

The recombinant BPTI/Kunitz-type inhibitor rShPI-1A from the Caribbean sea anemone S. helianthus has been crystallized and its structure has been refined to 2.5 Å resolution and compared with known X-ray structures of BPTI-like Kunitz-type inhibitors.
Keywords: protease inhibitors, BPTI/Kunitz-type domains
Abstract
The BPTI/Kunitz-type inhibitor family includes several extremely potent serine protease inhibitors. To date, the inhibitory mechanisms have only been studied for mammalian inhibitors. Here, the first crystal structure of a BPTI/Kunitz-type inhibitor from a marine invertebrate (rShPI-1A) is reported to 2.5 Å resolution. Crystallization of recombinant rShPI-1A required the salt-induced dissociation of a trypsin complex that was previously formed to avoid intrinsic inhibitor aggregates in solution. The rShPI-1A structure is similar to the NMR structure of the molecule purified from the natural source, but allowed the assignment of disulfide-bridge chiralities and the detection of an internal stabilizing water network. A structural comparison with other BPTI/Kunitz-type canonical inhibitors revealed unusual ϕ angles at positions 17 and 30 to be a particular characteristic of the family. A significant clustering of ϕ and ψ angle values in the glycine-rich remote fragment near the secondary binding loop was additionally identified, but its impact on the specificity of rShPI-1A and similar molecules requires further study.
1. Introduction
The BPTI/Kunitz-type inhibitor family (Pfam PF00014; Finn et al., 2010 ▶) includes some of the most extensively studied canonical serine protease inhibitors, which have been isolated from invertebrate to mammalian species (Laskowski et al., 2000 ▶; Kunitz & Northrop, 1936 ▶; Delfín et al., 1996 ▶). Canonical BPTI/Kunitz-type inhibitors are small protein domains (of around 6 kDa) with a compact hydrophobic core structure containing a central β-sheet and three conserved disulfide bridges (Antuch et al., 1993 ▶; Scheidig et al., 1997 ▶; Czapinska et al., 2000 ▶). The extreme stability of the target enzyme interaction, which is characterized by dissociation constants ranging from 10−13 to 10−7 M, results from a substrate-like interaction of the convex exposed binding loop with the highly complementary concave protease active site (Laskowski et al., 2000 ▶). The association energy depends mainly on the residue at position P1, supported by further residues within the primary binding site and the secondary binding loop of the inhibitor (Scheidig et al., 1997 ▶; Laskowski et al., 2000 ▶; Czapinska et al., 2000 ▶). The majority of the structures that have been determined to study the inhibitory activity are of bovine pancreatic trypsin inhibitor (BPTI; Huber et al., 1974 ▶; Burgering et al., 1997 ▶; Scheidig et al., 1997 ▶). We have previously reported the isolation of ShPI-1, a protease inhibitor from the Caribbean sea anemone Stichodactyla helianthus that shares the common structure of the BPTI/Kunitz-type family (PDB entry 1shp; Antuch et al., 1993 ▶) but exhibits an unusually broad specificity (Delfín et al., 1996 ▶). The recent high-level expression of a recombinant variant of ShPI-1 (rShPI-1A; Gil et al., 2011 ▶) enabled its structural investigation by X-ray crystallography. Although the rShPI-1A structure closely resembles the previously determined NMR structure of ShPI-1 purified from the natural source (PDB entry 1shp; Antuch et al., 1993 ▶), an internal water-stabilization network as well as the disulfide-bridge chiralities were revealed that had not previously been identified, thereby extending the structural characterization of this multifunctional invertebrate inhibitor.
2. Materials and methods
2.1. Protein production
Recombinant rShPI-1A was expressed and purified as described by Gil et al. (2011 ▶) (Table 1 ▶). The product includes additional residues at the N-terminus (Glu−3, Ala−2, Glu−1 and Ala0) and at the C-terminus (Leu56 and Gly57). Two different approaches for concentration of rShPI-1A buffered in 20 mM Tris–HCl pH 8.0, 1.0 M NaCl (buffer 1) were tested: lyophilization and re-solution in water (solution A) and concentration by ultrafiltration (solution B) using Centricon CM-3 concentration devices (Millipore). Protein concentration was determined from the absorbance at 280 nm (∊0.1% 280 nm = 0.52). Activity was confirmed by inhibition of bovine pancreatic trypsin (EC 3.4.21.4; Sigma) as described by Erlanger et al. (1961 ▶). Far-UV (190–260 nm) circular-dichroism (CD) spectroscopy and dynamic light-scattering (DLS) measurements were performed as described by Redecke et al. (2009 ▶) using a J-715 spectropolarimeter (Jasco) and a Spectroscatterer 201 (Molecular Dimensions), respectively. Molecular masses were calculated from hydrodynamic radii using SpectroSize (Fischer et al., 2004 ▶). The theoretical R h of natural ShPI-1 was estimated from its monomeric NMR structure (Antuch et al., 1993 ▶) using HYDROPRO (García de la Torre et al., 2000 ▶).
Table 1. Protein-production information.
| Source organism | S. helianthus strain KM71H |
| Expression vector | pZErO-2.1 |
| Expression host | Pichia pastoris |
| Complete amino-acid sequence of the construct produced | EAEASICSEPKKVGRCKGYFPRFYFDSETGKCTPFIYGGCGGNGNNFETLHQCRAICRALG |
2.2. Crystallization
Hanging-drop vapour-diffusion crystallization trials (1 µl protein solution in buffer 1 plus 1 µl reservoir solution equilibrated against 500 µl reservoir solution) were performed with rShPI-1A solutions A (4.0 mM) and B (3.5 mM) applying the reservoir buffer conditions previously reported for the crystallization of homologous BPTI/Kunitz-type domains in the Biological Macromolecule Crystallization Database (BMCD; Tung & Gallagher, 2009 ▶). In addition, binary complexes formed after incubation of rShPI-1A solutions A and B with equimolar concentrations of bovine pancreatic trypsin (EC 3.4.21.4; Sigma) for 1 h at 298 K were screened for crystal growth applying identical conditions as used for the free inhibitors. Crystals measuring up to 0.4 mm in size grew within one week at 288 K only in setups containing trypsin complexes of rShPI-1A sample A or B with a reservoir solution consisting of 0.1 M Tris–HCl pH 8.5, 1.7 M ammonium sulfate, 6%(w/v) glycerol. No crystallization conditions were identified for uncomplexed inhibitor solutions A and B.
2.3. Data collection and processing
A single protein crystal grown in a setup containing the binary trypsin complex formed using rShPI-1A solution A was mounted in a capillary. Diffraction data were collected at 293 K to 2.5 Å resolution using an in-house Rigaku RU-200 rotating-anode X-ray generator operated at 50 kV and 100 mA and equipped with a MAR Research image-plate detector. Data were reduced using MOSFLM (Leslie & Powell, 2007 ▶) and SCALA (Winn et al., 2011 ▶). Full details are presented in Table 2 ▶.
Table 2. Data-collection and processing statistics.
Values in parentheses are for the outer shell.
| Diffraction source | Rigaku RU-200 rotating anode |
| Wavelength (Å) | 1.5418 |
| Temperature (K) | 293 |
| Detector | 180 mm MAR Research image plate |
| Crystal-to-detector distance (mm) | 120 |
| Rotation range per image (°) | 1.5 |
| Total rotation range (°) | 129 |
| Exposure time per image (s) | 240 |
| Space group | P43212 |
| Unit-cell parameters (Å) | a = b = 37.16, c = 114.98 |
| Mosaicity (°) | 0.48 |
| Resolution range (Å) | 35.36–2.50 (2.64–2.50) |
| Total No. of reflections | 15197 |
| No. of unique reflections | 3127 |
| Completeness (%) | 99.0 (100) |
| Multiplicity | 4.9 (5.0) |
| 〈I/σ(I)〉 | 5.8 (3.1) |
| R r.i.m. † | 0.111 (0.235) |
| Overall B factor from Wilson plot (Å2) | 30.2 |
Estimated R r.i.m. = R merge[N/(N − 1)]1/2, where N is the data multiplicity.
2.4. Structure solution and refinement
The structure was solved by molecular replacement with MOLREP (Winn et al., 2011 ▶) using the NMR coordinates of natural ShPI-1 (Antuch et al., 1993 ▶; PDB entry 1shp) as a search model. The structure was refined with REFMAC v.5.2.0019 (Murshudov et al., 2011 ▶) and by manual intervention employing Coot (Emsley et al., 2010 ▶). The electron density was well defined by the rShPI-1A model, but features corresponding to a bound trypsin molecule were not detected. Furthermore, the Matthews coefficient of 3.27 Å3 Da−1 (Matthews, 1968 ▶) corroborated the presence of one rShPI-1A molecule in the asymmetric unit. MolProbity (Chen et al., 2010 ▶) was used for structure validation and Ramachandran analysis. Asn41 and Asn44 were outside the allowed regions of the Ramachandran plot, but were well defined in the electron-density map. The equivalent residues exhibited less favourable ϕ/ψ angles in all known structures of BPTI (Czapinska et al., 2000 ▶). No further geometric conflicts were detected. Refinement statistics are summarized in Table 3 ▶. Calculations of the torsion angles, the intramolecular hydrogen bonds and of the average NMR structure of natural ShPI-1 as well as the structural alignment with 12 BPTI/Kunitz-type domains were performed with WHAT IF (Vriend, 1990 ▶).
Table 3. Structure refinement.
Values in parentheses are for the outer shell.
| Resolution range (Å) | 28.80–2.50 (2.57–2.50) |
| Completeness (%) | 98.4 |
| σ cutoff | None |
| No. of reflections, working set | 2955 (210) |
| No. of reflections, test set | 135 (6) |
| Final R cryst | 0.190 (0.258) |
| Final R free | 0.221 (0.373) |
| No. of non-H atoms | |
| Protein | 444 |
| Ions (Cl−) | 3 |
| Waters | 28 |
| Total | 475 |
| R.m.s. deviations | |
| Bonds (Å) | 0.012 |
| Angles (°) | 1.402 |
| Average B factors (Å2) | |
| Protein | 21.4 |
| Ions (Cl−) | 46.0 |
| Waters | 28.0 |
| Ramachandran plot (%) | |
| Favoured regions | 94.7 |
| Additionally allowed | 3.5 |
| Outliers | 1.8 |
3. Results and discussion
Depending on the protein concentration and on the concentration technique applied, purified rShPI-1A aggregates in solution (Fig. 1 ▶ a). Nonspecific self-association during ultrafiltration has previously been attributed to mechanical stress on the protein at the membrane surface (Cromwell et al., 2006 ▶). A locally high protein concentration compared with the bulk solution favours concentration-dependent aggregation. During lyophilization, however, the ionic strength increases with the reduced volume of the solution, which is suggested to prevent the formation of large rShPI-1A aggregates, as observed for BPTI (Lafont et al., 1994 ▶). Neither the freezing process during lyophilization nor the aggregation state affected the native secondary structure of the inhibitor, as shown by the almost identical far-UV CD spectra obtained for both samples, which displayed the curve progression expected for rShPI-1A (Gil et al., 2011 ▶; Supplementary Fig. S11). Oligomeric rShPI-1A in the lyophilized sample (sample A) disassembled on complex formation with trypsin into monomeric enzyme–inhibitor complexes, while the ultrafiltered sample B remained highly heterogeneous (Fig. 1 ▶ b). Crystals grown using the monomeric complex setup appeared to be optimized regarding size and quality by visual inspection (Supplementary Fig. S21). Thus, a single crystal from this setup was used for diffraction data collection. Interestingly, the asymmetric unit contained only free rShPI-1A, not the expected inhibitor–trypsin complex, suggesting that crystallization was a consequence of salt-induced complex dissociation induced by the precipitant solution. A comparable crystallization chaperone effect has been reported for other canonical serine protease inhibitors, e.g. eglin C in the presence of subtilisin DY (Betzel et al., 1993 ▶), but has not been reported to date for the crystallization of canonical BPTI/Kunitz-type inhibitors. The X-ray structure of rShPI-1A (Fig. 2 ▶ a) is highly similar to the NMR structure of the molecule purified from the natural source (average backbone r.m.s.d. 0.89 Å; Antuch et al., 1993 ▶). Thus, the native fold was not affected by the additional residues at the N- and C-termini. Backbone r.m.s.d.s of more than 1.7 Å are restricted to residues Pro6, Arg11, Lys13 and Glu24. The increased B factors of Pro6 and Glu24 reflect the enhanced flexibility of the N-terminal residues and of the small loop connecting the two β-strands where these residues are located. In contrast, the deviations of Arg11 and Lys13 are attributed to the involvement of both residues in strong crystal contacts which reduce the overall flexibility of the canonical loop in the crystal structure compared with the NMR model, as indicated by the low B factors. A preferred left-handed conformation was assigned to the disulfide bridges Cys3–Cys53 and Cys28–Cys49, while Cys12–Cys36 adopts the unusual right-handed conformation (Fig. 2 ▶ a) common in BPTI/Kunitz-type inhibitors (Czapinska et al., 2000 ▶). The coexistence of both chiralities at Cys12–Cys36 previously observed in BPTI structures was not detected (Otting et al., 1993 ▶; Czapinska et al., 2000 ▶). The impact of disulfide-bond chiralities on the conformation and the flexibility of the binding loop in BPTI/Kunitz-type domains is still under discussion (Petersen et al., 1996 ▶; Czapinska et al., 2000 ▶). rShPI-1A is stabilized by an internal network of only three buried water molecules, which are located in a tetrahedral coordination highly similar to those in BPTI (Fig. 2 ▶ b; Deisenhofer & Steigemann, 1975 ▶; Wlodawer et al., 1984 ▶). The fourth internal water molecule is present, but is shifted by the side chain of Asn39 into a slightly different position compared with that in BPTI, preventing a contribution to the network. However, the overall rShPI-1A stability is compensated by a direct interaction between Pro6 and Asn39.
Figure 1.

(a) DLS analysis of the rShPI-1A aggregation state in solution after concentration by different techniques. Lyophilization (sample A) induced rShPI-1A oligomerization, while large inhomogeneous aggregates are formed by ultrafiltration (sample B). For monomeric ShPI-1 a theoretical hydrodynamic radius (R h) of 1.6 nm was calculated. (b) In the presence of equimolar concentrations of trypsin, rShPI-1A disaggregates in solution A (4.0 mM) owing to complex formation, while the heterogeneous radius distribution in solution B (3.5 mM) is not affected. DLS analysis of free trypsin is shown for comparison. The colour code corresponds to the relative frequency of particles characterized by a specific radius in solution, with dark red being the highest and blue the lowest.
Figure 2.
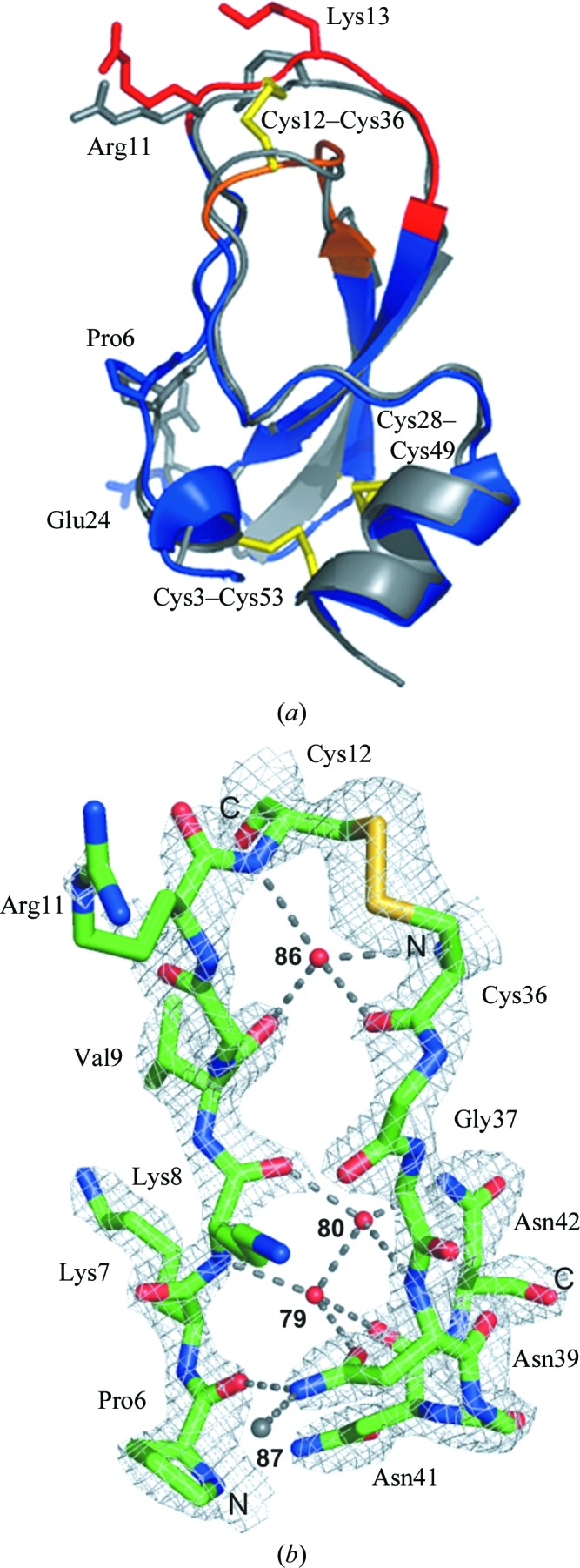
(a) Superposition of the three-dimensional structure of free rShPI-1A (blue) and the average NMR structure of ShPI-1 purified from the natural source (grey). The canonical (P3–P3′) and secondary (Ile32–Gly37) binding loops are highlighted in red and orange, respectively, while the conserved disulfide bridges are shown in yellow stick representation. Residues with backbone r.m.s.d.s of more than 1.7 Å are labelled. (b) Internal water-coordination sites near the binding loops of rShPI-1A. The shifted water molecule (87) observed in rShPI-1A is shown in grey. Dashed lines represent water-mediated hydrogen bonds. The unusal right-handed conformation of the Cys12–Cys36 disulfide bridge is well defined by the electron density (grey mesh, 2σ).
Comparison of rShPI-1A with all crystal structures of BPTI/Kunitz-type canonical domains currently annotated in the PDB (Fig. 3 ▶ a) confirms the structural conservation, including the intramolecular hydrogen-bond network. Significant Cα deviations (>1 Å) are restricted to flexible loop regions (Fig. 3 ▶ b). ϕ angles unusual for β-sheet structures at the nonconserved residues Pro17 and Pro30 of ShPI-1 have previously been associated with its reduced thermostability compared with BPTI (Antuch et al., 1993 ▶). However, we identified unusual ϕ angles at the equivalent positions in all canonical BPTI/Kunitz-type domains independent of the residue that was present (Supplementary Fig. S3a 1). Thus, a contribution of this characteristic feature of canonical BPTI/Kunitz-type inhibitors to the structural restraints for the highly twisted antiparallel β-sheet is more likely than a correlation with the different stability of these inhibitors, which requires further investigation. Moreover, a specific ϕ/ψ-angle clustering was detected for equivalent residues that compose the glycine-rich remote fragment (Supplementary Figs. S3b and S3c), which has been linked to the specificity and binding affinity of BPTI/Kunitz-type domains by restricting the available inhibitor conformations (Pritchard & Dufton, 1999 ▶). However, further studies are required in order to investigate the contribution of this region to the broad specificity of ShPI-1, which has previously been associated with a defensive role in S. helianthus (Delfín et al., 1996 ▶).
Figure 3.

(a) Structure-based multiple sequence alignment of BPTI/Kunitz-type canonical domains. The domain names and the PDB codes are shown on the left, while the sequence identity (%), the number of equivalent/total compared Cα atoms and the resulting r.m.s.d. values are displayed on the right. The canonical (P3–P3′) and secondary binding loops (residues 32–37 in rShPI-1A) are highlighted in boxes. (b) Cα deviations (Å) of the analyzed BPTI/Kunitz-type canonical domains compared with rShPI-1A (from Ala0 to Arg54 in rShPI-1A numbering). Line identifiers correspond to the associated sequences of the BPTI/Kunitz-type domains shown in (a).
Supplementary Material
PDB reference: rShPI-1A, 3ofw
Supplementary material file. DOI: 10.1107/S1744309112039085/en5515sup1.pdf
Acknowledgments
This work was partially supported by the International Foundation for Science (IFS), Sweden (grant F4086) and the Deutscher Akademischer Austausch Dienst (DAAD). CB and LR thank the German Federal Ministry of Education and Research (BMBF) for financial support (grants 01KX0806 and 01KX0807). We are indebted to Professor Dr Hahn for his support in starting this research and Professor Dr J. Díaz and Dr M. Mansur for their contributions to the heterologous expression of rShPI-1A. We acknowledge Dr T. Bergfors (Biomedical Center, Uppsala University, Sweden) for helpful advice during the crystallization experiments.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: EN5515).
References
- Antuch, W., Berndt, K. D., Chávez, M. A., Delfín, J. & Wüthrich, K. (1993). Eur. J. Biochem. 212, 675–684. [DOI] [PubMed]
- Betzel, C., Dauter, Z., Genov, N., Lamzin, V., Navaza, J., Schnebli, H. P., Visanji, M. & Wilson, K. S. (1993). FEBS Lett. 317, 185–188. [DOI] [PubMed]
- Burgering, M. J., Orbons, L. P., van der Doelen, A., Mulders, J., Theunissen, H. J., Grootenhuis, P. D., Bode, W., Huber, R. & Stubbs, M. T. (1997). J. Mol. Biol. 269, 395–407. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cromwell, M. E. M., Hilario, E. & Jacobson, F. (2006). AAPS J. 8, E572–E579. [DOI] [PMC free article] [PubMed]
- Czapinska, H., Otlewski, J., Krzywda, S., Sheldrick, G. M. & Jaskólski, M. (2000). J. Mol. Biol. 295, 1237–1249. [DOI] [PubMed]
- Deisenhofer, J. & Steigemann, W. (1975). Acta Cryst. B31, 238–250.
- Delfín, J., Martínez, I., Antuch, W., Morera, V., González, Y., Rodríguez, R., Márquez, M., Saroyán, A., Larionova, N., Díaz, J., Padrón, G. & Chávez, M. (1996). Toxicon, 34, 1367–1376. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Erlanger, B. F., Kokowsky, N. & Cohen, W. (1961). Arch. Biochem. Biophys. 95, 271–278. [DOI] [PubMed]
- Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., Gavin, O. L., Gunasekaran, P., Ceric, G., Forslund, K., Holm, L., Sonnhammer, E. L., Eddy, S. R. & Bateman, A. (2010). Nucleic Acids Res. 38, D211–D222. [DOI] [PMC free article] [PubMed]
- Fischer, H., Polikarpov, I. & Craievich, A. F. (2004). Protein Sci. 13, 2825–2828. [DOI] [PMC free article] [PubMed]
- García De La Torre, J., Huertas, M. L. & Carrasco, B. (2000). Biophys. J. 78, 719–730. [DOI] [PMC free article] [PubMed]
- Gil, D. F., García-Fernández, R., Alonso-del-Rivero, M., Lamazares, E., Pérez, M., Varas, L., Díaz, J., Chávez, M. A., González-González, Y. & Mansur, M. (2011). FEMS Yeast Res. 11, 575–586. [DOI] [PubMed]
- Huber, R., Kukla, D., Bode, W., Schwager, P., Bartels, K., Deisenhofer, J. & Steigemann, W. (1974). J. Mol. Biol. 89, 73–101. [DOI] [PubMed]
- Kunitz, M. & Northrop, J. H. (1936). J. Gen. Physiol. 19, 991–1007. [DOI] [PMC free article] [PubMed]
- Lafont, S., Veesler, S., Astier, J. P. & Boistelle, R. (1994). J. Cryst. Growth, 143, 249–255. [DOI] [PubMed]
- Laskowski, M. Jr, Qasim, M. A. & Lu, S. M. (2000). Protein–Protein Recognition, edited by C. Kleanthous, pp. 228–279. Oxford University Press.
- Leslie, A. G. W. & Powell, H. R. (2007). Evolving Methods for Macromolecular Crystallography, edited by R. J. Read & J. L. Sussman, pp. 41–51. Dordrecht: Springer.
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otting, G., Liepinsh, E. & Wüthrich, K. (1993). Biochemistry, 32, 3571–3582. [DOI] [PubMed]
- Petersen, L. C., Bjørn, S. E., Olsen, O. H., Nordfang, O., Norris, F. & Norris, K. (1996). Eur. J. Biochem. 235, 310–316. [DOI] [PubMed]
- Pritchard, L. & Dufton, M. J. (1999). J. Mol. Biol. 285, 1589–1607. [DOI] [PubMed]
- Redecke, L., Binder, S., Elmallah, M. I., Broadbent, R., Tilkorn, C., Schulz, B., May, P., Goos, A., Eich, A., Rübhausen, M. & Betzel, C. (2009). Free Radic. Biol. Med. 46, 1353–1361. [DOI] [PubMed]
- Scheidig, A. J., Hynes, T. R., Pelletier, L. A., Wells, J. A. & Kossiakoff, A. A. (1997). Protein Sci. 6, 1806–1824. [DOI] [PMC free article] [PubMed]
- Tung, M. & Gallagher, D. T. (2009). Acta Cryst. D65, 18–23. [DOI] [PubMed]
- Vriend, G. (1990). J. Mol. Graph. 8, 52–56. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wlodawer, A., Walter, J., Huber, R. & Sjölin, L. (1984). J. Mol. Biol. 180, 301–329. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: rShPI-1A, 3ofw
Supplementary material file. DOI: 10.1107/S1744309112039085/en5515sup1.pdf