

The myelin protein P2 is a peripheral membrane protein functional in lipid bilayer binding and stacking. In order to study the fine details of P2 structure and function, 14 point mutants of human P2 were generated and crystallized; a total of eight different crystal forms were obtained, some of which diffracted to atomic resolution.
Keywords: myelin, P2 protein, membrane binding, mutants, fatty acid-binding proteins
Abstract
The myelin sheath is a multilayered membrane that surrounds and insulates axons in the nervous system. One of the proteins specific to the peripheral nerve myelin is P2, a protein that is able to stack lipid bilayers. With the goal of obtaining detailed information on the structure–function relationship of P2, 14 structure-based mutated variants of human P2 were generated and produced. The mutants were designed to potentially affect the binding of lipid bilayers by P2. All mutated variants were also crystallized and preliminary crystallographic data are presented. The structural data from the mutants will be combined with diverse functional assays in order to elucidate the fine details of P2 function at the molecular level.
1. Introduction
The formation of the lipid-rich myelin sheath around axons is crucial to the normal development and function of the vertebrate nervous system. Myelin is a tightly packed membrane multilayer that carries a specific set of proteins that are important for the formation of myelin and for its interactions with the axon (Snipes & Suter, 1995 ▶). A number of neurological diseases, both inherited and autoimmune, are related to myelin abnormalities or degeneration. Myelin-specific proteins are centrally implicated in such diseases, which include peripheral neuropathies and multiple sclerosis (Martin et al., 1992 ▶; Kerlero de Rosbo et al., 1993 ▶; Suter & Scherer, 2003 ▶).
One of the myelin-specific proteins in peripheral nerves is P2, a small 15 kDa protein of the fatty-acid binding protein (FABP) family. P2 is localized into specific areas in compact myelin (Trapp et al., 1984 ▶) and is a peripheral membrane protein (Sedzik et al., 1985 ▶). P2 has been estimated to comprise up to 15% of the total myelin protein (Greenfield et al., 1973 ▶), but its function has remained elusive.
Previously, we have determined the crystal structure of recombinant human P2 in complex with bound palmitate (PDB entry 2wut; Majava et al., 2010 ▶). We also showed that P2 is able to stack lipid bilayers (Suresh et al., 2010 ▶), possibly in synergy with the myelin basic protein, and that it affects lipid-membrane dynamics (Knoll et al., 2010 ▶). While the details of P2–membrane interactions are unclear, the crystal structure suggests the involvement of two opposite faces of P2, both with a high positive charge, in binding between two membranes. Owing to the specific properties of P2 as an FABP and in stacking lipid bilayers, we have a further interest in elucidating the molecular details of the interactions of P2 with lipid bilayers. As one step towards this goal, we designed a panel of mutants based on the human wild-type P2 crystal structure that can be used to study P2 structure and function.
2. Materials and methods
2.1. Mutagenesis and protein production
For mutagenesis, the expression vector for wild-type human P2 (Majava et al., 2010 ▶) containing the human P2 full-length cDNA in the pTH-27 vector (Hammarström et al., 2006 ▶) was used as a template in the QuikChange protocol (Stratagene). The primers used in the mutagenesis procedure are listed in Table 1 ▶. All mutant constructs were sequenced to confirm the presence of the desired mutation. Protein expression and purification (Table 1 ▶) were carried out exactly as previously described for the wild-type protein (Majava et al., 2010 ▶). Briefly, expression was performed in Escherichia coli using autoinduction (Studier, 2005 ▶); after purification of the affinity-tagged protein on Ni–NTA, the His tag was cleaved using TEV protease (van den Berg et al., 2006 ▶). After cleavage of the tag, one extra glycine residue remained at the N-terminus of the recombinant P2 before the starting methionine. The final purification step consisted of size-exclusion chromatography and concentration in 20 mM HEPES pH 7.5, 150 mM NaCl, 10% glycerol.
Table 1. Recombinant protein-production information and primers used for human P2 mutagenesis.
| Source organism | Homo sapiens |
| Expression vector | pTH-27 |
| Expression host | E. coli Rosetta (DE3) |
| Expression protocol | Autoinduction (4 h at 310 K followed by 48 h at 291 K) |
| Primers used for mutagenesis | |
| N2D | 5′-ATTTTCAGGGCATGAGCGACAAATTCCTGGGCACC-3′ |
| K3N | 5′-ATTTTCAGGGCATGAGCAACAATTTCCTGGGCACC-3′ |
| K21Q | 5′-GAACTTTGACGATTACATGCAGGCTCTGGGTGTGGGGTTAG-3′ |
| L27D | 5′-AAGCTCTGGGTGTGGGGGATGCCACCAGAAAACTGGG-3′ |
| R30Q | 5′-GGGTGTGGGGTTAGCCACCCAGAAACTGGGAAATTTGGCCA-3′ |
| K31Q | 5′-GTGGGGTTAGCCACCAGACAGCTGGGAAATTTGGCCAAA-3′ |
| L35S | 5′-AGCCACCAGAAAACTGGGAAATTCGGCCAAACCCAC-3′ |
| P38G | 5′-AACTGGGAAATTTGGCCAAAGGCACTGTGATCATCAGCAAG-3′ |
| K45S | 5′-GCCAAACCCACTGTGATCATCAGCAAGAGCGGAGATATTATAACTA-3′ |
| F57A | 5′-ATATTATAACTATACGAACTGAAAGTACCGCTAAAAATACAGAAATCTCCTTCAAGCTAG-3′ |
| K65Q | 5′-AAAAATACAGAAATCTCCTTCCAGCTAGGCCAGGAATTTGAAG-3′ |
| R88Q | 5′-AAGAGCATCGTAACCCTGCAGCAGGGATCACTGAATCAAGTGCAG-3′ |
| K112Q | 5′-ATAAAGAGAAAGCTAGTGAATGGGCAGATGGTAGCGGAATGTAAAATGAAG-3′ |
| K120S | 5′-AAAATGGTAGCGGAATGTAAAATGAGCGGCGTGGTGTGCA-3′ |
2.2. Crystallization
All of the human P2 mutants were crystallized by sitting-drop vapour diffusion on MRC crystallization plates (Molecular Dimensions). The protein concentration for crystallization was approximately 5 mg ml−1; drops consisting of 0.5 µl protein solution and 0.5 µl well solution were equilibrated against 50 µl well solution. Crystallization experiments were carried out in parallel at 291 and 277 K. The optimized crystallization conditions for each mutant are listed in Table 2 ▶.
Table 2. Crystallization conditions for the crystals used for data collection for the human P2 mutants.
| Mutant | Well solution | Temperature (K) |
|---|---|---|
| N2D | 36% PEG 6000, 0.1 M citrate pH 5.5 | 277 |
| K3N | 35% PEG 6000, 0.1 M citrate pH 5.0 | 277 |
| K21Q | 38% PEG 6000, 0.1 M citrate pH 5.0 | 277 |
| L27D | 38% PEG 6000, 0.1 M citrate pH 5.0 | 291 |
| R30Q | 3.6 M ammonium sulfate, 10% glycerol | 291 |
| K31Q | 32% PEG 6000, 0.1 M citrate pH 5.0 | 277 |
| L35S | 3.6 M ammonium sulfate, 0.1 M HEPES pH 7.0 | 291 |
| P38G | 40% PEG 1000, 0.1 M citrate pH 5.0 | 277 |
| K45S | 45% PEG 550 MME, 0.1 M citrate pH 5.0 | 291 |
| F57A | 42% PEG 6000, 0.1 M bis-tris pH 6.0 | 277 |
| K65Q | 40% PEG 1000, 0.1 M citrate pH 4.5 | 291 |
| R88Q | 40% PEG 1000, 0.1 M citrate pH 5.0 | 291 |
| K112Q | 3.2 M ammonium sulfate, 10% glycerol | 291 |
| K120S | 30% PEG 6000, 0.1 M citrate pH 5.0 | 277 |
2.3. Data collection and processing
Diffraction data were collected on several occasions, using slightly different wavelengths, on the X12 crystallography beamline of the EMBL Hamburg Outstation at the DORIS storage ring of DESY. Data collection was carried out under a stream of gaseous nitrogen at 100 K. Except for the F57A data set, a Rayonics 225 detector was used; for F57A, the detector used was a MAR CCD 165. Diffraction data were processed using XDS (Kabsch, 2010 ▶) and XDSi (Kursula, 2004 ▶) and the data statistics are listed in Table 3 ▶. The structures were solved by molecular replacement with Phaser (McCoy et al., 2007 ▶; Adams et al., 2010 ▶) using the crystal structure of wild-type human P2 (PDB entry 2wut; Majava et al., 2010 ▶) as a model.
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Mutant | N2D | K3N | K21Q | L27D | R30Q | K31Q | L35S |
|---|---|---|---|---|---|---|---|
| Wavelength (Å) | 0.97 | 0.91 | 0.97 | 0.97 | 1.0 | 0.91 | 1.0 |
| Crystal-to-detector distance (mm) | 200 | 250 | 250 | 270 | 300 | 200 | 210 |
| Rotation range per image (°) | 0.5 | 0.5 | 0.5 | 0.4 | 1.0 | 0.3 | 0.5 |
| Total rotation range (°) | 90 | 140 | 90 | 120 | 150 | 120 | 160 |
| Exposure time per image (s) | 20 | 30 | 30 | 60 | 60 | 10 | 20 |
| Space group | P41212 | P41212 | P41212 | P41212 | P212121 | P41212 | P41212 |
| Unit-cell parameters | |||||||
| a (Å) | 58.3 | 64.5 | 58.0 | 64.9 | 58.6 | 58.2 | 65.8 |
| b (Å) | 58.3 | 64.5 | 58.0 | 64.9 | 76.0 | 58.2 | 65.8 |
| c (Å) | 101.7 | 101.2 | 101.2 | 100.9 | 100.6 | 102.2 | 101.3 |
| α = γ (°) | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| β | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| Resolution range (Å) | 30–1.65 (1.70–1.65) | 20–2.70 (2.77–2.70) | 30–2.30 (2.36–2.30) | 30–2.81 (2.88–2.81) | 20–3.00 (3.08–3.00) | 20–1.80 (1.85–1.80) | 20–2.00 (2.05–2.00) |
| Total No. of reflections | 99921 | 67814 | 41115 | 46757 | 43544 | 159455 | 191753 |
| No. of unique reflections | 21068 | 6311 | 8082 | 5609 | 9332 | 16817 | 15619 |
| Completeness† (%) | 97.4 (78.9) | 99.5 (100) | 99.2 (97.6) | 98.8 (98.0) | 98.4 (95.5) | 98.7 (99.8) | 99.8 (98.2) |
| Multiplicity | 4.7 (2.0) | 10.7 (11.2) | 5.1 (4.7) | 8.3 (7.9) | 4.7 (4.6) | 9.5 (9.2) | 12.3 (9.7) |
| 〈I/σ(I)〉 | 15.2 (1.5) | 14.5 (3.1) | 11.1 (2.2) | 12.7 (3.4) | 9.0 (1.2) | 19.0 (2.5) | 19.8 (1.7) |
| R meas ‡ | 0.090 (0.703) | 0.181 (0.838) | 0.153 (0.846) | 0.156 (0.717) | 0.142 (1.885) | 0.117 (0.981) | 0.104 (1.574) |
| CC1/2 § (%) | 99.8 (62.5) | 99.7 (95.0) | 99.5 (70.9) | 99.6 (94.7) | 99.7 (47.9) | 99.9 (78.0) | 99.9 (84.8) |
| Overall B factor from Wilson plot (Å2) | 19 | 37 | 32 | 38 | 81 | 22 | 41 |
| Protein monomers per asymmetric unit | 1 | 1 | 1 | 1 | 2 | 1 | 1 |
| Solvent content (%) | 57 | 65 | 57 | 65 | 67 | 57 | 66 |
| Phaser Z-score¶ | 27 | 45 | 26 | 41 | 28 | 24 | 31 |
| Mutant | P38G | K45S | F57A | K65Q | R88Q | K112Q | K120S |
|---|---|---|---|---|---|---|---|
| Wavelength (Å) | 1.03 | 1.03 | 1.10 | 1.03 | 0.91 | 1.0 | 0.91 |
| Crystal-to-detector distance (mm) | 200 | 250 | 65 | 110/220 | 200 | 180 | 320 |
| Rotation range per image (°) | 0.25 | 0.4 | 0.5 | 0.5/1.0 | 0.25 | 0.5 | 0.5 |
| Total rotation range (°) | 180 | 180 | 180 | 113/120 | 125 | 150 | 115 |
| Exposure time per image (s) | 20 | 30 | 10 | 30/10 | 10 | 20 | 30 |
| Space group | P212121 | C2 | C2 | P43 | P41212 | P41212 | P41212 |
| Unit-cell parameters | |||||||
| a (Å) | 55.5 | 120.1 | 112.8 | 83.0 | 64.9 | 65.7 | 63.7 |
| b (Å) | 65.5 | 63.6 | 36.1 | 83.0 | 64.9 | 65.7 | 63.7 |
| c (Å) | 82.2 | 84.5 | 31.2 | 77.9 | 264.4 | 101.2 | 101.4 |
| α = γ (°) | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| β | 90 | 130.1 | 96.9 | 90 | 90 | 90 | 90 |
| Resolution range (Å) | 20–1.80 (1.85–1.80) | 20–2.20 (2.26–2.20) | 20–1.27 (1.30–1.27) | 20–1.20 (1.23–1.20) | 40–1.80 (1.85–1.80) | 25–1.80 (1.85–1.80) | 20–2.90 (2.98–2.90) |
| Total No. of reflections | 142607 | 76070 | 93246 | 787155 | 389891 | 248391 | 35641 |
| No. of unique reflections | 25915 | 22051 | 31894 | 154301 | 53492 | 21102 | 4976 |
| Completeness† (%) | 91.0 (58.4) | 88.2 (49.4) | 96.8 (77.4) | 93.9 (63.3) | 99.4 (96.9) | 99.6 (96.0) | 99.6 (97.4) |
| Multiplicity | 5.5 (3.7) | 3.4 (2.8) | 2.9 (1.8) | 5.1 (2.1) | 7.3 (5.9) | 11.8 (11.2) | 7.2 (7.3) |
| 〈I/σ(I)〉 | 21.8 (2.2) | 11.1 (1.7) | 8.2 (1.2) | 19.6 (1.4) | 19.8 (2.0) | 30.3 (2.1) | 10.5 (1.8) |
| R meas ‡ | 0.054 (0.664) | 0.092 (0.728) | 0.092 (0.781) | 0.044 (0.785) | 0.085 (0.841) | 0.063 (1.262) | 0.144 (1.445) |
| CC1/2 § (%) | 99.9 (77.0) | 99.7 (75.3) | 99.6 (43.5) | 100 (56.2) | 99.9 (67.3) | 100 (83.4) | 99.8 (85.9) |
| Overall B factor from Wilson plot (Å2) | 32 | 43 | 17 | 17 | 27 | 34 | 72 |
| Protein monomers per asymmetric unit | 2 | 3 | 1 | 4 | 3 | 1 | 1 |
| Solvent content (%) | 51 | 55 | 41 | 45 | 60 | 66 | 64 |
| Phaser Z-score¶ | 29 | 41 | 10 | 47 | 33 | 32 | 32 |
In cases of low completeness in the highest resolution shell, the cause is usually data collection on a square detector, where data were processed all the way to the corners.
R
meas, the redundancy-independent R factor (Diederichs & Karplus, 1997 ▶), is defined as 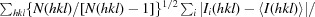
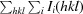 .
.
CC1/2 is the correlation coefficient between two random half data sets, as described by Karplus & Diederichs (2012 ▶).
The shown Z-score is that for the Phaser translation function upon finding the last monomer in the asymmetric unit.
3. Results and discussion
Based on the crystal structure of human P2 and its predicted membrane-binding surfaces on opposite faces of the protein (Majava et al., 2010 ▶), we generated a panel of 14 point mutants for structure–function studies. The goal is to use these mutants to combine the functional data from various membrane-binding assays with high-resolution structural data. This in turn will elucidate the fine detail of the function of P2 in myelin membrane stacking.
We mutated both basic surface residues as well as hydrophobic residues predicted to enter into the hydrophobic layer upon bilayer binding. The locations of the mutated residues in the wild-type P2 structure are depicted in Fig. 1 ▶. All of the generated mutant variants were successfully produced and purified on a large scale and all of them also produced diffraction-quality crystals (Fig. 2 ▶, Table 3 ▶). Crystals were formed under two basic conditions: high PEG concentrations around pH 5 and high ammonium sulfate concentrations at neutral pH. Despite the limited variability in the crystallization conditions, a number of different crystal forms were observed, which is a sign of different packing induced by the surface mutations.
Figure 1.

Stereoview of the locations of the mutated residues in the human wild-type P2 crystal structure (PDB entry 2wut; Majava et al., 2010 ▶). The bound palmitate molecule inside P2 is shown in white. The mutated residues are highlighted in yellow and labelled. The predicted membrane-binding surfaces are at the top and bottom in this view.
Figure 2.

Crystals of human P2 mutants. The scale bar at the bottom right applies to all images.
Interestingly, the best crystals, which were of the K65Q mutant, diffracted X-rays to atomic resolution (1.2 Å), while our previous data from the wild-type protein only extended to 1.8 Å resolution (Majava et al., 2010 ▶). The K65Q crystals belonged to the same space group as the wild-type protein, but the a and b unit-cell dimensions were >10% shorter, indicating much tighter packing. The F57A mutant also diffracted to better than 1.3 Å resolution. Even for those data sets for which 〈I/σ(I)〉 in the highest-resolution shell was below 2, the correlation coefficient between random half data sets (CC1/2; Karplus & Diederichs, 2012 ▶) indicated significant information content (Table 3 ▶). If required in the future, higher resolution data can be collected from some of the mutants.
The structures of all mutants have now been solved by molecular replacement (Table 3 ▶) and the crystal structures are currently under refinement and detailed analysis. The mutants are simultaneously being used for functional studies of the human P2 protein both in vitro and in vivo.
Acknowledgments
This study was financially supported by the Academy of Finland, the Sigrid Juselius Foundation, the European Spallation Source and the Department of Biochemistry, University of Oulu. We wish to thank the excellent support at the EMBL Hamburg crystallography beamlines during this project.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Berg, S. van den, Löfdahl, P. A., Härd, T. & Berglund, H. (2006). J. Biotechnol. 121, 291–298. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- Greenfield, S., Brostoff, S., Eylar, E. H. & Morell, P. (1973). J. Neurochem 20, 1207–1216. [DOI] [PubMed]
- Hammarström, M. Woestenenk, E. A., Hellgren, N., Härd, T. & Berglund, H. (2006). J. Struct. Funct. Genomics, 7, 1–14. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Karplus, P. A. & Diederichs, K. (2012). Science, 336, 1030–1033. [DOI] [PMC free article] [PubMed]
- Kerlero de Rosbo, N., Milo, R., Lees, M. B., Burger, D., Bernard, C. C. & Ben-Nun, A. (1993). J. Clin. Invest. 92, 2602–2608. [DOI] [PMC free article] [PubMed]
- Knoll, W., Natali, F., Peters, J., Nanekar, R., Wang, C. & Kursula, P. (2010). Spectroscopy, 24, 585–592.
- Kursula, P. (2004). J. Appl. Cryst. 37, 347–348.
- Majava, V., Polverini, E., Mazzini, A., Nanekar, R., Knoll, W., Peters, J., Natali, F., Baumgärtel, P., Kursula, I. & Kursula, P. (2010). PLoS One, 5, e10300. [DOI] [PMC free article] [PubMed]
- Martin, R., McFarland, H. F. & McFarlin, D. E. (1992). Annu. Rev. Immunol. 10, 153–187. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Sedzik, J., Blaurock, A. E. & Hoechli, M. (1985). J. Neurochem. 45, 844–852. [DOI] [PubMed]
- Snipes, G. J. & Suter, U. (1995). J. Anat. 186, 483–494. [PMC free article] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Suresh, S., Wang, C., Nanekar, R., Kursula, P. & Edwardson, J. M. (2010). Biochemistry, 49, 3456–3463. [DOI] [PubMed]
- Suter, U. & Scherer, S. S. (2003). Nature Rev. Neurosci. 4, 714–726. [DOI] [PubMed]
- Trapp, B. D., Dubois-Dalcq, M. & Quarles, R. H. (1984). J. Neurochem. 43, 944–948. [DOI] [PubMed]