13 | 118.12 (14) |
| C3—C6—C9 | 109.33 (12) | C15—C14—H14 | 120.9 |
| C8—C6—C9 | 108.57 (13) | C13—C14—H14 | 120.9 |
| C7—C6—C9 | 109.07 (14) | C14—C15—C16 | 120.67 (15) |
| C6—C7—H7A | 109.5 | C14—C15—H15 | 119.7 |
| C6—C7—H7B | 109.5 | C16—C15—H15 | 119.7 |
| H7A—C7—H7B | 109.5 | C15—C16—C11 | 120.24 (14) |
| C6—C7—H7C | 109.5 | C15—C16—H16 | 119.9 |
| H7A—C7—H7C | 109.5 | C11—C16—H16 | 119.9 |
| H7B—C7—H7C | 109.5 |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N4—H4A···N2i | 0.90 (1) | 2.23 (1) | 3.1195 (17) | 172 (2) |
| N4—H4B···O1ii | 0.90 (1) | 2.39 (1) | 3.241 (2) | 160 (2) |
| C14—H14···N4iii | 0.93 | 2.54 | 3.403 (2) | 155 |
Symmetry codes: (i) x, −y+1/2, z+1/2; (ii) x, −y−1/2, z+1/2; (iii) −x+1, y−1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: BT6847).
References
- Abonia, R., Castillo, J., Insuasty, B., Quiroga, J., Nogueras, M. & Cobo, J. (2010). Eur. J. Org. Chem. 33, 6454–6463.
- Abonia, R., Rengifo, E., Quiroga, J., Insuasty, B., Cobo, J. & Nogueras, M. (2004). Tetrahedron, 60, 8839–8843.
- Abonia, R., Rengifo, E., Quiroga, J., Insuasty, B., Sanchez, A., Cobo, J., Low, J. N. & Nogueras, M. (2002). Tetrahedron Lett. 43, 5617–5620.
- Ahmed, M. S. M., Kobayashi, K. & Mori, A. (2005). Org. Lett. 7, 4487–4489. [DOI] [PubMed]
- Bruker (1999). SMART and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Guerrero, M., Pons, J., Parella, T., Font-Bardia, M., Calvet, T. & Ros, J. (2009). Inorg. Chem. 48, 8736–8750. [DOI] [PubMed]
- Low, J. N., Cobo, J., Abonia, R., Quiroga, J. & Glidewell, C. (2004). Acta Cryst. C60, o194–o195. [DOI] [PubMed]
- Quiroga, J., Portilla, J., Abonia, R., Insuasty, B., Nogueras, M. & Cobo, J. (2008). Tetrahedron Lett. 49, 6254–6256.
- Schutznerová, E., Popa, I., Krystof, V., Koshino, H., Trávnícek, Z., Hradil, P. & Cankar, P. (2012). Tetrahedron, 68, 3996–4002.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812042791/bt6847sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812042791/bt6847Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report