

Despite its high affinity for NADPH, AKR1a4 crystallized in the apo form, which is very rare for aldo-keto reductase enzymes.
Keywords: aldo-keto reductases, AKR, AKR1a4, NADPH, aldehyde reductases
Abstract
Aldo-keto reductase 1a4 (AKR1a4; EC 1.1.1.2) is the mouse orthologue of human aldehyde reductase (AKR1a1), the founding member of the AKR family. As an NADPH-dependent enzyme, AKR1a4 catalyses the conversion of d-glucuronate to l-gulonate. AKR1a4 is involved in ascorbate biosynthesis in mice, but has also recently been found to interact with SMAR1, providing a novel mechanism of ROS regulation by ATM. Here, the crystal structure of AKR1a4 in its apo form at 1.64 Å resolution as well as the characterization of the binding of AKR1a4 to NADPH and P44, a peptide derived from SMAR1, is presented.
1. Introduction
Aldo-keto reductase 1a4 (AKR1a4) is the mouse equivalent of human aldehyde reductase (AKR1a1) and pig aldehyde reductase (AKR1a2). Aldehyde reductases are monomeric enzymes of ∼325 residues (∼36 kDa) that catalyze the NADPH-dependent reduction of a wide variety of endogenous and exogenous aldehydes to their corresponding alcohols (Carbone et al., 2008 ▶; El-Kabbani et al., 1994 ▶; Flynn, 1982 ▶; Spite et al., 2007 ▶). AKR1a4 was first cloned from mouse embryonic tissue (Allan & Lohnes, 2000 ▶) and appears to be expressed ubiquitously in adult mice.
Although suspected for many years, the role of AKR1a4 in ascorbate biosynthesis has only very recently been confirmed (Gabbay et al., 2010 ▶). Unlike primates (including humans), mice can biosynthesize ascorbic acid or vitamin C. The first step in ascorbate synthesis involves conversion of d-glucuronate to l-gulonate. Similar to other AKR enzymes, the reduction of d-glucuronate follows a Bi-Bi ordered catalytic mechanism in which the cofactor NADPH binds first and leaves last (Faucher et al., 2008 ▶). The binding of NADPH induces a conformational change in the enzyme, which in turn allows the formation of a mature binding pocket for the substrate. During catalysis the 4-pro-R hydride is transferred from the NADPH nicotinamide ring to the carbonyl of the substrate, which is followed by the transfer of a proton from a tyrosine (Tyr50) to the negatively charged carbonyl O atom (Ye et al., 2001 ▶).
Recently, AKR1a4 has been shown to be inhibited by SMAR1, a protein involved in regulation of the cell cycle and apoptosis (Singh et al., 2010 ▶). This appears to our knowledge to be the first case of AKR enzyme modulation through protein–protein interaction. In this model, SMAR1 was suggested to bind to AKR1a4 and to inhibit its activity. Upon SMAR1 phosphorylation in response to oxidative stress, the complex dissociates, resulting in AKR1a4 activation. Inhibition of AKR1a4 was also observed in the presence of a 44-amino-acid peptide (P44) derived from SMAR1 (Singh et al., 2010 ▶).
Here, we report the high-resolution crystal structure of AKR1a4 in its apo form at 1.64 Å resolution. This model represents the highest resolution structure of an aldehyde reductase. The most unexpected result is the crystallization of AKR1a4 in the apo form despite its high affinity for NADPH. Crystallization of an AKR enzyme in an apo form is a very rare event. The high affinity of natural substrates such as NADPH appears to prevent AKR enzymes from crystallizing in the apo form. In addition to the structural work, we characterized the interaction of AKR1a4 with NADPH as well as with a SMAR1 peptide (P44) using isothermal titration calorimetry.
2. Materials and methods
2.1. Cloning, expression and purification of AKR1a4
The AKR1a4 cDNA was purchased from Open Biosystems (catalogue No. MMM1013-7514285). AKR1a4 was subcloned in pGEX4T3 utilizing BamHI–EcoRI cloning sites with the primers 5′-CCCGGATCCATGACGGCCTCCAGTGTCCTCC and 5′-CCGAATTCTCAGTATGGGTCATTAAAGGGATAC. Escherichia coli BL21 cells were transformed with pGEX4T3-AKR1a4 and the protein was expressed in ZYM-5052 auto-induction medium (Studier, 2005 ▶). Cells were allowed to grow at 310 K for 3.5 h; the temperature was then lowered to 291 K and maintained for 16 h. The cells were harvested by centrifugation (5000 rev min−1, 45 min, 277 K) and resuspended in 1× PBS buffer. The cell suspension was supplemented with 0.75 mg ml−1 lysosyme and incubated for 30 min on ice. The cells were then disrupted by three freeze–thaw cycles in liquid nitrogen and sonication. The cell lysate was centrifugated at 30 000g for 30 min at 277 K. The supernatant was applied onto a Sepharose 4B column and ∼12 mg pure GST-fusion protein was purified from 100 ml bacterial culture. The GST-AKR1a4 fusion protein was cleaved using thrombin for 2 d at 277 K during dialysis (20 mM Tris pH 8, 10 mM NaCl, 1 mM β-mercaptoethanol) and the fragments were purified by anion-exchange chromatography on a Mono Q column using an increasing salt gradient. About 5 mg highly purified AKR1a4 was routinely obtained from 100 ml bacterial culture. The pure AKR1a4 was concentrated to 10 mg ml−1 before crystallization.
2.2. Crystallization and X-ray analysis
Preliminary crystallization attempts were performed by the hanging-drop method using an incomplete matrix of conditions known as the AKR screen (Faucher, Pereira de Jesus-Tran, Cantin, Couture et al., 2006 ▶) empirically derived from published crystallization conditions of AKRs. The conditions of the AKR screen are listed in Table 1 ▶. Preliminary crystals were obtained in AKR screen condition Nos. 4 and 6 at 295 K. Crystals were optimized by slight modification of the initial pH and by removing the sodium acetate additive. The crystal used for the X-ray diffraction experiment grew within three weeks using drops composed of 1 µl protein solution plus 1 µl crystallization condition (26% PEG 4000, 0.1 M sodium citrate pH 5.8). Typical crystals grew as thin but very large plates with approximate dimensions of 0.05 × 0.6 × 0.8 mm and had to be broken down in order to mount them on a cryoloop. Crystals used for X-ray diffraction were cryopreserved in mother liquor supplemented with 15% ethylene glycol and were flash-cooled in liquid nitrogen. A complete data set was collected on beamline 23ID-B at the Advanced Photon Source (Argonne National Laboratory) at 100 K and a wavelength of 1.0332 Å. Data-collection statistics are summarized in Table 2 ▶. The data were indexed in the monoclinic space group P2, with unit-cell parameters a = 56.94, b = 92.62, c = 70.10 Å, β = 106.16°. The space group was determined to be P21 according to systematically absent reflections. Calculation of the Matthews coefficient (2.47 Å3 Da−1; Matthews, 1968 ▶) suggested the presence of two molecules in the asymmetric unit.
Table 1. Crystallization conditions of the AKR screen.
| Precipitant | Buffer, pH | Additive | |
|---|---|---|---|
| 1 | 20% PEG 4000 | 0.1M sodium citrate pH 5.6 | 0.1M lithium sulfate |
| 2 | 20% PEG 4000 | 0.1M sodium citrate pH 5.6 | 0.2M sodium citrate |
| 3 | 23% PEG 4000 | 0.1M sodium citrate pH 5.6 | 0.2M ammonium acetate |
| 4 | 23% PEG 4000 | 0.1M sodium citrate pH 5.6 | 0.2M sodium acetate |
| 5 | 26% PEG 4000 | 0.1M sodium citrate pH 5.6 | 0.2M ammonium sulfate |
| 6 | 26% PEG 4000 | 0.1M sodium citrate pH 5.6 | 0.2M sodium acetate |
| 7 | 29% PEG 4000 | 0.1M sodium citrate pH 5.6 | 0.2M ammonium acetate |
| 8 | 29% PEG 4000 | 0.1M sodium citrate pH 5.6 | 0.2M ammonium sulfate |
| 9 | 20% PEG 4000 | 0.1M MES pH 6.4 | 0.2M ammonium acetate |
| 10 | 20% PEG 4000 | 0.1M MES pH 6.4 | 0.2M ammonium sulfate |
| 11 | 23% PEG 4000 | 0.1M MES pH 6.4 | 0.2M sodium citrate |
| 12 | 23% PEG 4000 | 0.1M MES pH 6.4 | 0.2M ammonium sulfate |
| 13 | 26% PEG 4000 | 0.1M MES pH 6.4 | 0.1M lithium sulfate |
| 14 | 26% PEG 4000 | 0.1M MES pH 6.4 | 0.2M ammonium acetate |
| 15 | 29% PEG 4000 | 0.1M MES pH 6.4 | 0.1M lithium sulfate |
| 16 | 29% PEG 4000 | 0.1M MES pH 6.4 | 0.2M sodium acetate |
| 17 | 20% PEG 4000 | 0.1M HEPES pH 7.5 | 0.2M ammonium sulfate |
| 18 | 20% PEG 4000 | 0.1M HEPES pH 7.5 | 0.2M sodium acetate |
| 19 | 23% PEG 4000 | 0.1M HEPES pH 7.5 | 0.1M lithium sulfate |
| 20 | 23% PEG 4000 | 0.1M HEPES pH 7.5 | 0.2M sodium acetate |
| 21 | 26% PEG 4000 | 0.1M HEPES pH 7.5 | 0.2M ammonium acetate |
| 22 | 26% PEG 4000 | 0.1M HEPES pH 7.5 | 0.2M sodium citrate |
| 23 | 29% PEG 4000 | 0.1M HEPES pH 7.5 | 0.1M lithium sulfate |
| 24 | 29% PEG 4000 | 0.1M HEPES pH 7.5 | 0.2M sodium citrate |
Table 2. Summary of data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Wavelength () | 1.033 |
| Resolution () | 201.64 (1.681.64) |
| Space group | P21 |
| Unit-cell parameters (, ) | a = 56.94, b = 92.62, c = 70.10, = 106.16 |
| Total reflections | 302500 (18223) |
| Unique reflections | 84168 (6163) |
| Multiplicity | 3.6 (3.0) |
| Completeness (%) | 98.5 (97.7) |
| I/(I) | 19.7 (8.0) |
| R meas † (%) | 5.2 (17.5) |
| R merge ‡ (%) | 4.4 (14.4) |
| Wilson B (2) | 10.2 |
| Refinement | |
| R cryst | 0.148 (0.203) |
| R free § | 0.178 (0.257) |
| R.m.s.d. from ideal bond lengths () | 0.007 |
| R.m.s.d. from ideal bond angles () | 1.128 |
| Average B factors (2) | |
| All atoms | 13.3 |
| Protein | 11.1 |
| Solvent | 24.9 |
| Ramachandran plot (%) | |
| Most favoured regions | 98.74 |
| Allowed regions | 1.26 |
| Disallowed regions | 0 |
| PDB code | 4gac |
R meas is the redundancy-independent R factor (on intensities; Diederichs Karplus, 1997 ▶).
R
merge = 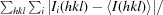
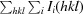 , where I(hkl) is the average value of the intensity of reflection hkl in the data set and Ii(hkl) is the intensity of the ith observation of that reflection.
, where I(hkl) is the average value of the intensity of reflection hkl in the data set and Ii(hkl) is the intensity of the ith observation of that reflection.
R free is calculated for a test set of reflections (5%) which were not included in the refinement.
2.3. Structure determination and refinement
The diffraction images were integrated using XDS and scaled using XSCALE (Kabsch, 2010 ▶). The structure of apo AKR1a4 was solved by molecular replacement with the MOLREP software from the CCP4 suite (Winn et al., 2011 ▶) using the amino-acid sequence of AKR1a4 and the coordinates of apo m17αHSD (PDB entry 2he8; Faucher, Pereira de Jésus-Tran, Cantin, Luu-The et al., 2006 ▶) as a search model, in which the three mobile loops were removed and amino acids were converted to alanines. The resulting electron-density map was well defined for the two molecules per asymmetric unit as visualized in Coot (Emsley & Cowtan, 2004 ▶). The initial model was submitted to rigid-body refinement and one cycle of simulated annealing at 3000 K followed by energy minimization and B-factor refinement. The model was subsequently refined using phenix.refine (Adams et al., 2002 ▶) by simple energy minimization followed by isotropic B-factor refinement and water updating. Between rounds of refinement, the model was corrected by manual rebuilding. Two citrate ions and two ethylene glycol molecules were added to the AKR1a4 model and used in refinement. The quality of the model was verified using PROCHECK (Laskowski et al., 1993 ▶) and Coot (Emsley & Cowtan, 2004 ▶). The final model comprises residues 2–325 for both monomers A and B, while the loop regions between residues 128–130, 215–222 and 303–309 are missing in monomer B only. Final refinement statistics are shown in Table 2 ▶.
2.4. Binding-affinity measurements
Isothermal titration calorimetry (ITC) was used to measure the binding affinity of NADPH and SMAR1 peptide P44 to AKR1a4 in the presence or absence of NADPH. ITC experiments were performed at 303 K and consisted of 29 injections of 10 µl each with 300 s equilibration time between injections. The AKR1a4 concentration was 38.8 µM (as measured by amino-acid analysis), that of P44 was 40 µM and NADPH was present at various concentrations in 10 mM Tris pH 8.0. Fitting a single-site binding isotherm to the data led to the enthalpy of binding (ΔH) and the dissociation constant (K d).
3. Results and discussion
3.1. Structure of AKR1a4
AKR enzymes adopt the well known triosephosphate isomerase (β/α)8-barrel motif or TIM barrel (Banner et al., 1975 ▶). This fold consists of a cylindrical core of eight parallel β-strands that form a β-barrel surrounded by eight α-helices running antiparallel to the strands. The C-terminal part of the β-barrel delineates the active site in which the substrate and the nicotinamine ring of NADPH interact. The α-helices are connected to the strands by a short loop on the N-terminal side of the β-barrel, while the connecting loops on the C-terminal side of the β-barrel, especially loops A, B and C, are much longer. Finally, the N-terminal opening of the β-barrel is closed by an antiparallel β-sheet formed by two β-strands and its connecting loop.
We have crystallized and determined the structure of AKR1a4 in the apo form at 1.64 Å resolution. As expected, AKR1a4 displays a very well conserved (α/β)8-barrel fold (Fig. 1 ▶) typical of the AKR superfamily. The three mobile loops (loops A, B and C), which were omitted in the search model, were clearly visible in the difference electron-density map after molecular replacement and were manually rebuilt. More than 98.5% of the amino acids are located in the most favourable region of the Ramachandran plot. The structure was refined to a crystallographic R factor of 0.148 (R free = 0.178). The final model lacks only the first methionine of monomer A and residues 1–2, 128–130, 215–222 and 303–309 of monomer B. It is noteworthy that monomer A appears to be well stabilized by the crystal packing, which explains why the three mobile loops were clearly visible in its electron-density map; these loops point towards a solvent channel in monomer B. The overall r.m.s.d. between the two monomers is 0.308 Å. The model also includes two citrate ions, two ethylene glycol and 991 water molecules. Surprisingly, no electron density for the cofactor NADPH was observed in either of the two monomers. This contrasts with most mammalian AKR structures determined to date. Usually, AKR binding to NADPH is so strong that it is observed in the structures without any addition of NADPH to the purified protein; the NADPH molecule comes from the expression cells. This makes the current model very rare (only one other example exists) among known AKR structures. Atomic coordinates and structure factors have been deposited in the PDB as entry 4gac.
Figure 1.

AKR1a4 structure and sequence alignment. The overall crystal structure of AKR1a4 (monomer A). Mobile loops are labelled in the left panel, while secondary-structure elements are identified in the right panel. Secondary structures are labelled according to the nomenclature adopted by El-Kabbani et al. (1994 ▶). Briefly, α-helices are designated H, β-strands are designated S and strands of the N-terminal β-sheet are designated N.
3.2. Structural comparison with other aldehyde reductases
AKR1a4 is the third member of the AKR1a family to be crystallized after the human and porcine enzymes (AKR1a1 and AKR1a2, respectively). All but one of the aldehyde reductase structures known to date is in complex with at least the cofactor. Only AKR1a1 (PDB entry 2alr; El-Kabbani et al., 1994 ▶) was solved in the apo form. AKR1a4 is very similar to its human counterpart, sharing 95% homology and displaying an r.m.s.d. of 0.44 Å calculated on Cα atoms. However, the structure of apo AKR1a1 lacks most of the mobile loops, similar to monomer B in our AKR1a4 model. Thus, AKR1a4 chain A is indeed the first complete apo aldehyde reductase structure to display all three mobile loops A, B and C.
3.3. Characterization of NADPH binding
In an attempt to understand why an apo AKR1a4 crystal was obtained (even in the presence of NADPH), we measured the affinity (K d) of NADPH for the enzyme by ITC. We obtained a K d of 144.6 ± 35.4 nM (Fig. 2 ▶), indicating very tight binding. Structural comparison with an AKR1a2 ternary complex (PDB entry 3cv7; Carbone et al., 2008 ▶) showed that all 16 amino acids involved in hydrogen bonding to the cofactor NADPH are conserved in AKR1a4. Thus, we conclude that the crystallization conditions must have favoured the formation of the apo-form crystal.
Figure 2.

ITC binding experiment for the binding of NADPH to AKR1a4. The upper panel shows the heat changes with each injection as a function of time. The lower panel shows a plot of the calculated enthalpies per injection versus the molar ratio of ligand and target (squares). The ITC thermogram was fitted to a single-site binding model. ΔH NADPH = −6080 ± 14.4 cal mol−1, K d,NADPH = 144.6 ± 35.4 nM. 1 cal = 4.186 J.
3.4. Characterization of P44 binding
Recently, AKR1a4 was shown to interact with SMAR1, a protein involved in regulation of the cell cycle and apoptosis (Singh et al., 2010 ▶). The activity of AKR1a4 was reported to be inhibited in the presence of a SMAR1-derived peptide (P44; TAWRRKQRGQSLAVKSFSRRTPSSSSYSASETM). To reveal the potential structural basis of the suggested inhibition, we attempted cocrystallization of AKR1a4 in the presence of either a truncated SMAR1 (160–350) protein or P44 without success (both with and without NADPH). In order to make sure that P44 physically interacts with AKR1a4, we decided to assess P44 binding to AKR1a4 by ITC. Our results (data not shown) clearly showed that there was no interaction between AKR1a4 and P44. Because of the Bi-Bi ordered mechanism displayed by aldehyde reductase, we further assessed the binding of P44 to AKR1a4 in the presence of saturating amounts of NADPH. Again, no binding of P44 to AKR1a4 was detected (data not shown). The observed inhibition of AKR1a4 by P44 in the study of Singh et al. (2010 ▶) probably stems from undetermined interactions between P44 and the cytosolic extract used in the assay.
Supplementary Material
PDB reference: AKR1a4, 4gac
Acknowledgments
This work was supported by a research grant to ZJ from Canadian Institutes of Health Research. We thank Kim Munroe for technical assistance with ITC experiments. FF is supported by an FRSQ postdoctoral fellowship and ZJ is a Killam Research Fellow and holds a Canada Research Chair in Structural Biology.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Allan, D. & Lohnes, D. (2000). Mech. Dev. 94, 271–275. [DOI] [PubMed]
- Banner, D. W., Bloomer, A. C., Petsko, G. A., Phillips, D. C., Pogson, C. I., Wilson, I. A., Corran, P. H., Furth, A. J., Milman, J. D., Offord, R. E., Priddle, J. D. & Waley, S. G. (1975). Nature (London), 255, 609–614. [DOI] [PubMed]
- Carbone, V., Chung, R., Endo, S., Hara, A. & El-Kabbani, O. (2008). Arch. Biochem. Biophys. 479, 82–87. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- El-Kabbani, O., Green, N. C., Lin, G., Carson, M., Narayana, S. V. L., Moore, K. M., Flynn, T. G. & DeLucas, L. J. (1994). Acta Cryst. D50, 859–868. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Faucher, F., Cantin, L., Luu-The, V., Labrie, F. & Breton, R. (2008). Biochemistry, 47, 13537–13546. [DOI] [PubMed]
- Faucher, F., Pereira de Jésus-Tran, K., Cantin, L., Couture, J.-F., Legrand, P. & Breton, R. (2006). International Conference on the Crystallization of Biological Macromolecules (ICCBM-11), Quebec, Canada.
- Faucher, F., Pereira de Jésus-Tran, K., Cantin, L., Luu-The, V., Labrie, F. & Breton, R. (2006). J. Mol. Biol. 364, 747–763. [DOI] [PubMed]
- Flynn, T. G. (1982). Biochem. Pharmacol. 31, 2705–2712. [DOI] [PubMed]
- Gabbay, K. H., Bohren, K. M., Morello, R., Bertin, T., Liu, J. & Vogel, P. (2010). J. Biol. Chem. 285, 19510–19520. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., Moss, D. S. & Thornton, J. M. (1993). J. Mol. Biol. 231, 1049–1067. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Singh, S., Sreenath, K., Pavithra, L., Roy, S. & Chattopadhyay, S. (2010). Int. J. Biochem. Cell Biol. 42, 1105–1114. [DOI] [PubMed]
- Spite, M., Baba, S. P., Ahmed, Y., Barski, O. A., Nijhawan, K., Petrash, J. M., Bhatnagar, A. & Srivastava, S. (2007). Biochem. J. 405, 95–105. [DOI] [PMC free article] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Ye, Q., Hyndman, D., Green, N., Li, X., Korithoski, B., Jia, Z. & Flynn, T. G. (2001). Proteins, 44, 12–19. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: AKR1a4, 4gac