

Abstract
A novel class of isochroman dopamine analogues, 1, originally reported by Abbott Laboratories, had greater than 100-fold selectivity for D1-like vs. D2-like receptors. We synthesized a parallel series of chroman compounds, 2, and showed that repositioning the oxygen in the heterocyclic ring reduced potency and conferred D2-like receptor selectivity to these compounds. In silico modeling supported the hypothesis that the altered pharmacology for 2 was due to potential intramolecular hydrogen bonding between the oxygen in the chroman ring and the meta-hydroxyl of the catechol moiety. This interaction realigns the catechol hydroxyl groups and disrupts key interactions between these ligands and critical serine residues in TM5 of the D1-like receptors. This hypothesis was tested by the synthesis and pharmacological evaluation of a parallel series of carbocyclic compounds, 3. Our results suggest that when the potential for intramolecular hydrogen bonding is removed, D1-like receptor potency and selectivity is restored.
Keywords: catechol, D1-selective agonist, dopamine, drug design, intramolecular hydrogen bond
Introduction
Dopamine is an important neurotransmitter involved in several brain neuronal pathways, including reward circuitry, cognitive function, locomotion, and prolactin release. It also has several peripheral actions, including proper kidney functioning. Dopaminergic dysfunction can have a profound effect on the human body, perhaps some of the most well recognized being Parkinson’s disease and schizophrenia. Drug addiction, obesity, depression, and other mood and cognitive disorders also are directly linked to improper functioning of dopaminergic neurotransmission.[1] Elucidation of the physiological roles of the dopamine receptor subtypes is a main driving force behind the synthesis of compounds that act as selective agonists or antagonists at these sites.[2] Such selective agents not only could yield a greater understanding of dopamine neuropharmacology, but also could potentially be employed as novel therapies.
All dopamine receptors belong to the seven transmembrane G-protein-coupled family of receptors (GPCRs) consisting of seven hydrophobic transmembrane alpha helices.[3] The numerous actions of dopamine are mediated by five types of receptors, divided into two main families: the D1-like family, and the D2-like family.[4] The D1-like family includes the D1 and D5 receptors and, through coupling with Gαs/Gαolf proteins, increases the production of cAMP by activating adenylate cyclase. The D2-like family, consisting of the D2, D3, and D4 receptors, is coupled t o Gαi/Gαo proteins and decreases the activity of adenylate cyclase, or couples to other signaling pathways.
The native ligand binding site (orthosteric site) is located in a hydrophobic region surrounded by the seven transmembrane regions. Based on deletion mutations and molecular modelling studies of the D1 receptor active site, Asp 103(3.32) in TM 3 is most likely responsible for binding the protonated nitrogen of the dopamine ethylamine side chain, whereas Ser 198(5.42), Ser 199(5.43), and Ser 202(5.46) (in TM 5) are involved in binding to the catechol hydroxyls.[5-7] The putative binding pocket of the D1 receptor also contains an accessory binding region, deduced from the high affinity of compounds containing a phenyl substituent at the beta side chain position (β-phenyldopamine, Figure 1).[8, 9] Similarly, the D2 receptor has an aspartate residue in TM 3 (ASP 114) involved in binding the protonated amine. Two or three serines (Ser 193, 194, 197) in transmembrane helix 5 are critical for binding the catechol moiety through hydrogen bonding; however, there is no analogous accessory region to accommodate β-phenyl substituents in the D2-like receptors. By analyzing the structures of known D1-like selective agonist ligands and comparing them with known D2-like selective ligands, it is apparent that a catechol moiety is crucial to conferring D1-like potency and selectivity, whereas several non-catechol agonist molecules possess D2-like selectivity. It can thus be presumed that the hydrogen bonding network in the D1-like receptors is more complex and less permissive than that in the D2-like receptors.[10]
Figure 1.
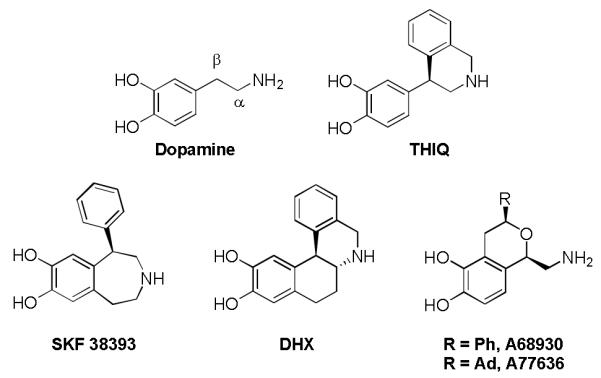
Dopamine and D1-like Selective Agonists
Figure 1 depicts several classes of compounds known to show selectivity toward the D1-like receptors, including 1-phenyl-3-benzazepines (SKF 38393),[11] 4-phenyl-1,2,3,4-tetrahydroisoquinolines (THIQ),[12] benzo[a]phenanthridines (DHX),[13] and isochromans (A68390).[14, 15] The compounds in this last family, first synthesized by Abbott laboratories in the early 1990’s, are extremely potent and selective D1-like dopamine agonists with high intrinsic activity. Although these compounds do not have a β-phenyl moiety, the spatial and electronic and/or hydrophobic characteristics that mimic the β-phenyl moiety are still present in the series. Interestingly, large non-aromatic substituents, such as the adamantyl (A77636, 1c), may be substituted for the phenyl (A68930, 1d) in these molecules to provide structures with high potency and D1-like selectivity.
An attempt by our laboratory to develop a new D1-like selective template based on an oxygen bioisostere of the Abbott series of molecules was surprisingly unsuccessful. As depicted in Figure 2, we constructed chroman analogues, 2a-c, 2e, with substituents analogous to the Abbott isochroman series, 1a-d. Our chroman series did not yield compounds that were either potent or selective for the D1-like receptors, as we had anticipated, but did offer important new insights into the poorly understood hydrogen-bonding networks of the D1-like dopamine receptors. Importantly, the chroman series possesses a potential intramolecular hydrogen bond, as shown in Figure 3, which can force a specific orientation of the catechol hydroxyl groups. We hypothesized that this orientation potentially changes the alignment of the molecule in the D1-like binding pocket, decreasing its affinity and activity. There is no controversy in the literature as to whether such a bond exists, but only on the best way to measure its strength. Estacio et al.[16] have calculated intramolecular H bond enthalpies for ortho-methoxyphenol using the ortho-para method and three theory levels, and obtained values ranging from 9.8-11.6 kJ/mol. Varfolomeev et al.[17] have recently presented both experimental and computational evidence that such an intramolecular hydrogen bond is nearly the exclusive conformation of ortho-methoxyphenol in infinite dilution. In the relatively hydrophobic interior of the receptor one would therefore expect such an intramolecular H bond to be highly favorable.
Figure 2.
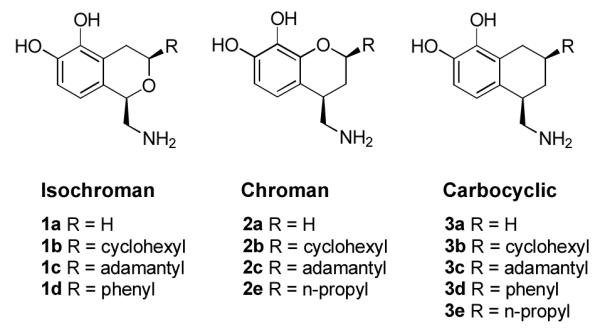
Bicyclic dopamine analogues evaluated in this study
Figure 3.
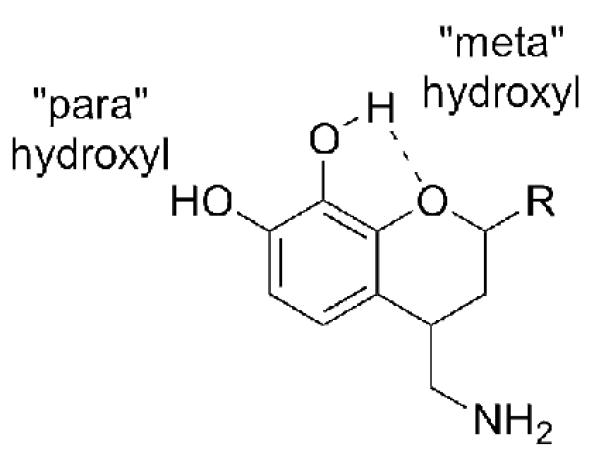
Intramolecular hydrogen bond is only possible with the catechol moiety in the chroman series, 2
To test the hypothesis that this intramolecular hydrogen bond is responsible for the unexpected pharmacology of the chroman series, the carbocyclic series of compounds, 3a-e, also was synthesized and evaluated for activity at D1-like and D2-like receptors. We now present both the synthesis and pharmacological evaluation of the chroman and carbocyclic series compared with parallel analysis of the isochroman series. Our discussion further considers the putative hydrogen bonding network in the D1 receptor.
Results
Chemistry: Chroman Series
The chroman compounds were synthesized from the common intermediate 9 (Scheme 1). Treating pyrogallol with ethyl acetoacetate in neat sulfuric acid with cooling yielded 7,8-dihydroxy-4-methylcoumarin, 4. The yields were low, but both starting materials are inexpensive and large quantities (100 g) could be quickly produced. Both the catechol protection (5) and the 4-methyl oxidation (6) proceeded in good yield. The choice of O-benzyl protection proved a good one in that it survived the harsh selenium dioxide oxidation conditions. Reductive amination then introduced the benzylamino side chain (7) in modest yield.[18] If crude 6 was first purified by chromatography, however, yields up to 80% could be obtained. For further elaboration of the molecule, we were forced to include an additional N-benzyl group (8), because of the remaining relatively acidic amine proton.
Scheme 1. Synthesis of 9.
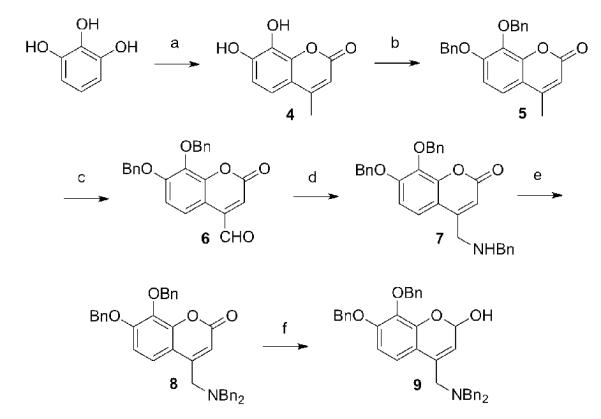
Reagents and conditions: (a) ethyl acetoacetate, H2SO4, 25%; (b) BnBr, K2CO3, DMF, 85%; (c) SeO2, xylenes, 150 °C, 12h, 61%; (d) BnNH2, NaBH3CN, 50-80%; (e) BnBr, K2CO3, DMF, 100 °C, 90%; (f) DiBAlH, CH2Cl2, −78 °C, 72%.
At this point, we attempted to introduce aliphatic substituents at the 2-position of 8. Treatment of the tetrabenzyl lactone 8 with cyclohexylmagnesium chloride resulted in double addition and ring opening. Similar results were obtained with phenyl lithium and phenyl magnesium bromide in either anhydrous THF or diethyl ether. Treatment with the corresponding ceriummagnesium complex,[19] selective for monoaddition[20] to lactones was unsuccessful, as was attempted olefination with Tebbe reagent.[21] Therefore, the lactone was reduced to the lactol 9. For this reaction, DiBAl-H in a solution of dichloromethane was the reagent of choice. The unstable lactol 9 did not require purification before further reaction. Deoxygenation of such lactol systems with boron trifluoride diethyl etherate (BF3·OEt2) results in formation of oxonium ions, which in turn may be reduced with a hydride source[22] or trapped with an appropriate nucleophile.[23, 24] A deep red color was formed, indicative of the oxonium ion, but attempts to trap it with a variety of electrophilic reagents such as cyclohexylMgCl, phenylMgBr, adamantylMgCl, or adamantylZnCl at either 0 °C or −78 °C yielded numerous products. Triethylsilane did prove to be an effective hydride source to yield the unsubstituted 10 (Scheme 2). We were then successful in attaching an allyl group (11) in good yield by this methodology.[24, 25] Catalytic hydrogenation then afforded the reduced propyl compound 2e. Optimal yields were obtained when a large excess (4.5 molar eq.) of palladium was used. Use of the hydrochloride salt of the amine gave the best results.
Scheme 2. Synthesis of 2a-c, e.

Reagents and conditions: (a) BF3·Et2O, Et3SiH, CH2Cl2, 0 °C; (b) BF3·Et2O, allylTMS, CH2Cl2, 0 °C; (c) CyMgCl or AdMgBr; (d) Mitsunobu conditions; (e) H2, Pd/C, EtOH, 1 atm.
For the introduction of other substituents to the common intermediate 9, the synthesis was altered slightly. Adding a large excess (10 eq.) of the appropriate organomagnesium reagent to lactol 9 resulted in monoaddition and ring-opened diol 12.[26] These compounds did not require isolation and, when subjected to Mitsunobu conditions,[27] cyclized smoothly to the desired tetrabenzylchromans 13. The yields were about 50% for both compounds over two steps, which were deemed acceptable. The catechols (2a, 2b, and 2c) were obtained employing the same hydrogenation methodology used for the propylcatechol 2e.
Having successfully synthesized several 2-alkyl-substituted compounds, we attempted to produce the 2-phenyl compound by this same approach (Scheme 3). Addition of phenylmagnesium bromide to 9 produced the expected diol 14 in a good yield. Unfortunately, numerous attempts to cyclize this compound by the Mitsunobu reaction failed. Variations in temperature, order of addition, and type of phosphine (Bu3P, Ph3P) all resulted in complex mixtures that appeared to arise from deoxygenation of the activated allylic-benzylic alcohol.
Scheme 3. Attempted synthesis of phenyl-substituted chroman.
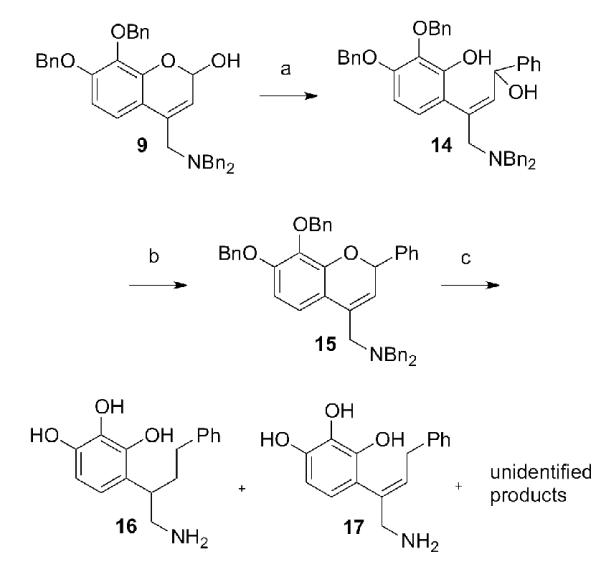
Reagents and conditions: (a) PhMgBr, 85%; (b) SOCl2, pyridine, 0 °C, 62%; (c) H2, catalyst (see text).
This problem was successfully circumvented by replacing the problematic alcohol with a chlorine atom and performing a nucleophilic base-promoted cyclization, all in one step, to provide 15. The most important factor for the successful generation of 15 was cooling of the reaction mixture, because HCl generated by the reaction catalyzed cleavage of the ether linkage at slightly elevated temperatures. Unfortunately, only ring-opened products (16 and 17) were obtained from attempts to hydrogenate this system. The benzylic ether could not be preserved, even when attempting hydrogenations with a variety of different catalysts, including Pd/C, Pt/C, Pd black, Lindlar catalyst, Wilkinson’s catalyst, Pearlman’s catalyst, and Adams catalyst.[28-30] Variations in pressure (1-4 atm), catalyst ratios, and solvents always produced complex mixtures of products. Various amines are sometimes also used to “poison” (deactivate) hydrogenation catalysts to alter their selectivity.[31, 32] Addition of triethylamine or pyridine (varying amounts) to palladium black, palladium, or Pearlman’s catalyst also produced mixtures containing unreacted starting material. Finally, transfer hydrogenation with a large excess of diimide, generated from potassium azodicarboxylate, yielded no products, even at elevated temperatures.[33]
At this point, overwhelming evidence pointed to the extreme instability of the 2-phenylchroman skeleton. In hindsight, this was to be expected considering that in this particular ring system, it is highly probable that neighboring group participation of the pendant phenyl moiety favors the ring opening. Therefore, further efforts to prepare this compound were abandoned.
Chemistry: Carbocyclic Series
Unfortunately, in the carbocyclic series, there appeared to be no tractable way to incorporate the aliphatic or aromatic functionality at a late stage in the synthesis that would allow the divergent use of a common intermediate; therefore, the substituent had to be incorporated at the very beginning of each synthesis. Both the unsubstituted and phenyl-substituted carbocyclic compounds have been reported previously, although they were not pharmacologically evaluated for selectivity at dopamine receptor subtypes. The phenyl compound 3d was made according to the procedure reported by Schoenleber and colleagues.[34] We were able to synthesize the unsubstituted compound 3a more efficiently than previously published,[35] however, as described below.
As depicted in Scheme 4, the first step in the synthesis of 3a was the formation of paraconic acid 18 from commercially available 2,3-dimethoxybenzaldehyde and succinic anhydride.[36] The pure, crystalline paraconic acid was then heated to effect ring opening and decarboxylation to afford 19. The reaction typically began to yield side products before all of the starting material was consumed. The starting paraconic acid and product butenoic acid have pKas that differ by nearly one pH unit and thus were separated by careful titration, with the non-decarboxylated paraconic acid easily recovered. Unsaturated acid 19 was then catalytically hydrogenated, and polyphosphoric acid was used to form tetralone 21, giving a nearly quantitative yield over two steps. Trimethylsilylcyanide and BF3·OEt2 were allowed to react with the tetralone to add the nitrile to the carbonyl and dehydrate the resulting protected alcohol in one step.[37, 38] The unsaturated nitrile 22 was reduced with H2 over Raney nickel to the aminomethyl tetralin 23, which was O,O-demethylated and crystallized from methanol-ethyl acetate to afford 3a as the hydrobromide salt.
Scheme 4. Synthesis of 3a.

Reagents and conditions: (a) ZnCl2, NEt3, CH2Cl2, 74%; (b) 180 °C, 65%; (c) H2, 5% Pd/C, EtOH, quant.; (d) PPA, 80 °C, 98%; (e) TMSCN, BF3·Et2O, toluene, 84%; (f) H2, Raney Ni, NH4OH, MeOH, 49%; (g) 1. BBr3, CH2Cl2, 2. MeOH, 99%.
The aliphatically-substituted compounds were prepared in a fashion similar to the patented procedure for the phenyl compound, with several key differences (Scheme 5). An aldol reaction between 2,3-dimethoxybenzaldehyde and the ethyl ester of the appropriate substituted acetic acid yielded benzylic alcohols 25, but which were completely resistant to dehydration, most likely due to an intramolecular hydrogen bond. The alcohols were thus converted to their benzylic chlorides by treatment with thionyl chloride. These chloroesters were dehalogenated and reduced to alcohols 26 in one step with LiAlH4.[39] The resulting primary alcohols were then efficiently mesylated, followed with nucleophilic substitution by cyanide ion to afford nitriles 28. Hydrolysis of the nitriles proved non-trivial, however, and could not be achieved under a variety of stringent reaction conditions. Katsuri and colleagues have described the difficulties in hydrolyzing sterically crowded nitriles in both acidic and basic conditions.[40, 41]
Scheme 5.

Synthesis of 3b, c, and e. Reagents and conditions: (a) 1. LDA, THF, 2. 2,3-dimethoxybenzaldehyde; (b) 1. SOCl2, benzene, 2. LiAlH4, ether; (c) MsCl, NEt3, THF; (d) NaCN, DMSO; (e) 1. DiBAlH, toluene, 2. CrO3, aq. H2SO4 (Jones’ reagent), acetone; (f) PPA, 180 °C; (g) 1. TMSCN, ZnI2, CH2Cl2, 2. LiAlH4, Et2O, 3. HCl (conc.); (h) EtOH, reflux; (i) 4 atm H2, PtO2, EtOH; (j) 1. BBr3, CH2Cl2, 2. MeOH.
We therefore treated the nitriles 28 with DiBAl-H to provide the intermediate aldehydes.[42] Isolation of the aldehydes proved quite difficult, as they quickly decomposed. After confirming the structures by mass spectrometry and 1H NMR, the crude materials were used directly in the next reaction without further purification. The aldehydes were oxidized to the carboxylic acids 29 with Jones’ reagent[43] and then easily closed to the tetralones 30 with polyphosphoric acid. It was discovered during thesynthesis of the adamantyl carboxylic acid that prolonged treatment with Jones’ reagent could actually yield the tetralone directly from the aldehyde, explaining the low yield of the isolated acid.
After treatment of the tetralones 30 with trimethylsilylcyanide and zinc iodide to make the TMS-protected alcohol, the cyano functionalities were immediately reduced to the primary amines and the alcohol moieties were simultaneously deprotected with LiAlH4. The hydrochloride salts of aminoalcohols 31 were dehydrated by reflux in ethanol with a trace amount of 2N ethanolic HCl added to catalyze the reaction, and unsaturated amines 32 were then reduced catalytically to afford the desired cis isomers of the saturated aminomethyl compounds 33. This material was carried forward to the final O,O-demethylation step. The catechols 3, isolated as their hydrobromide salts, were off-white solids, and were submitted for pharmacological evaluation.
NMR evidence supports the fact that the newly synthesized series of compounds are in fact the cis diastereomers. 2D NOESY studies show the coupling of the diaxial protons (data not shown). In the carbocyclic series, a quartet around 1.0 ppm with a high J value (approx. 12 Hz) is present, and is the signal for the axial hydrogen on carbon-2 (identified by COSY). The two neighboring axial protons, as well as its geminal neighbor, split the signaling proton equally with the large coupling constant, typical of both diaxial and geminal splitting.
Pharmacology
The dopamine D1-like and D2-like receptor affinities of compounds 1a-d, 2a-c, 2e, and 3a-e were evaluated in competition binding assays using porcine striatal tissue homogenates. Standard antagonist ligands for D1-like and D2-like receptors, SCH 23390 and chlorpromazine, respectively, also were assessed for comparison to the new compounds (Table 1). All test compounds were full agonists at the cloned human D1 receptor (data not shown).
Table 1.
Binding Affinities at D1-like and D2-like Dopamine Receptors
| Porcine Striatal Binding,[a] Ki, (nM) | |||
|---|---|---|---|
| Ligand | D1-like | D2-like | Fold D1-like Selectivity |
| DHX[b] | 10 ± 0.8 | 370 ± 10 | 37 |
| 1a | 80 ± 9 | 1960 ± 140 | 24.5 |
| 1b | 3.4 ± 0.6 | 920 ± 140 | 270 |
| 1c | 3.9 ± 0.6 | 1860 ± 180 | 480 |
| 1d | 2.6 ± 0.27 | 240 ± 45 | 92 |
| 2a | 9100 ± 1300 | 290 ± 40 | 0.032 |
| 2b | 770 ± 100 | 4600 ± 810 | 6.0 |
| 2c | 4200 ± 760 | 6700 ± 460 | 1.6 |
| 2e | 3100 ± 810 | 810 ± 150 | 0.26 |
| 3a | 1100 ± 70 | 2000 ± 250 | 1.8 |
| 3b | 40 ± 2.5 | 3500 ± 700 | 88 |
| 3c | 220 ± 36 | 12200 ± 1530 | 55 |
| 3d | 23 ± 4.3 | 770 ± 60 | 33 |
| 3e | 270 ± 50 | 2500 ± 320 | 9.3 |
| SCH-23390 | 0.79 ± 0.1 | N.D, | |
| chlorpromazine | N.D. | 3.2 ± 0.5 | |
All results shown are the mean ± SEM for at least three independent experiments
Dihydrexidine
Discussion
The present work evaluated a series of closely related bioisosteres for binding affinity at D1- and D2-like dopamine receptors in porcine striatal homogenates. In an effort to compare the three series of compounds accurately, the Abbott isochromans 1, were assessed in parallel with the newly synthesized chroman 2, and carbocyclic series 3. The results of the porcine striatal binding assays are summarized in Table 1. It was surprising to us that unsubstituted isochroman 1a analogue displayed reasonable D1:D2 receptor subtype selectivity, being modestly D1-like selective (24-fold) despite lacking a substituent engaging the accessory binding region. Chroman 2a, by contrast, although having low affinity, actually showed selectivity for D2-like receptors. Also surprising was the relatively low affinity of the unsubstituted chroman and carbocyclic compounds for the D1-like receptors (Ki > 1 μM) when compared to the unsubstituted isochroman 1a. Based on previous reports for the isochromans, we hypothesized that the hydrophobic substitutions on each of the analogues would increase D1 receptor affinity by engaging the receptor accessory binding region.[14, 15] The results of the receptor binding studies support our hypothesis, revealing that all of the substituents increase D1-like receptor affinity. The increases in affinity for the isochromans are most pronounced (>20 fold), whereas more modest increases in D1-like affinity were observed for the chroman (2-10-fold) and the carbocyclic (4-25-fold) series of compounds.
The unsubstituted chroman 2a was actually selective for D2-like receptors, with very poor affinity at D1-like receptors. It is our hypothesis that this poor D1-like receptor affinity is a consequence of an intramolecular hydrogen bond that disrupts the crucial hydrogen-bond network necessary within the D1 receptor binding site. For the other chroman compounds 2b-e, each of the substitutions provided an increase in D1-like selectivity through a combination of increased D1-like affinity and decreased D2-like affinity. For example, the cyclohexyl substituted 2b shows 6-fold selectivity for D1-like receptors. Presumably, the benefit of having the cyclohexyl group in the accessory binding region of the D1-like receptor compensates to some extent for the disruption of the hydrogen bonding network caused by the intramolecular hydrogen bond. When there is no possibility of this intramolecular hydrogen bond in the carbocyclic 3b, the D1-like selectivity is increased to 88-fold, with recovery of significant D1-like affinity.
The same pattern is present among the adamantyl series as well (1c, 2c, 3c). Figure 4 depicts each of these three molecules docked into our in silico-activated human dopamine D1 receptor homology model. As described in the supporting information, an in silico activated model of the β2 adrenergic receptor was first generated. A homology model was then constructed from this receptor and, using unbiased routines, the ligands were docked and the structures of the resulting complexes were optimized using energy minimization and molecular dynamics (MD) simulations. Panel A shows 1c, a very high affinity D1-like ligand, participating in a likely hydrogen bonding network. The meta hydroxyl of the catechol moiety is involved in a hydrogen bond with Ser 198. The para hydroxyl is hydrogen bonding to Ser 202, which in turn hydrogen bonds to Thr 108. These results are consistent with a study of DHX and its mono-hydroxy analogues in the D1 receptor containing Ser-Ala point mutations.[44, 45]
Figure 4.

Simulated binding poses of 1c (Panel A), 2c (Panel B), and 3c (Panel C) illustrating how the heterocyclic oxygen atom in the chroman disrupts the hydrogen bonding network in the D1 receptor. The view is within the membrane, looking from helices 3 and 5, with helix 4 hidden to allow a better view of the hydrogen bonding networks. The aspartate in TM3 is to the left of the molecule, with the three TM5 serines at the bottom right in each panel. Thr 108 is at the very bottom, and Asn292 (6.55 in TM6) is at the top right.
When 2c undergoes equivalent docking and unconstrained molecular dynamics simulations in our D1 receptor model, a different hydrogen bond network is formed (Figure 4, Panel B). The meta hydroxyl moiety is not available to interact with the protein residues because it is tightly held in an intramolecular hydrogen bond to the heterocyclic oxygen atom. This disruption alters the binding of the ligand in the receptor, as reflected in its low binding affinity at D1-like receptors. Note that both Ser 198 and Ser 202 engage the para-OH of the ligand. By contrast, the binding of 3c in the receptor model establishes a hydrogen bonding network identical to that of 1c (Panel C). This observation directly supports our hypothesis and is validated by its nearly 20-fold higher binding affinity at D1-like receptors compared to 2c. Although we cannot be certain that these illustrations show the exact docked poses for the ligands we studied, they do illustrate how the intramolecular hydrogen bond in the chroman affects a potential hydrogen bonding scheme, and importantly, the proposed docking modes are consistent with our experimental receptor binding and potency results. Even though we had initially hypothesized, based entirely on chemical principles, that intramolecular hydrogen bonding in the chromans was responsible for their unexpected pharmacology, it was gratifying to observe the altered hydrogen bonding pattern in the unbiased docking results.
We do acknowledge that the carbocyclic series does not fully recover D1 affinity or selectivity and that the heterocyclic oxygen atom in the isochroman series may play a role in D1 receptor binding, in addition to the catechol moiety. One potential explanation is that the heterocyclic oxygen in the isochromans is interacting with a polar residue in the D1-like orthosteric binding site. Without a heterocyclic oxygen, the carbocyclic lacks this additional interaction. This hypothesis was explored through sitedirected mutagenesis of the human D1 receptor with no evidence that any of the mutated residues interacted with the heterocyclic oxygen atom (data not shown).
Another, perhaps more plausible explanation, is that intramolecular hydrogen bonding occurs between the heterocyclic oxygen atom and the hydrogens on the amine nitrogen. Such an interaction would reduce the degrees of rotational freedom, and energetically favor an orientation of the aminomethyl side chain that is more complementary to the binding site. There is no possibility for this hydrogen bond in the carbocyclic molecules, and thus their flexible aminomethyl side chains can adopt various conformations, many of which are presumably not favorable for interaction with the Asp 103 in the binding site. An intramolecular hydrogen bond between the isochroman oxygen and the side chain amino group of the ligand could stabilize the active binding orientation, decreasing entropy and likely also would offset the energy required to desolvate the ligand when it enters the receptor binding site.
Such an intramolecularly hydrogen bonded side chain of 1d would have a conformation that is essentially superimposable on an octahydrobenz[h]isoquinoline ring system. We have recently synthesized and evaluated such a benz[h]isoquinoline compound[46] and discovered that, compared to 3d, it possesses a nearly 4-fold increase in D1-like affinity, a D1-like selectivity increase from 33-fold to 73-fold, and a nearly 3-fold increase in potency. The significant increases of affinity, selectivity, and potency over 3d are consistent with the hypothesis of an intramolecular hydrogen bond.
Conclusion
We have analyzed three analogous bicyclic dopamine agonist series for their differential ability to bind to D1-like and D2-like receptors. It is well-known that D1-like-selective agonists require a catechol moiety to bind and to activate fully the D1-like receptors.[47] We have synthesized a series of catechol-containing chroman compounds that do not bind well to the D1-like receptors due to a hypothesized intramolecular hydrogen bond that we speculate interferes with the interaction between the catechol moiety and residues in the receptor responsible for binding. In essence, this intramolecular hydrogen bond destroys the functional characteristics of the ligand catechol moiety, so that for the purposes of ligand-receptor interactions, the ligand effectively possesses only one OH group. Unbiased docking studies with our homology model of the activated D1 receptor are consistent with the pharmacological data, illustrating disruption of the catecholserine hydrogen bonding network that is observed for potent compounds in the isochroman series. When a carbocyclic analogue was synthesized that lacked the ability to intramolecularly hydrogen bond, both the D1-like selectivity and the model’s hydrogen bond network were largely restored, supporting our hypothesis and leading to new insights into the complex hydrogen bonding network of the D1-like receptors. With this new information, it may be possible to design noncatechol compounds with similar hydrogen bonding abilities that would be more bioavailable and metabolically stable. Such molecules would be much improved drug candidates to treat disorders where dopamine D1 receptor activation would be therapeutic.
Experimental Details
Chemistry
General
All reagents were commercially available (Aldrich, Alfa Aesar) and were used without further purification unless otherwise indicated. Dry THF was distilled immediately before use from benzophenone-sodium under argon. Column chromatography was carried out using SiliCycle SiliaFlash P60 silica gel (230-400 mesh). J.T. Baker flexible thin layer chromatography sheets (silica gel IB2-F) were used to monitor reaction progress. Melting points were determined using a Mel-Temp apparatus and are reported as uncorrected values. 1NMR spectra were recorded using a 300 MHz Bruker ARX300 NMR spectrometer or 500 MHz Bruker DRX500 NMR spectrometer, as noted. Chemical shifts are reported in δ values (ppm) relative to an internal reference (0.03%, v/v) of tetramethylsilane (TMS) in CDCl3, except where noted. Abbreviations used to report NMR peaks are as follows: bs = broad singlet, d = doublet, dd = doublet of doublets, m = multiplet, q = quartet, s = singlet, t = triplet. Electrospray ionization analyses were carried out on a FinniganMAT LCQ Classic (ThermoElectron Corp, San Jose, CA) mass spectrometer system. The low-resolution electron impact (EI) and chemical ionization (CI) studies were carried out using a Hewlett-Packard Engine (Hewlett-Packard Company, Wilmington, DE) mass spectrometer. Elemental analyses were performed by the Purdue University Microanalysis Laboratory and all compounds reported possess ≥ 95% purity. All reactions were carried out under an argon atmosphere, unless noted otherwise.
7,8-Dihydroxy-4-methylchromen-2-one, 4
Pyrogallol, (50.0 g, 0.39 mol) and ethylacetoacetate (50 mL, 0.39 mol) were combined in a 500 mL three-neck round bottom flask equipped with mechanical stirring. The flask was immersed in an ice bath and conc sulfuric acid (80 mL) was added dropwise over 30 min. The reaction mixture was stirred an additional 2 h and poured onto ice. The resulting solid was collected, filtered, and dried for 30 min under a stream of argon gas. Recrystallization twice from hot MeOH yielded the desired product (18.7 g , 25%); mp > 240 °C (lit.[48] mp 232 °C). 1H NMR (500 MHz, CDCl3): δ 7.07 (d, 1H, J = 8.7 Hz), 6.80 (d, 1H, J = 8.7 Hz), 6.06 (s, 1H), 4.90 (bs, 2H), 2.38 (s, 3H). MS (ESI): [M+H]+ = 193.
7,8-Dibenzyloxy-4-methylchromen-2-one, 5
A solution of 4 (10.1 g, 52.59 mmol) in DMF (120 mL) was filtered through a fritted glass funnel to remove a small amount of insoluble material. Benzyl bromide (13.75 mL, 115.6 mmol) was added, followed by potassium carbonate (73 g, 325 mesh), and the mixture was magnetically stirred for 1 h at room temperature. Dichloromethane (500 mL) was added and the reaction mixture was filtered through a pad of Celite. The filtrate was washed with water (3 × 500 mL), dried over MgSO4, filtered, and concentrated to dryness. The resulting oil was dissolved in CH2Cl2 (10 mL) and Et2O was slowly added, just to turbidity. Stirring was continued until a white precipitate was produced and then more Et2O (400 mL) was added. The white solid was collected by filtration, dried under argon, and placed on a vacuum pump for 2 h. The product weighed 16.63 g (85%); mp 149–153 °C (lit.[49] mp 157 °C). 1H NMR (500 MHz, CDCl3): δ 7.41 (m, 8H), 7.25 (m, 3H), 7.16 (d, 1H, J = 8.70 Hz), 6.18 (s, 1H), 5.25 (s, 2H), 5.13 (s, 2H), 2.43 (s, 3H). MS (ESI): [M+H]+ = 373. Anal. calcd for C24H20O4: C 77.40, H 5.41, found: C 77.48, H 5.53.
7,8-Dibenzyloxy-2-oxo-2H-chromene-4-carboxaldehyde, 6
A 500 mL round bottom flask was charged with 5 (8.9 g, 23.9 mmol), selenium dioxide (3.9 g, 35.14 mmol), and 151 mL of xylenes (mixed), and the mixture was stirred and heated at 150 °C for 12 h. The reaction mixture was allowed to cool to room temperature and was filtered through a pad of Celite, which was further washed with Et2O. Hexane was added to the filtrate and a yellow precipitate formed, which was collected by filtration. The filtrate was concentrated by rotary evaporation and additional product was precipitated with hexane. The combined yellow solid was recrystallized from cold CH2Cl2-Et2O to yield aldehyde (5.63 g, 61%) that was sufficiently pure for the next step. A small sample was dissolved in CH2Cl2 and passed through a short silica column (1:1 hexane:CH2Cl2). The pure fractions were combined and concentrated to yield an analytically pure sample; mp 117-118°C. 1H NMR (300 MHz, CDCl3): δ 10.05 (s, 1H), 8.25 (d, 1H, J = 9.0 Hz), 7.37 (m, 10H), 6.97 (d, 1H, J = 9.0 Hz), 6.72 (s, 1H), 6.22 (s, 2H), 5.18 (s, 2H). MS (ESI): [M+H]+ = 387. Anal. calcd for C24H18O5: C 74.60, H 4.70, found C 74.27, H 4.80.
4-(N-Benzylaminomethyl)-7,8-dibenzyloxychromen-2-one, 7
A solution of 6 (24.3 g, 62.9 mmol) in CHCl3 (280 mL) was filtered through a glass fritted funnel to remove a small amount of insoluble material. Benzylamine (8.73 mL, 79.59 mmol) and sodium carbonate (2.1 g) were added to the dark solution, which was stirred for 14 h at room temperature. The mixture was then filtered through a pad of Celite into a 500 mL round bottom flask, and the filtrate was cooled on an ice bath. Dry MeOH (50 mL) was added, followed by portionwise addition of NaBH3CN (4.27 g, 67.95 mmol) over 30 min. The pH was monitored with moist litmus paper while the reaction mixture was kept slightly acidic using conc HCl. After stirring for 2 h at room temperature, the reaction was concentrated to dryness. The resulting solid was partitioned between 300 mL CH2Cl2 and a solution of saturated NaHCO3 (200 mL). The aqueous layer was further extracted with CH2Cl2 (2 × 100 mL), and the combined organic extracts were washed with H2O (200 mL), brine (100 mL), dried over MgSO4, filtered, and concentrated to dryness. The resulting oil was placed under an aspirator vacuum, which induced solidification. The solid was recrystallized from a minimal amount of CH2Cl2 and excess hot MeOH to yield 17.5 g (58%) of product as colorless needles; mp 129-130 °C. Yields of up to 80% were obtained if chromategraphically pure 6 was used. 1H NMR (500 MHz, d6-DMSO): δ7.41 (m, 16H), 7.24 (d, 1H, J = 9.0 Hz), 7.17 (d, 1H, I = 9.0 Hz), 6.41 (s, 1H), 5.27 (s, 2H), 5.08 (s, 2H), 3.87 (s, 2H), 3.78 (s, 2H). MS (ESI): [M+H]+ = 648. Anal. calcd for C31H27NO4: C 77.97, H 5.70, N 2.93, found: C 77.66, H 5.59, N 2.63.
7,8-Dibenzyloxy-4-N,N-dibenzylaminomethylchromen-2-one, 8
In a 1 L round bottom flask, 16.69 g (34.98 mmol) of 7, 200 mL DMF, and benzyl bromide (8.65 mL, 72.72 mmol) were heated together on an oil bath for a few minutes until the starting material had dissolved. Potassium carbonate (325 mesh, 55 g) was then added and the mixture was heated at 110 °C with stirring until TLC indicated complete disappearance of starting material. Dichloromethane (500 mL) was added to the reaction mixture, which was then filtered through a pad of Celite. The solution was washed with water (3 × 500 mL) and the combined water washes were back extracted with CH2Cl2 (2 × 100 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated to afford the desired product containing a small amount of DMF. Addition of Et2O (500 mL) produced a white precipitate that was filtered and washed with additional Et2O (300 mL). A total of 17.88 g (90%) of product sufficiently pure for the next step was obtained. A small amount of material was recrystallized by vapor diffusion (MeOH, CH2Cl2/Et2O); mp 159-161 °C. 1H NMR (500 MHz, CDCl3): δ7.48 (m, 2H), 7.33 (m, 19H), 6.82 (d, 1H, J = 9.0 Hz), 6.63 (s, 1H), 5.17 (s, 2H), 5.16 (s, 2H), 3.63 (s, 2H), 3.63 (s, 4H). MS (ESI): [M+H]+ = 568. Anal. calcd for C38H33NO4 (0.5 eq. MeOH): C 79.22, H 6.04, N 2.40, found: C 79.57, H 5.88, N 2.08.
7,8-Dibenzyloxy-4-N,N-dibenzylaminomethyl-2H-chromen-2-ol, 9
A solution of 8 (14.1 g, 24.85 mmol) in CH2Cl2 (700 mL) in a 1 L flask was flushed with argon for 10 min. The flask was then cooled to −78 °C and DiBAl-H (37.27 mL, 1M in hexane, 37.27 mmol) was added over 10 min. After 2 h the starting material was consumed and ethyl acetate (200 mL) was added to quench the reaction. The solution was removed from the dry ice/acetone bath and poured into a 2 L flask containing 300 mL of saturated Rochelle’s salt solution. The emulsion was stirred vigorously until the layers separated (~1 h). The organic layer was separated and washed with brine (2 × 200 mL), dried over MgSO4, filtered, and concentrated to dryness. Chromatography (9:1 hexane:EtOAc) afforded a yellow oil that solidified under a high vacuum (10.19 g, 72%). A small amount was recrystallized from methanol-ether-hexane to afford a white solid; mp 90-94 °C. 1H NMR (500 MHz, CDCl3): δ 7.31 (m, 20H), 7.04 (d, 1H, J = 8.7 Hz), 6.53 (d, 1H, J = 8.7 Hz), 5.93 (m, 2H), 5.12 (s, 2H), 5.08 (s, 2H), 3.61 (d, 2H, J = 13.5 Hz); 3.53 (d, 2H, J = 13.5 Hz); 3.40 (d, 1H, J = 14.0 Hz), 3.36 (d, 1H, J = 14.0 Hz), 2.48 (d, J = 7.4 Hz). MS (ESI): [M+H]+ = 570. Anal. calcd for C38H35NO4 (0.5 eq. MeOH): C 78.95, H 6.37, N 2.39, found C 78.66, H 6.26, N 2.37.
N,N-Dibenzyl-N-(7,8-dibenzyloxy-2H-chromen-4-ylmethyl)amine hydrochloride, 10
A solution of 9 (3.88 g, 6.81 mmol) in CH2Cl2 (70 mL) in a 250 mL round bottom flask was placed on an ice bath. Triethylsilane (2.14 mL, 13.40 mmol) was added, followed by dropwise addition of BF3•OEt2 (1.7 mL, 13.42 mmol), during which the solution turned dark. The ice bath was removed and the reaction mixture was stirred for 1 h, after which it was again placed on an ice bath. A saturated solution of NH4Cl was added (100 mL) and the crude mixture was extracted with CH2Cl2 (3 × 150 mL). The organic layers were combined and washed with brine (200 mL), dried over MgSO4, filtered, and concentrated to dryness. Column chromatography (15:3:2 hexane:CH2Cl2:acetone) afforded the desired product as a colorless oil (3.21 g, 85%). The hydrochloride salt was prepared by dissolving the product in a minimal amount of CH2Cl2, neutralizing with 1M HCl in dry EtOH, and precipitating with Et2O; mp 160–163 °C. 1H NMR (500 MHz, CDCl3 (free base): δ 7.33 (m, 20H), 6.90 (d, 1H, J = 8.7 Hz), 6.44 (d, 1H, J = 8.7 Hz), 5.81 (bs, 1H), 5.09 (s, 2H), 5.02 (s, 2H), 4.71 (d, 2H, J = 6.0 Hz), 3.55 (s, 4H), 3.29 (s, 2H). MS (ESI): [M+H]+ = 554. Anal. calcd for C38H36ClNO3: C 77.34, H 6.15, N 2.37, found: C 76.98, H 6.29, N, 2.41.
7,8-Dihydroxy-4-aminomethylchroman hydrochloride, 2a
Absolute ethanol (220 mL) and 10 (2.8 g, 4.74 mmol) were stirred vigorously for 5 min and the solution was then filtered through a fritted glass funnel into a 500 mL round bottom flask equipped with a stirring bar. The flask was briefly flushed with argon and 1.9 g of 10% Pd on carbon (dry) was added. The flask was capped with a rubber septum and hydrogen gas was passed through it for 20 min. A balloon filled with hydrogen was then attached and the contents of the flask were stirred at room temperature for 24 h under an atmosphere of hydrogen. The crude suspension was filtered through a pad of Celite that had been previously washed with absolute EtOH. After filtration the pad was washed with an additional 500 mL of EtOH. The dark filtrate was concentrated to dryness and placed under a high vacuum overnight. The resulting black solid was dissolved in MeOH (20 mL) followed by slow addition of Et2O. Vigorous stirring and scratching with a spatula induced formation of a black gummy precipitate. The tan solution was decanted away from the black precipitate into another flask. This process was continued three times until an offwhite precipitate began to form upon addition of Et2O. Excess Et2O was added to ensure complete precipitation of the product. Throughout the whole process a gentle stream of argon was passed through the flask to prevent oxidation. The precipitate was filtered, dried under a stream of argon, and placed under a high vacuum for 12 h. Recrystallization by vapor diffusion (MeOH-Et2O) three times, yielded an analytically pure sample (329 mg, 30%); mp 235-238 °C (dec.). 1H NMR (500 MHz, CD3OD): δ 6.53 (d, 1H, J = 8.5 Hz), 6.42 (d, 1H, J = 8.5 Hz), 4.22 (m, 2H), 3.26 (d, 1H), 3.09 (m, 2H), 2.12 (m, 1H), 1.93 (m, 1H). MS (ESI): [M+H]+ = 196, [M-NH3] = 179. Anal. calcd for C10H14ClNO3: C 51.84, H 6.09, N 6.05, found: C 51.48, H 5.95, N 5.70.
N,N-Dibenzyl-N-(2-allyl-7,8-dibenzyloxy-2H-chromen-4-ylmethyl)-amine hydrochloride, 11
A solution of 9 (2.43 g, 4.32 mmol) and allyltrimethylsilane (1.38 mL, 8.64 mmol) in CH2Cl2 (60 mL) was placed on an ice bath, and BF3•OEt2 (1.09 mL, 8.64 mmol) was added through a syringe. The deep red solution was stirred at room temperature for 2 h and then quenched with 100 mL of saturated NaHCO3. The organic layer was separated and the aqueous layer was further extracted with CH2Cl2 (3 × 80 mL). The organic fractions were combined, dried over MgSO4, filtered, and concentrated to dryness. The crude product was chromatographically purified using 10% EtOAc in hexane to afford a clear oil (2.31 g, 90%). The hydrochloride salt was prepared by dissolving the product in a minimal amount of 1:1 CH2Cl2:EtOH solution, neutralizing with 1M HCl in dry EtOH, and precipitating with Et2O; mp 127-130 °C. 1H NMR (300 MHz, CDCl3): δ 7.43 (m, 20H), 6.58 (d, 2H, J = 8.7 Hz), 6.46 (d, 1H, J = 8.7 Hz), 5.94 (m, 1H), 5.17 (m, 2H), 5.14 (s, 2H), 5.09 (s, 2H), 4.97 (m, 1H), 4.20 (bs, 4H), 3.88 (bs, 2H), 2.61 (bs, 2H). MS (ESI): [M+H]+ = 594. Anal. calcd for C41H40ClNO3: C 78.14, H 6.40, N 2.22, found: C 77.83, H 6.76, N 1.88.
4-Aminomethyl-2-propylchroman-7,8-diol hydrochloride, 2e
In a method analogous to the procedure for the synthesis of 2a, 11 (760 mg, 1.21 mmol) was converted to the title compound. An analytically pure sample was obtained by vapor diffusion recrystallization (MeOH-Et2O) three times to yield a total of 110 mg (33%) of the catechol hydrochloride; mp 245-255 °C. 1H NMR (500 MHz, CD3OD): δ 6.57 (d, 1H, J = 8.5 Hz), 6.42 (d, 1H, J = 8.5 Hz), 3.98 (q, 1H, J = 6.0), 3.48 (dd, 1H, J = 3.5, 13.0 Hz), 3.27 (bs, 1H), 3.01 (dd, 1H, J = 9, 13 Hz), 2.19 (dd, 1H, J = 6.0, 13.0 Hz), 1.86 (m, 1H), 1.66 (m, 2H), 1.53 (m, 2H), 1.03 (t, 3H, J = 7.0 Hz). MS (ESI): [M+H]+ = 238. Anal. calcd for C13H20ClNO3: C 57.04, H 7.36, N 5.12, found: C 56.65, H 7.50, N 5.09.
N,N-Dibenzyl-N-(7,8-dibenzyloxy-2-cyclohexyl-2H-chromen-4-ylmethyl)amine, 13b
A solution of 9 (3.08 g, 6.67 mmol) in 30 mL of dry THF in a 250 mL round bottom flask was placed on an ice bath and cyclohexylmagnesium chloride (2M, 34 mL, 68 mmol) was added slowly. The reaction mixture was stirred at room temperature for 1 h, placed on the ice bath once more and carefully quenched with ice (100 g). The crude material was partitioned between CH2Cl2 (300 mL) and saturated NH4Cl (200 mL). The organic layer was separated and the aqueous layer was further extracted with CH2Cl2 (3 × 120 mL). The organic fractions were combined, dried over MgSO4, filtered, and concentrated to dryness. The crude material was dried under a high vacuum for 3 h, and was then dissolved in 20 mL of dry THF. The solution was added by syringe to a 0 °C THF solution (40 mL) containing DEAD (1.2 mL, 7.67 mmol) and tributylphosphine (1.89 mL, 7.67 mmol) under an argon atmosphere. The orange solution was stirred at room temperature for 2 h, quenched with water (100 mL), and then extracted with Et2O (3 × 150 mL) and CH2Cl2 (100 mL). The organic fractions were combined, dried over MgSO4, filtered, and concentrated to dryness. Warm benzene (50 mL) was added to the resulting solid and the mixture was stirred for 10 min. The slurry was then filtered through a fritted funnel and the filtrate concentrated once more. Column chromatography (15:3:2 hexane:CH2Cl2:acetone) afforded the desired product as a clear oil (2.12 g, 51%). The hydrochloride salt was prepared by dissolving the product in a minimal amount of CH2Cl2-EtOH (1:1), neutralizing with 1M HCl in dry EtOH, and precipitating with Et2O; mp 132-134 °C. 1H NMR (500 MHz, CDCl3): δ7.34 (m, 20H), 6.89 (d, 1H, J = 8.5 Hz), 6.4 (d, 1H, J = 8.5 Hz), 5.74 (d, 1H, J = 3.0 Hz), 5.02 (m, 4H), 4.59 (bs, 1H), 3.55 (s, 4H), 3.29 (s, 2H), 1.90 (d, 1H, J = 10.5 Hz), 1.63 (m, 5H), 1.18 (m, 5H). MS (ESI): [M+H]+ = 636. Anal. calcd for C44H46ClNO3: C 78.61, H 6.90, N 2.08, found: C 78.66, H 6.94, N 2.02.
4-Aminomethyl-2-cyclohexylchroman-7,8-diol hydrochloride, 2b
In a method analogous to the synthesis of 2a above, 12b (3.65 g, 5.43 mmol) was converted to the title compound. A total of 1.26 g (70%) of the catechol hydrochloride was obtained. An analytically pure sample was obtained after four vapor diffusion recrystallizations (MeOH-Et2O). Yields for the analytically pure samples were usually 20-30%; mp 180 °C (solvent release) and 240 °C (dec.). 1H NMR (500 MHz, CD3OD): δ 6.57 (d, 1H, J = 8.5 Hz), 6.41 (d, 1H, J = 8.5 Hz), 3.76 (q, 1H, J = 5.5 Hz), 3.49 (dd, 1H, J = 3.5, 13.0 Hz), 3.24 (bs, 1H), 3.13 (dd, 1H, J = 9.0, 13.0 Hz), 2.18 (dd, 1H, J = 6.0, 13.0 Hz), 2.05 (d, 1H, J = 12.0 Hz), 1.8 (m, 5H), 1.54 (q, 1H, J = 12.0 Hz), 1.31 (m, 5H). MS (EI): [M+H]+ = 278, 261 (M-NH3). Anal. calcd for C16H24ClNO3 (0.11 eq. MeOH): C 60.97, H 7.76, N 4.41, found: C 60.74, H 7.38, N 4.44.
N,N-Dibenzyl-N-(2-adamant-2-yl-7,8-dibenzyloxy-2H-chromen-4-ylmethyl)amine hydrochloride, 13c
Magnesium (16 g, 658 mmol) and 1-bromoadamantane were placed in a 3-neck round bottom flask flushed with argon. Et2O (70 mL) was added and the slurry was stirred vigorously with a magnetic stirrer. The reaction was initiated by addition of 70 μL of MeMgBr and a small crystal of iodine along with brief heating to reflux. The organomagnesium solution was stirred at room temperature for 30 min and diluted with 200 mL of dry Et2O, after which it was placed on a dry ice/acetonitrile bath. A solution of 9 (4.7 g, 8.25 mmol) in 25 mL of dry THF was added dropwise to the grey slurry. The flask was stirred at room temperature for 1 h, placed in an ice bath, and then quenched with ice (100 g). The crude material was partitioned between EtOAc (300 mL) and saturated NH4Cl (250 mL). The organic layer was separated and the aqueous layer was further extracted with EtOAc (3 × 120 mL). The organic fractions were combined, dried over MgSO4, filtered, and concentrated to dryness. The resulting solid was dissolved in 300 mL of CH2Cl2, the solution was filtered, and the filtrate kept on ice. The product solution was then dried over MgSO4, filtered, concentrated to dryness, and placed under high vacuum for 3 h. The residue was dissolved in 15 mL of dry THF and then added by syringe to a 0 °C THF solution (80 mL) containing 3.8 mL of DEAD (24.2 mmol) and triphenylphosphine (6.8 g, 25.93 mmol) under argon at 0 °C. The solution was stirred at room temperature until TLC indicated complete consumption of starting material (1 h). Ice water (200 mL) was added slowly and the crude mixture was extracted into Et2O (2 × 200 mL) and CH2Cl2 (200 mL). The organic fractions were combined, dried over MgSO4, filtered, and concentrated to dryness. Purification by column chromatography (20:1 hexane:EtOAc) yielded a clear oil (3.06 g, 54%). The hydrochloride salt was obtained by dissolving the oil in a minimal amount of 1:1 CH2Cl2-EtOH and neutralizing with 1M HCl in anhyd EtOH. The product was then precipitated with Et2O, filtered, and dried under high vacuum; mp 155-158 °C. 1H NMR (500 MHz, CD3OD): δ 7.32 (m, 21H), 6.43 (d, 1H, J = 8.5 Hz), 6.20 (bs, 2H), 5.03 (m, 4H), 4.29 (m, 5H), 4.18 (bs, 2H), 3.21 (s, 3H), 1.74 (m, 12H). MS (EI): [M+H]+ = 691. Anal. calcd for C48H52ClNO3 (1eq. MeOH): C 77.60, H 7.44, N 1.85, found: C 77.85, H 7.18, N 1.91.
2-Adamant-1-yl-4-aminomethylchroman-7,8-diol hydrochloride, 2c
In a method analogous to the synthesis of 2a above, 12c (2.85 g, 3.92 mmol) was converted into the title compound. A total of 1.33 g (92%) of the crude catechol hydrochloride was obtained. The pink solid was dissolved in MeOH and treated with decolorizing carbon and filtered through a pad of Celite. Slow addition of Et2O with vigorous stirring induced formation of a tan precipitate that was collected by filtration under argon. An analytically pure sample was obtained after four vapor diffusion recrystallizations (MeOH-Et2O); (512 mg, 36%); mp 210-220 °C. 1H NMR (500 MHz, CD3OD): δ 6.57 (d, 1H, J = 8.5 Hz), 6.41 (d, 1H, J = 8.5 Hz), 3.48 (m, 2H), 3.30 (bs, 2H), 3.00 (dd, 1H, J = 9.0, 13.0 Hz), 2.23 (dd, 1H, J = 6.0, 13.0 Hz), 2.02 (bs, 3H), 1.89 (d, 3H, J = 12.0 Hz), 1.77 (m, 9H), 1.89 (q, 1H, J = 12.0 Hz). MS (ESI): [M+H]+ = 330. Anal. calcd for C20H28ClNO3: C 65.65, H 7.71, N 3.83, found: C 65.28, H 8.02, N 3.92.
2,3-Dibenzyloxy-6-[1-[(N,N-dibenzylamino)methyl]-3-phenylpropenyl]phenol, 14
Phenyl magnesium bromide (1M in THF, 60 mL, 60 mmol) was added dropwise to a solution of 9 (4.85 g, 8.51 mmol) in dry THF (40 mL) at 0 °C. The solution was removed from the ice bath, stirred for 1 h, and quenched with ice (50 mL). The crude reaction was partitioned between EtOAc (150 mL) and saturated NH4Cl (100 mL). The aqueous layer was extracted with EtOAc (2 × 150 mL), the organic layers were combined, dried over MgSO4, filtered, and concentrated to dryness. The crude product was chromatographically purified (15:3:2 hexane:CH2Cl2:acetone) to yield the product as a white solid (4.6 g, 83%); mp 146-149 °C. 1H NMR (300 MHz, CDCl3): δ7.31 (m, 30H), 6.60 (d, 1H, J = 8.5 Hz), 6.47 (d, 1H, J = 8.5 Hz), 5.91 (d, 1H, J = 9.6 Hz), 5.12 (m, 5H), 3.69 (d, 2H, J = 13.5 Hz), 3.52 (d, 2H, J = 13.5 Hz), 3.32 (d, 1H, J = 13.5 Hz), 3.19 (d, 1H, J = 13.2 Hz), 2.19 (bs; 1H). MS (ESI): [M+H]+ = 648. Anal. calcd for C44H41NO4: C 81.58, H 6.38, N 2.16, found: C 81.54, H 6.26, N 2.30.
N,N-Dibenzyl-N-(7,8-dibenzyloxy-2-phenyl-2H-chromen-4-yl-methyl)amine hydrochloride, 15
A stirring solution of 14 (5.13 g, 7.92 mmol) in pyridine (42 mL) in a 250 mL round bottom flask was cooled to 0 °C. Thionyl chloride (3.5 mL, 18.12 mmol) was slowly added dropwise, during which the solution turned deep red. After 5 min, TLC indicated complete reaction, and CH2Cl2 (300 mL) was added. The red solution was poured into 600 mL of a cold solution of 1M HCl and the aqueous layer was extracted with CH2Cl2 (2 × 300 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated to dryness. Column chromatography (hexane → 15:3:2 hexane:CH2Cl2:acetone) yielded the desired product as a yellow oil (3.09 g, 62%). The hydrochloride salt was obtained by dissolving the oil in a minimal amount of 1:1 CH2Cl2-EtOH solution and neutralizing with 1M ethanolic HCl solution. The product was then precipitated with Et2O, filtered, and dried under high vacuum; mp 162-165 °C. 1H NMR: (300 MHz, CDCl3): δ 7.31 (m, 30H), 6.93 (d, 1H, J = 8.7 Hz), 6.42 (d, 1H, J = 8.7 Hz), 5.91 (d, 1H, J = 3.6 Hz), 5.85 (d, 1H, J = 3.6 Hz), 5.06 (d, 2H, J = 3.0 Hz), 4.85 (d, 2H, J = 9.0 Hz), 3.58 (s, 4H), 3.37 (s, 2H). MS (ESI): [M+H]+ = 630. Anal. calcd for C44H40ClNO3: C 79.32, H 6.05, N 2.10, found: C 79.25, H 6.07, N 2.08.
2-(2,3-Dimethoxyphenyl)-5-oxo-tetrahydrofuran-3-carboxylic acid, 18
To a flame-dried 3-neck flask fitted with a condenser and dried addition funnel was added anhydrous, powdered zinc chloride (25.0 g, 0.184 mol). To this solid was added 100 mL CH2Cl2, followed by 2,3-dimethoxybenzaldehyde (15.3 g, 0.092 mol) and succinic anhydride (13.8 g, 0.138 mol). Triethylamine (25.6 mL, 0.184 mol) was added dropwise to the flask with rapid stirring and the mixture was heated at reflux for 4 days. The reaction was cooled to room temperature and poured over ice-cold 6N HCl. The organic component was extracted with EtOAc (3 × 250 mL), which was then washed with 2 N HCl (1 × 250 mL), and brine (1 × 250 mL). The product was extracted into satd NaHCO3 (4 × 200 mL) until TLC indicated no product remaining in the organic layer. The aqueous layer was washed with CH2Cl2 (1 × 200 mL) and acidified with conc. HCl. The white, milky solution was extracted with CH2Cl2 (3 × 250 mL), dried over Na2SO4, and evaporated under vacuum to afford a pale yellow solid (18.0 g, 73.6%) that was recrystallized from EtOAc-hexanes; mp 129-130 °C (lit.[50] mp 132 °C). 1H NMR: (300 MHz, CDCl3): δ 7.01 (t, 1H, J = 7.5 Hz), 6.89 (dd, 1H, J = 2.5, 8.1 Hz), 6.82 (dd, 1H, J = 2.5, 8.1 Hz), 5.73 (d, 1H, J = 6.6 Hz), 3.81 (s, 3H), 3.80 (s, 3H), 3.44 (dt, 1H, J = 6.6, 8.5 Hz), 2.90 (d, 2H, J = 8.5 Hz). MS (EI): [M+H]+ = 266.
4-(2,3-Dimethoxyphenyl)but-3-enoic acid, 19
Recrystallized 18 (8.6 g, 0.032 mol) was placed into a one-neck round bottom flask and the flask was heated for 6 h on a 180 °C oil bath. Carbon dioxide was observed bubbling out of the dark brown liquid. After 6 h, the reaction was cooled to room temperature and dissolved in CH2Cl2. The product and any unreacted starting material were extracted into 2 N NaOH (3 × 100 mL). The pKa of the butenoic acid is approximately 4.2, whereas the pKa of paraconic acid 18 is approximately 3.6, so the two compounds are separable by titration. The aqueous extract was therefore carefully acidified with 2 N HCl, with monitoring by a calibrated pH meter. At pH 4.0 the solution became very cloudy and was extracted with CH2Cl2. The titration was repeated until there was no turbidity at pH 4.0. Unreacted starting material could be recovered by acidifying to pH 3.0 and extracting with CH2Cl2. The initial organic extracts were dried over Na2SO4 and evaporated under reduced pressure to yield pure 19 that solidified under reduced pressure to provide a yellow solid (4.7 g, 65.2%) that was used without further purification; mp 84-86 °C (no lit.[51] mp reported). 1H NMR: (300 MHz, CDCl3): δ 7.08 (dd, 1H, J = 1.2, 8.0 Hz), 6.99 (t, 1H, J = 8.0 Hz), 6.80 (m, 2H), 6.29 (dt, 1H, J = 7.2, 15.9 Hz), 3.84 (s, 3H), 3.78 (s, 3H), 3.32 (dd, 2H, J = 1.2, 7.2 Hz). MS (ESI): [M+Na]+ = 245.
4-(2,3-Dimethoxyphenyl)butanoic acid, 20
A 500 mL Parr hydrogenation flask containing 0.6 g of 10% Pd/C and 19 (3.7 g, 0.017 mol) dissolved in absolute EtOH was pressurized with H2 and shaken at 2 atm H2 for 2 h. The contents were filtered through Celite, the filtrate was evaporated, and the resulting oil was dried under high vacuum to yield a grey solid (3.7 g, quant. yield). The solid was recrystallized from EtOAc-hexanes to afford fine white needles (2.2 g, 59.5%); mp 58-59 °C (lit.[52] mp 58.5-60 °C) 1H NMR: (300 MHz, CDCl3): δ 6.97 (t, 1H, J = 8 Hz), 6.76 (d, 1H, J = 8 Hz), 6.75 (d, 1H, J = 8 Hz), 3.84 (s, 3H), 3.80 (s, 3H), 2.67 (t, 2H, J = 7 Hz), 2.37 (t, 2H, J = 7 Hz), 1.92 (p, 2H, J = 7 Hz). MS (ESI): [M+Na]+ = 247.
5,6-Dimethoxy-3,4-dihydronaphthalen-1(2H)-one, 21
A dry, mechanically-stirred flask charged with 15 g polyphosphoric acid was heated on a 60 °C oil bath for 20 min. Finely powdered 20 (1.0 g, 4.46 mmol) was added in small portions into the center of the stirring vortex. After 30 min the reaction was a rust color and no starting material remained (TLC). The reaction was quenched by pouring over ice with vigorous stirring, whereupon the desired product crystallized. The crystals were filtered and washed with water to yield pearly off-white plates (900 mg, 97.9%); mp 103-104 °C (lit.[52] mp 104–105 °C) 1H NMR: (300 MHz, CDCl3): δ 7.79 (d, 1H, J = 8.7), 6.81 (d, 1H, J = 8.7), 3.86 (s, 3H), 3.75 (s, 3H), 2.89 (t, 2H, J = 6.3 Hz), 2.53 (t, 2H, J = 6.3 Hz), 2.05 (p, 2H, J = 6.3 Hz). MS (EI): [M]+ = 206.
5,6-Dimethoxy-3,4-dihydronaphthalene-1-carbonitrile, 22
TMSCN (1.42 mL, 10.7 mmol) was added dropwise to a slurry of 21 (1.7 g, 8.25 mmol) in freshly distilled toluene (25 mL). After stirring for 10 min, BF3•OEt2 (1.57 mL, 12.38 mmol) was added all at once, producing an immediate color change from yellow to brown. The reaction was stirred at room temperature for 3 h, until no starting material remained (TLC). The reaction was quenched by pouring over ice water (30 mL) with vigorous stirring. Et2O (20 mL) was added to this mixture, the layers were separated, and the aqueous layer was extracted twice more with Et2O and once with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and evaporated under reduced pressure to yield a tan solid (1.7 g, 96%) that could be recrystallized from MeOH to yield fine, colorless needles in 84% over three crops; mp 138-140 °C (lit.[35] mp 137-139 °C). 1H NMR: (300 MHz, CDCl3): δ 7.18 (d, 1H, J = 8 Hz), 6.80 (d, 1H, J = 8 Hz), 6.75 (t, 1H, J = 4.6 Hz), 3.88 (s, 3H), 3.76 (s, 3H), 2.87 (t, 2H, J = 8 Hz), 2.44 (m, 2H). MS (EI): [M]+ = 215.
(5,6-Dimethoxy-1,2,3,4-tetrahydronaphthalen-1-yl)methanamine, 23
A solution of 22 (1.25 g, 5.81 mmol) in MeOH (30 mL) was added to a Parr hydrogenation flask containing 0.5 g Raney-nickel catalyst in 10 mL MeOH. To this suspension was added 5 mL NH4OH before pressurizing the vessel with 4 atm H2 and shaking for 16 h. The reaction was carefully filtered through Celite and the filtrate evaporated under reduced pressure. The residue was dissolved in CH2Cl2 and extracted three times with 2N HCl. The combined aqueous layers were basified with 2N NaOH and extracted with Et2O. The ether layers were acidified with 2N ethanolic HCl and filtered to yield the HCl salt of the amine as a white powder (730 mg, 49%); mp 225-227 °C (dec.) (lit.[35] mp 249-251 °C). 1H NMR: (300 MHz, D2O): δ 6.98 (d, 1H, J = 8 Hz), 6.87 (d, 1H, J = 8 Hz), 3.76 (s, 3H), 3.66 (s, 3H), 3.02-2.90 (m, 3H), 2.69-2.56 (m, 2H), 1.78-1.61 (m, 4H). MS (ESI): [M+H]+ = 222; [M+H]+-NH3 = 205.
5-(Aminomethyl)-5,6,7,8-tetrahydronaphthalene-1,2-diol hydrobromide, 3a
A solution of 180 mg of 23 (0.814 mmol) dissolved in CH2Cl2 was placed into a flame-dried flask with magnetic stirring and cooled to −78 °C. A 1.0 M solution of boron tribromide in CH2Cl2 (2.5 mL) was then slowly added dropwise to the flask as the solution gradually became cloudy. The reaction was stirred at −78 °C for one hour and allowed to warm to room temperature overnight. MeOH (25 mL) was added to quench the reaction, followed by evaporation under reduced pressure. The brown solid residue was washed with MeOH and evaporated three additional times to remove any HBr. The residue was dried under high vacuum to yield the HBr salt as a brown solid (0.222 g, 99%) that could be recrystallized from MeOH-EtOAc; mp 203-205 °C (dec.) (lit.[35] mp 211-213 °C). 1H NMR: (300 MHz, D2O): δ 6.62 (d, 1H, J = 8 Hz), 6.57 (d, 1H, J = 8 Hz), 3.11-2.95 (m, 3H), 2.59-2.40 (m, 2H), 1.70-1.53 (m, 4H). MS (ESI): [M+H]+ = 194; [M+H]+-NH3 = 177.
Ethyl 2-cyclohexyl-3-(2,3-dimethoxyphenyl)-3-hydroxypropanoate, 25b
A flame-dried single-neck round bottom flask in a dry iceacetone bath was charged with 50 mL of freshly distilled dry THF, followed by addition of 33.1 mL of a 2.0M solution of lithium diisopropyl amide. A solution of 24b[53] (10.24 g, 0.0602 mol) dissolved in distilled THF (30 mL) was added dropwise over 15 min. The enolate solution was allowed to stir at −78 °C for 15 additional min, followed by the drop wise addition of asolution of 2,3-dimethoxybenzaldehyde (10.0 g, 0.0602 mol) in THF (75 mL). The reaction turned a bright yellow color and was allowed to warm to ambient temperature over the next 90 min. The reaction was quenched by the dropwise addition of water (30 mL). Approximately 50 mL Et2O were added, and the layers separated. The organic layer was washed vigorously with a satd solution of NaHSO3 (2 × 50 mL) to remove any unreacted benzaldehyde. The ether layer was dried over Na2SO4, filtered, and evaporated under reduced pressure to afford a dark yellow oil. After column chromatography (1:1 EtOAc:hexanes), the major product was isolated as a diastereomeric mixture of the title compound as a yellow oil (16.3 g, 80.3%). Diastereomers: 1H NMR: (300 MHz, CDCl3): δ 6.99 (t, 1H, J = 7.8), 6.88 (bd, 1H, J = 7.8), 6.83 (bd, 1H, J = 7.8), 5.26 (d, 0.3H, J = 3.9 Hz), 5.08 (d, 0.7H, J = 8.4 Hz), 3.95 (q, 2H, J = 6.0 Hz), 3.94 (s, 3H), 3.86 (s, 3H), 2.81 (dd, 0.7H, J = 3.9, 8.4 Hz), 2.67 (dd, 0.3 H, J = 3.9, 9.0 Hz), 2.04-1.68 (m, 7H), 1.32-1.08 (m, 4H), 1.04 (2 t, 3H, J = 6.0 Hz). MS (ESI): [M+Na]+ = 359. Anal. calcd for C19H28O5: C 67.83, H 8.39, found: C 67.46, H 8.48.
Ethyl 2-adamantyl-3-(2,3-dimethoxyphenyl)-3-hydroxypropanoate, 25c
In a procedure analogous to the synthesis of 25b above, 24c[54] (12.0 g, 0.0540 mol) was converted to the title compound. Column chromatography (1:2 EtOAc:hexanes) produced the title compound as an off-white inseparable mixture of solid diastereomers (19.5 g, 92.9%). An analytical sample was crystallized from EtOH; mp 90-91 °C. Diastereomers: 1H NMR: (300 MHz, CDCl3): δ 7.03-6.74 (m, 3H), 5.39 (d, 0.4H, J = 9.6 Hz), 5.09 (t, 0.4H, J = 9.6 Hz), 4.55 (d, 0.6H, J = 10.2 Hz), 4.00 (s, 1.2H), 3.99-3.71 (m, 2H), 3.84 (bs, 4.8H), 2.98 (d, 0.4H, J = 9.6 Hz), 2.69-2.65 (d, 0.6H, J = 10.2 Hz), 2.47 (bs, 0.6H), 2.12-1.61 (m, 15H), 1.05 (t, 1.8H, J = 7.2 Hz), 0.94 (t, 1.2H, J = 7.2 Hz). MS (ESI): [M+Na]+ = 411. Anal. calcd for C23H32O5: C 71.11, H 8.30, found: C 70.82, H 8.34.
Ethyl 2-((2,3-dimethoxyphenyl)(hydroxy)methyl)pentanoate, 25e
In a procedure analogous to the synthesis of 25b above, ethyl valerate (Aldrich, 4.48 mL, 0.0301 mol) was converted to the title compound. Although the resolution is unnecessary, column chromatography (1:1 EtOAc:hexanes), could resolve the title compound into its two diastereomers,. Both diastereomers were recovered as amber oils (5.5 g, 62.0%). Major diastereomer: 1H NMR: (300 MHz, CDCl3): δ 7.02 (t, 1H, J = 7.8), 6.92 (dd, 1H, J = 1.8, 7.8), 6.85 (dd, 1H, J = 1.8, 7.8), 4.99 (t, 1H, J = 5.7 Hz), 4.04 (q, 2H, J = 7.2 Hz), 3.92 (s, 3H), 3.86 (s, 3H), 3.35 (d, 1H, J = 5.7 Hz), 2.85-2.78 (m, 1H), 1.75-1.62 (m, 2H), 1.40-1.19 (m, 2H), 1.13 (t, 3H, J = 7.2 Hz), 0.86 (t, 3H, J = 7.0 Hz). MS (EI): [M]+ = 296. Anal. calcd for C16H24O5: C 64.84, H 8.16, found: C 65.03, H 8.04.
2-Cyclohexyl-3-(2,3-dimethoxyphenyl)propan-1-ol, 26b
Thionyl chloride (10.6 mL) was added to a solution of 25b (16.2 g, 0.048 mol) in 100 mL benzene. The reaction was stirred at room temperature for 2 h, followed by the removal of solvents by rotary evaporation. Toluene (15 mL) was added to the flask, followed by rotary evaporation to ensure that all of the thionyl chloride was removed. The resulting brown oil was dissolved in dry Et2O (75 mL) and slowly added dropwise to a suspension of 5.4 g LiAlH4 and 50 mL Et2O in a flame-dried, 3-neck flask, with magnetic stirring, The reaction flask was transferred to a 45 °C oil bath and the reaction was allowed to stir at reflux overnight. The reaction was cooled to room temperature and quenched by the slow, careful, dropwise addition of 5.4 mL of water, followed by the dropwise addition of 5.4 mL 15% aqueous NaOH, followed by the addition of 16.2 mL more water. This suspension was stirred at room temperature until solid granules formed that were removed by filtration. The filter cake was triturated with hot Et2O and filtered again. The filtrates were combined, dried over Na2SO4, filtered, and evaporated under reduced pressure. Column chromatography (1:2 EtOAc:hexanes) was needed to purify the major product, which was isolated as a dark yellow oil (10.4 g, 79.1%). 1H NMR: (300 MHz, CDCl3): δ 6.99 (t, 1H, J = 7.8 Hz), 6.79-6.73 (m, 2H), 3.86 (s, 3H), 3.85 (s, 3H), 3.53 (dd, 1H, J = 3.9, 11.7 Hz), 3.35 (dd, 1H, J = 3.9, 11.7 Hz), 2.69-2.66 (m, 2H), 1.89-1.05 (m, 11H). MS (ESI): [M+Na]+ = 301. Anal. calcd for C17H26O3: C 73.34, H 9.41, found: C 73.11, H 9.69.
2-Adamantyl-3-(2,3-dimethoxyphenyl)propan-1-ol, 26c
In a procedure analogous to the synthesis of 26b above, 25c (20.4 g, 0.0502 mol) was converted to the title compound. Column chromatography (1:2 EtOAc:hexanes) was again required to purify the product, which was isolated as a yellow oil (11.8 g, 71.5%). 1H NMR: (300 MHz, CDCl3): δ 7.00 (t, 1H, J = 7.8 Hz), 6.77 (d, 2H, J = 7.8 Hz), 3.86 (s, 3H), 3.85 (s, 3H), 3.72 (dd, 1H, J = 2.7, 12.0 Hz), 3.33 (dd, 1H, J = 3.3, 12.0 Hz), 2.73 (d, 2H, J = 7.8 Hz), 2.01 (bs, 3H), 1.81-1.62 (m, 12H), 1.15-1.10 (m, 1H). MS (EI): [M]+ = 330. Anal. calcd for C21H30O3: C 76.33, H 9.15, found: C 76.40, H 8.99.
2-(2,3-Dimethoxybenzyl)pentan-1-ol, 26e
In a procedure analogous to the synthesis of 26b above, 25e (12.1 g, 0.0384 mol) was converted to the title compound. Column chromatography (1:2 EtOAc:hexanes) was required to purify the title compound, which was isolated as a yellow oil (6.5 g, 70.9%). 1H NMR: (300 MHz, CDCl3): δ 7.00 (t, 1H, J = 8.1 Hz), 6.80-6.74 (m, 2H), 3.86 (s, 3H), 3.84 (s, 3H), 3.36 (d, 2H, J = 4.2 Hz), 2.67-2.64 (m, 2H), 1.73-1.68 (m, 1H), 1.45-1.36 (m, 4H), 0.93 (t, 3H, J = 7.4 Hz). MS (EI): [M]+ = 238. Anal. calcd for C14H22O3: C 70.56, H 9.30, found: C 70.29, H 8.95.
2-Cyclohexyl-3-(2,3-dimethoxyphenyl)propyl methanesulfonate, 27b
A solution of 26b (10.0 g, 0.0360 mol) in freshly distilled dry THF (200 mL) was stirred in a flame-dried flask on an ice bath. To this solution, 10.0 mL (0.072 mol) of triethylamine were added through a syringe. Methanesulfonyl chloride (5.6 mL, 0.072 mol) was added dropwise through a flame-dried addition funnel, over 15 min. The reaction was stirred at 0 °C for 2 h. Water (100 mL) and Et2O (100 mL) were added to the flask to quench the reaction, and the layers were separated. The aqueous layer was washed with Et2O (2 × 50 mL) and the combined organic layers were washed with brine (2 × 75 mL), dried over Na2SO4, filtered, and evaporated under reduced pressure. Column chromatography (1:2 EtOAc:hexanes) was used to purify the title compound, which was isolated as a brown oil (12.4 g, 96.7%) that slowly solidified upon standing and was recrystallized from EtOH; mp 43-45 °C. 1H NMR: (300 MHz, CDCl3): δ 6.99 (t, 1H, J = 7.8 Hz), 6.80 (dd, 1H, J = 1.2, 8.1 Hz), 6.75 (dd, 1H, J = 1.2, 7.5 Hz), 4.16-4.08 (m, 2H), 3.86 (s, 3H), 3.81 (s, 3H), 2.91 (s, 3H), 2.83 (dd, 1H, J = 5.1, 13.5 Hz), 2.51 (dd, 1H, J = 9.6, 13.5 Hz), 1.96-1.89 (m, 1H), 1.80-1.08 (m, 11H). MS (ESI): [M+Na]+ = 379. Anal. calcd for C18H28O5S: C 60.65, H 7.92, found: C 60.56, H 8.14.
2-Adamantyl-3-(2,3-dimethoxyphenyl)propyl methanesulfonate, 27c
In a procedure analogous to the synthesis of 27b above, 26c (13.0 g, 0.0394 mol) was converted to the title compound. Column chromatography (1:2 EtOAc:hexanes) was required to purify the title compound, which was isolated as a pale yellow powder (15.0 g, 93.3%) that could be crystallized from EtOH to yield colorless, cubic crystals; mp 96-98 °C 1H NMR: (300 MHz, CDCl3): δ 6.99 (t, 1H, J = 7.8 Hz), 6.78 (d, 2H, J = 7.8 Hz), 4.25 (dd, 1H, J = 3.0, 9.9 Hz), 4.05 (dd, 1H, J = 3.9, 9.9 Hz), 3.85 (s, 3H), 3.82 (s, 3H), 2.95 (dd, 1H, J = 2.7, 13.5 Hz), 2.82 (s, 3H), 2.47 (dd, 1H, J = 11.7, 13.5 Hz), 2.02 (bs, 3H), 1.77-1.55 (m, 13H). MS (ESI): [M+Na]+ = 431. Anal. calcd for C22H32O5S: C 64.68, H 7.89, found: C 64.36, H 7.80.
2-(2,3-Dimethoxybenzyl)pentyl methanesulfonate, 27e
In a procedure analogous to the synthesis of 27b above, 26e (6.00 g, 0.0250 mol) was converted to the title compound. Column chromatography (1:2 EtOAc:hexanes) was used to purify the title compound, which was isolated as a pale yellow oil (7.8 g, 97.5%). 1H NMR: (300 MHz, CDCl3): δ 6.98 (t, 1H, J = 8.1), 6.86 (dd, 1H, J = 1.5, 8.1), 6.75 (dd, 1H, J = 1.5, 8.1), 4.08 (ddd, 2H, J = 4.8, 7.2, 9.6 Hz), 3.86 (s, 3H), 3.81 (s, 3H), 2.95 (s, 3H), 2.71 (dd, 1H, J = 6.0, 13.5 Hz), 2.60 (dd, 1H, J = 8.4, 13.5 Hz), 2.11-2.06 (m, 1H), 1.48-1.35 (m, 4H), 0.91 (t, 3H, J = 7.4 Hz). MS (ESI): [M+Na]+ = 339. Anal. calcd for C15H24O5S: C 56.94, H 7.65, found: C 57.01, H 7.67.
3-Cyclohexyl-4-(2,3-dimethoxyphenyl)butanenitrile, 28b
NaCN (4.8 g, 0.0980 mol) was added all at once to a stirring solution of 27b (11.0 g, 0.0327 mol) in 75 mL DMSO. The reaction was stirred at 80 °C overnight, until all starting material was consumed. EtOAc (100 mL) and water (100 mL) were added to the reaction. The layers were separated, the aqueous phase was extracted with EtOAc (2 × 75 mL), and the combined organic layers were washed with water (2 × 100 mL) and brine (2 × 100 mL) to remove DMSO. The organic layer was concentrated to approximately 50 mL and the water and brine washes were repeated. The organic layer was dried over Na2SO4, filtered, and evaporated to dryness. Column chromatography (1:2 EtOAc:hexanes) was used to purify the title compound, which was isolated as a colorless oil that solidified into a colorless, amorphous material that was crystallized from EtOH (7.7 g, 86.7%); mp 48-52 °C. 1H NMR: (300 MHz, CDCl3): δ 7.00 (t, 1H, J = 7.8 Hz), 6.81 (dd, 1H, J = 1.5, 7.8 Hz), 6.74 (dd, 1H, J = 1.5, 7.8 Hz) 3.86 (s, 3H), 3.82 (s, 3H), 2.92 (dd, 1H, J = 4.5, 13.5 Hz), 2.44 (dd, 1H, J = 10.2, 13.5 Hz), 2.29-2.17 (m, 2H), 1.91-1.84 (m, 1H), 1.81-1.09 (m, 11H). MS (ESI): [M+H]+ = 288. Anal. calcd for C18H25NO2: C 75.22, H 8.77, N 4.87, found: C 75.52, H 8.52, N 5.08.
3-Adamantyl-4-(2,3-dimethoxyphenyl)butanenitrile, 28c
Following the method for the synthesis of 29b above, 27c (5.88g, 0.0144 mol) was converted to the nitrile. Column chromatography (1:2 EtOAc:hexanes) was used to purify the title compound, which was isolated as a colorless oil that crystallized as colorless radial crystals (3.47 g, 70.9%); mp 68-69 °C. 1H NMR: (300 MHz, CDCl3): δ 6.99 (t, 1H, J = 7.8 Hz), 6.81 (d, 2H, J = 7.8 Hz), 3.86 (s, 3H), 3.83 (s, 3H), 3.04 (dd, 1H, J = 2.1, 13.5 Hz), 2.41 (dd, 1H, J = 11.4, 13.5 Hz), 2.31 (ddd, 1H, J = 1.2, 3.6, 17.6 Hz), 2.15 (dd, 1H, J = 6.0, 17.6 Hz), 2.04 (bs, 3H), 1.85-1.60 (m, 13H). MS (ESI): [M+H]+ = 340. Anal. calcd for C22H29NO2: C 77.84, H 8.61, N 4.13, found: C 78.15, H 8.76, N 4.41.
3-(2,3-Dimethoxybenzyl)hexanenitrile, 28e
In a procedure analogous to the synthesis of 28b above, 27e (7.50 g, 0.0237 mol) was converted to the nitrile. Column chromatography (1:2 EtOAc:hexanes) was used to purify the title compound, which was isolated as a slightly yellow oil (4.3 g, 73.9%). 1H NMR: (300 MHz, CDCl3): δ 7.00 (t, 1H, J = 8.1), 6.81 (dd, 1H, J = 1.5, 8.1), 6.75 (dd, 1H, J = 1.5, 8.1), 3.86 (s, 3H), 3.82 (s, 3H), 2.81 (dd, 1H, J = 5.1, 13.2 Hz), 2.53 (dd, 1H, J = 9.3, 13.2 Hz), 2.25 (AB spin system, 2H), 2.09-2.01 (m, 1H), 1.51-1.36 (m, 4H), 0.94 (t, 3H, J = 7.4 Hz). MS (ESI): [M+H]+ = 248. Anal. calcd for C15H21NO2: C 72.84, H 8.56, N 5.66, found: C 72.60, H 8.31, N 5.70.
3-Cyclohexyl-4-(2,3-dimethoxyphenyl)butanoic acid, 29b
DiBAlH (32.1 mL of a 1.0 M solution) was added through a syringe to a stirring solution of 28b (5.0 g, 0.0178 mol) in 100 mL freshly distilled toluene cooled to −78 °C. The reaction was stirred on a dry ice/acetone bath for 2 h, and then on an ice bath for 1 additional hour. After the starting material was consumed, a 5% aqueous HCl solution (60 mL) was carefully added. The solution foamed and became cloudy, and was stirred at 0 °C for 30 min. The solution was extracted with Et2O (3 × 50 mL), the organic extract was dried over Na2SO4, filtered, and evaporated under reduced pressure to provide crude aldehyde as a brown oil. This oil was redissolved in acetone (100 mL) and Jones Reagent (25 g CrO3, 25 mL H2SO4, and 75 mL H2O, mixed at 0 °C) was slowly added with a pipette. As the reagent was added, the solution turned dark green, indicating the presence of aldehyde. Jones Reagent was added dropwise until the green color no longer appeared and the solution was a dark orange color (approximately 8 mL total were added). This orange solution was stirred at room temperature for 10 min, at which time a dark solid mass had formed in the bottom of the flask. Water (30 mL) was added to quench the reaction and dissolve the solid. The solution returned to a bright green color and was extracted with Et2O (3 × 50 mL). The Et2O layer was extracted with 1N NaOH (3 × 50 mL) and the aqueous extracts were acidified with conc. H2SO4. The acidic solution was extracted with Et2O (3 × 50 mL), dried over Na2SO4, filtered, and evaporated to yield the carboxylic acid as a dark amber oil (3.86 g, 60.2%) that could be used in the next step without purification. An analytical sample, purified by column chromatography (2:1 hexanes:EtOAc), crystallized as colorless needles; mp 80-82 °C. 1H NMR: (300 MHz, CDCl3): δ 6.96 (t, 1H, J = 7.8 Hz), 6.77-6.74 (m, 2H), 3.84 (s, 3H), 3.80 (s, 3H), 2.78 (dd, 1H, J = 5.7, 13.5 Hz), 2.45 (dd, 1H, J = 8.7, 13.5 Hz), 2.33 (dd, 1H, J = 7.2, 18.3 Hz), 2.25-2.13 (m, 2H), 1.76-1.06 (m, 11H). MS (EI): [M]+ = 306. Anal. calcd for C18H26O4: C 70.56, H 8.55, found: C 70.74, H 8.61.
3-Adamantyl-4-(2,3-dimethoxyphenyl)butanoic acid, 29c
In a procedure analogous to the synthesis of 29b above, 28c (6.30 g, 0.0187 mol) was converted to the title compound. The desired carboxylic acid was recovered as a brown oil (1.72 g, 25.8%) that solidified upon standing. The solid was recrystallized from EtOAc-hexanes to afford a light tan powder; mp 113-115 °C. The neutral organic layer was evaporated to yield a crude, tan solid, from which tetralone 30c could be crystallized from EtOAc as pale tan plates (1.69 g, 26.6% from nitrile 28c). 1H NMR: (300 MHz, CDCl3): δ 6.92 (t, 1H, J = 7.8 Hz), 6.76 (dd, 1H, J = 1.2, 7.8 Hz), 6.71 (dd, 1H, J = 1.2, 7.8 Hz), 3.82 (s, 6H), 2.91 (dd, 1H, J = 3.0, 13.2 Hz), 2.37 (dd, 1H, J = 6.9, 16.5 Hz), 2.26 (dd, 1H, J = 11.1, 13.2 Hz), 2.10 (dd, 1H, J = 4.2, 16.5 Hz), 1.99 (bs, 3H), 1.77-1.53 (m, 13H). MS (ESI): [M+Na]+ = 381. Anal. calcd for C22H30O4: (0.5 eq. H2O) C 71.90, H 8.50, found: C 72.06, H 8.42.
3-(2,3-Dimethoxybenzyl)hexanoic acid, 29e
In a procedure analogous to the synthesis of 29b above, 28e (2.20 g, 8.9 mmol) was converted to the carboxylic acid. The title compound was isolated as a pale yellow oil (1.85 g, 78.4%). 1H NMR: (300 MHz, CDCl3): δ 6.97 (t, 1H, J = 8.1), 6.78-6.74 (m, 2H), 3.85 (s, 3H), 3.78 (s, 3H), 2.75 (dd, 1H, J = 5.7, 13.5 Hz), 2.51 (dd, 1H, J = 7.5, 13.5 Hz), 2.30-2.18 (m, 3H), 1.46-1.31 (m, 4H), 0.90 (t, 3H, J = 7.4 Hz). MS (EI): [M]+ = 266. Anal. calcd for C15H22O4: C 67.64, H 8.33, found: C 67.71, H 7.96.
3-Cyclohexyl-5,6-dimethoxy-3,4-dihydronaphthalen-1(2H)-one, 30b
To a mechanically stirring flask of polyphosphoric acid (50 g) heated to 85 °C, carboxylic acid 29b (8.1 g, 0.026 mol) dissolved in minimal benzene (5 mL) was added. The resulting mixture was stirred and heated for 1 h during which time it turned from tan to dark red. With vigorous manual stirring, the dark red reaction mixture was poured over a mixture of 400 g of ice and 200 mL of water. The precipitate that formed was filtered, washed with water (3 × 75 mL), air dried, and then dissolved in EtOAc (200 mL). The organic solution was washed with water (50 mL), 0.5 N NaOH (50 mL), dried over anhydrous MgSO4, filtered, and concentrated to provide the title compound (6.1 g, 80.3%) as a fluffy solid that was recrystallized from EtOH to yield pale tan needles (4.1 g, 53.9%); mp 109-111 °C. 1H NMR: (300 MHz, CDCl3): δ 7.84 (d, 1H, J = 9.0 Hz), 6.87 (d, 1H, J = 9.0 Hz), 3.93 (s, 3H), 3.82 (s, 3H), 3.20 (dq, 1H, J = 1.9, 16.8 Hz), 2.69 (dq, 1H, J = 1.9, 16.5 Hz), 2.53 (dd, 1H, J = 11.1, 16.8 Hz), 2.32 (dd, 1H, J = 12.9, 16.5 Hz), 1.98-1.85 (m, 1H), 1.84-1.06 (m, 11H). MS (ESI): [M+H]+ = 289. Anal. calcd for C18H24O3 (0.5 eq. EtOH): C 73.28, H 8.74, found: C 73.36, H 8.36.
3-Adamantyl-5,6-dimethoxy-3,4-dihydronaphthalen-1(2H)-one, 30c
Following a procedure similar to that for 30b, 29c (1.5 g, 4.2 mmol) was converted to the title product as a crude solid (1.4 g, 95.8%) that was difficult to crystallize. Column chromatography (1:2 EtOAc:hexanes) afforded 30c as a white powdery solid (0.98 g, 67.8%); mp 189-190 °C. 1H NMR: (300 MHz, CDCl3): δ 7.82 (d, 1H, J = 8.7 Hz), 6.87 (d, 1H, J = 8.7 Hz), 3.92 (s, 3H), 3.83 (s, 3H), 3.27 (dt, 1H, J = 3.0, 16.8 Hz), 2.74 (dt, 1H, J = 2.7, 16.2 Hz), 2.42 (dd, 1H, J = 12.0, 16.8 Hz), 2.26 (dd, 1H, J = 13.8, 16.2 Hz), 2.03 (bs, 3H), 1.78-1.58 (m, 13H). MS (ESI): [M+H]+ = 341. Anal. calcd for C22H28O3: C 77.71, H 8.29, found: C 77.28, H 8.44.
5,6-Dimethoxy-3-propyl-3,4-dihydronaphthalen-1(2H)-one, 30e
Following a procedure similar to that for the synthesis of 30b, 29e (2.8 g, 0.011 mol) provided the title compound (2.6 g, 99.9%) as a fluffy solid that was recrystallized from EtOH to yield fine, tan needles (1.5 g, 57.5 %); mp 88-91 °C. 1H NMR: (300 MHz, CDCl3): δ 7.85 (d, 1H, J = 8.7 Hz), 6.87 (d, 1H, J = 8.7 Hz), 3.93 (s, 3H), 3.82 (s, 3H), 3.22 (ddd, 1H, J = 2.1, 3.9, 16.8 Hz), 2.70 (ddd, 1H, J = 1.8, 3.3, 16.2 Hz), 2.45 (dd, 1H, J = 10.2, 16.8 Hz), 2.25 (dd, 1H, J = 11.7, 16.2 Hz), 2.17-2.10 (m, 1H), 1.46-1.38 (m, 4H), 0.94-0.87 (m, 3H). MS (EI): [M]+ = 248. Anal. calcd for C15H20O3: C 72.55, H 8.12, found: C 72.15, H 8.50.
1-(Aminomethyl)-3-cyclohexyl-5,6-dimethoxy-1,2,3,4-tetrahydronaphthalen-1-ol hydrochloride, 31b
Tetralone 30b (4.1 g, 0.0142 mol) and 231 mg (0.725 mmol) of anhydrous zinc iodide were dissolved in 75 mL of CH2Cl2. TMSCN (2.9 mL, 0.022 mol) was added dropwise and the solution was heated at reflux for 20 h and monitored by IR for the loss of the C=O stretch. Additional ZnI2 could be added to speed the reaction. The mixture was cooled and concentrated, then dissolved in dry Et2O (10 mL) and added dropwise to a slurry of 1.9 g (0.051 mol) of LiAlH4 in anhydrous Et2O (60 mL). The reaction mixture was heated at reflux for 18 h, and then cooled to room temperature. To quench the reaction, 1.9 mL of water in 5 mL of THF was carefully added dropwise, followed by 1.9 mL of 15% aqueous NaOH, followed by an additional 5.7 mL of water. The reaction mixture was stirred for 30 min until a granular precipitate formed. The solid was filtered, the filter cake was triturated with hot ether, and filtered again. The filtrates were combined and acidified with conc. HCl (5 mL). A white precipitate formed that was collected by filtration to afford 2.93 g (64.6%) of off-white solid that was a mixture of diastereomers; mp 161-163 °C (dec.). Diastereomers: 1H NMR: (500 MHz, d6-DMSO): δ 8.02 (bs, 1.5H), 7.94 (bs, 1.5H), 7.26-7.20 (m, 1H), 6.98-6.90 (m, 1H), 4.01 (bs, 1H), 3.77 (s, 1.5H), 3.76 (s, 1.5H), 3.66 (s, 3H), 3.34-3.27 (m, 0.5H), 2.98-2.92 (m, 0.5H), 2.92-2.86 (m, 0.5H), 2.85-2.72 (m, 1.5H), 2.21 (dd, 0.5H, J = 11.6, 17.6 Hz), 2.09 (dd, 0.5H, J = 11.6, 16.9 Hz), 2.01 (bd, 0.5H, J = 12.9 Hz), 1.96 (bd, 0.5H, J = 12.9 Hz), 1.80-1.55 (m, 6H), 1.45-1.33 (m, 1H), 1.35-0.98 (m, 6H). MS (ESI): [M+Na]+ = 342. Anal. calcd for C19H30ClNO3: C 64.12, H 8.50, N 3.93, found: C 64.38, H 8.35, N 4.08.
1-(Aminomethyl)-3-adamantyl-5,6-dimethoxy-1,2,3,4-tetrahydronaphthalen-1-ol hydrochloride, 31c
Following the method used for synthesizing 31b, 30c (0.75 g, 2.2 mmol) was converted to the title compound (0.47 g, 52.6%) as a tan powder that was a mixture of diastereomers; mp 128-130 °C (dec.). Diastereomers: 1H NMR: (500 MHz, d6-DMSO): δ 7.83 (bs, 3H), 7.24 (bd, 1H, J = 8.7 Hz), 6.95 (bd, 1H, J = 8.7 Hz), 5.65-5.56 (2 bs, 1H), 3.77-3.76 (2 s, 3H), 3.67-3.66 (2 s, 3H), 2.96-2.92 (m, 1H), 2.84-2.77 (m, 2H), 2.31-2.22 (m, 1H), 2.12-1.96 (m, 4H), 1.79-1.30 (m, 13H). MS (ESI): [M+H-H2O]+ = 354. Anal. calcd for C23H34ClNO3: C 67.71, H 8.40, N 3.43, found: C 67.77, H 8.60, N 3.52.
1-(Aminomethyl)-5,6-dimethoxy-3-propyl-1,2,3,4-tetrahydronaphthalen-1-ol hydrochloride, 31e
In a fashion analogous to the synthesis of 31b, 30e (1.5 g, 6.1 mmol) was converted into a diastereomeric mixture of the title compound (1.13 g, 59.2%) as a white powdery solid; mp 150-152 °C (dec.). Diastereomers: 1H NMR: (500 MHz, d6-DMSO): 7.90 (bs, 3H), 7.27-7.21 (m, 1H), 6.97-6.93 (m, 1H), 5.65-5.56 (2 bs, 1H), 3.78-3.77 (2 s, 3H), 3.66 (2 s, 3H), 3.25 (d, 1H, J = 12.9 Hz), 2.98-2.91 (m, 2H), 2.80 (dd, 1H, J = 12.6, 19.5 Hz), 2.11-1.91 (m, 2H), 1.75-1.66 (m, 1H), 1.48-1.29 (m, 4H), 0.91 (t, 3H, J = 7.4 Hz). MS (ESI): [M+Na]+ = 302. Anal. calcd for C16H26ClNO3: C 60.85, H 8.30, N 4.43, found: C 60.72, H 8.32, N 4.52.
(3-Cyclohexyl-5,6-dimethoxy-3,4-dihydronaphthalen-1-yl)methanamine hydrochloride, 32b
Ethanol (25 mL), 31b (2.37 g, 6.67 mmol), and 2 drops of 2N ethanolic HCl were placed into a singlenecked round bottom flask. The flask was fitted with a reflux condenser and the solution was heated at 80 °C and magnetically stirred for 14 h. The solvent was removed by rotary evaporation and the colorless oily residue dissolved in Et2O (30 mL) and allowed to stand at room temperature, during which time the product crystallized as a pale tan solid (1.64 g, 72.9%). The solid could be recrystallized from EtOH as tan needles; mp 148-152 °C (dec.). 1H NMR: (300 MHz, CDCl3): δ 8.57 (bs, 3H), 6.90 (d, 1H, J = 8.7 Hz), 6.73 (d, 1H, J = 8.7 Hz), 6.14 (d, 1H, J = 3.9 Hz), 3.94 (bd, 1H, J = 4.8 Hz), 3.84 (s, 3H), 3.77 (s, 3H) 2.85 (dd, 1H, J = 6.9, 16.0 Hz), 2.72 (dd, 1H, J = 9.3, 16.0 Hz), 2.20 (bs, 1H), 1.80-1.05 (m, 11H). MS (ESI): [M+H]+ = 302. Anal. calcd for C19H28ClNO2: C 67.54, H 8.35, N 4.15, found: C 67.19, H 8.31, N 4.14.
(3-Adamantyl-5,6-dimethoxy-3,4-dihydronaphthalen-1-yl)methanamine hydrochloride, 32c
Ethanol (25 mL), 31c (300 mg, 0.74 mmol), and 2 drops of 2N ethanolic HCl were placed into a singlenecked round bottom flask. The flask was fitted with a reflux condenser and the solution was heated at 80 °C and magnetically stirred overnight. The solvent was removed by rotary evaporation and the colorless oily residue was dissolved in Et2O (10 mL) and extracted with 2N HCl (3 × 10 mL). An insoluble oil formed at the interface of the two layers each time, and was recovered separately. The acidic aqueous layers were combined, washed with Et2O, and basified with conc. aqueous NaOH. The basified solution was extracted with Et2O (3 × 15 mL), dried over Na2SO4, filtered, and evaporated to yield a colorless residue from which the title compound crystallized after the addition of minimal Et2O (69 mg, 24.0%). The recovered oil layer was dissolved in EtOH and evaporated under reduced pressure to dryness. The residue could be crystallized from Et2O to yield the desired product as an off-white powder (164 mg, 57.1%); mp 174-177 °C (dec.). 1H NMR: (300 MHz, d6-DMSO): δ 8.14 (bs, 3H), 6.99 (d, 1H, J = 8.1 Hz), 6.85 (d, 1H, J = 8.1 Hz), 6.05 (bs, 1H), 3.84 (s, 2H), 3.79 (s, 3H), 3.68 (s, 3H), 2.80 (dd, 1H, J = 6.9, 15.9 Hz), 2.61-2.52 (m, 1H), 1.93 (bs, 3H), 1.70-1.48 (m, 13H). MS (ESI): [M+H]+ = 354. Anal. calcd for C23H32ClNO2: C 70.84, H 8.27, N 3.59, found: C 70.48, H 8.46, N 3.66.
(5,6-Dimethoxy-3-propyl-3,4-dihydronaphthalen-1-yl)methanamine hydrochloride, 32e
Ethanol (25 mL), 31e (150 mg, 0.48 mmol), and 1 drop of 2N ethanolic HCl were placed into a singlenecked round bottom flask. The flask was fitted with a reflux condenser and the solution was heated to 80 °C and magnetically stirred overnight. The solvent was removed by rotary evaporation and the white solid residue was dissolved in minimal EtOH and Et2O was added dropwise. A small amount of a granular white solid precipitated immediately and was removed by filtration. Additional Et2O was added to the filtrate and large white crystals slowly formed and were collected by filtration to give the title compound (82 mg, 58.2%); mp 126-128 °C (dec.). 1H NMR: (300 MHz, d6-DMSO): δ 8.18 (bs, 3H), 7.02 (d, 1H, J = 8.4 Hz), 6.87 (d, 1H, J = 8.4 Hz), 5.97 (d, 1H, J = 3.6 Hz), 3.81 (bs, 2H), 3.80 (s, 3H), 3.66 (s, 3H), 2.85 (dd, 1H, J = 6.0, 15.3 Hz), 2.49-2.28 (m, 2H), 1.48-1.29 (m, 4H), 0.90 (t, 3H, J = 6.9 Hz). MS (ESI): [M+H]+ = 262. Anal. calcd for C16H24ClNO2: C 64.53, H 8.12, N 4.70, found: C 64.14, H 8.14, N 4.80.
Cis-(3-cyclohexyl-5,6-dimethoxy-1,2,3,4-tetrahydronaphthalen-1-yl)methanamine hydrochloride, 33b
The alkene hydrochloride 32b (0.10 g, 0.30 mmol) was dissolved in EtOH (5 mL) and placed in an Ace hydrogenation bomb along with platinum (IV) oxide catalyst (0.15 g). The vessel was pressurized to 4 atm H2 and shaken for 16 h. The solution was filtered through a pad of Celite to remove the catalyst, and evaporated under reduced pressure to produce the hydrochloride salt of the desired cis saturated amine as a white solid. This solid was crystallized from EtOH-Et2O to yield 33b (0.097 g, 97.0%) as a white crystalline powder; mp 242-244 °C (dec.). 1H NMR: (300 MHz, d6-DMSO): δ 7.87 (bs, 3H), 7.02 (d, 1H, J = 9.0 Hz), 6.87 (d, 1H, J = 9.0 Hz), 3.75 (s, 3H), 3.56 (s, 3H), 3.41 (bd, 1H, J = 10 Hz), 3.02 (bs, 1H), 2.91-2.75 (m, 2H), 2.21-2.14 (m, 1H), 2.11-2.04 (m, 1H), 1.88-0.98 (m, 13H). MS (ESI): [M+H]+ = 304. Anal. calcd for C19H30ClNO2: C 67.54, H 8.35, N 4.15, found: C 67.19, H 8.31, N 4.14.
Cis-(3-adamantyl-5,6-dimethoxy-1,2,3,4-tetrahydronaphthalen-1-yl)methanamine hydrochloride, 33c
Following the prior method for the synthesis of 33b, 32c (120 mg, 0.308 mmol) was converted exclusively to the cis reduced product as pale tan crystals (118 mg, 97.5%); mp 184-186 °C (dec.). 1H NMR: (500 MHz, d6-DMSO): δ 7.98 (bs, 3H), 7.02 (d, 1H, J = 8.6 Hz), 6.86 (d, 1H, J = 8.6 Hz), 3.75 (s, 3H), 3.65 (s, 3H), 3.42 (bd, 1H, J = 9.4 Hz), 3.01-2.94 (m, 1H), 2.85 (bd, 1H, J = 15.9 Hz), 2.79 (dd, 1H, J = 9.4, 12.0 Hz), 2.23-2.15 (m, 2H), 1.98 (bs, 3H), 1.71-1.50 (m, 12H), 1.14-1.06 (m, 1H), 0.96 (q, 1H, J = 12.1 Hz). MS (ESI): [M+H]+ = 356. Anal. calcd for C23H34ClNO2: C 70.48, H 8.74, N 3.57, found: C 70.13, H 8.81, N 3.57.
Cis-(5,6-dimethoxy-3-propyl-1,2,3,4-tetrahydronaphthalen-1-yl)-methanamine hydrochloride, 33e
Following the prior method for the synthesis of 33b, 32e (205 mg, 0.688 mmol) was converted exclusively to the cis isomer of the title compound as a white crystalline powder (108 mg, 52.4%); mp 236-237 °C (dec.). 1H NMR: (500 MHz, d6-DMSO): δ 8.04 (bs, 3H), 7.03 (d, 1H, J = 8.6 Hz), 6.87 (d, 1H, J = 8.6 Hz), 3.75 (s, 3H), 3.65 (s, 3H), 3.40 (dd, 1H, J = 4.5, 14.5 Hz), 3.10-3.02 (m, 1H), 2.87 (dd, 1H, J = 4.0, 18.0 Hz), 2.77 (dd, 1H, J = 9.5, 14.0 Hz), 2.13-2.01 (m, 2H), 1.55-1.49 (m, 1H), 1.49-1.26 (m, 4H), 1.01 (q, 1H, J = 12.0 Hz), 0.94 (t, 3H, J = 7.0 Hz). MS (ESI): [M+H]+ = 264. Anal. calcd for C16H26ClNO2 (1.0 eq. H2O): C 60.46, H 8.88, N 4.41, found: C 60.66, H 8.63, N 4.29.
Cis-5-(aminomethyl)-7-cyclohexyl-5,6,7,8-tetrahydronaphthalene-1,2-diol hydrobromide, 3b
A magnetically stirring solution of 33b (50 mg, 0.147 mmol) in CH2Cl2 (15 mL) in a flame-dried flask, was cooled to −78 °C. A 1.0 M solution of boron tribromide in CH2Cl2 (0.45 mL) was then slowly added dropwise to the flask as the solution gradually became cloudy. The reaction was stirred at −78 °C for 1 h and then at room temperature for an additional 90 min. The flask was cooled back to −78 °C and MeOH (2 mL) was added to quench the reaction, followed by evaporation under reduced pressure, keeping the water bath temperature below 40 °C. The brown solid residue was washed with MeOH and evaporated three additional times to remove any residual boronate esters. The residue was dried under high vacuum to yield a tan foam solid that was easily powdered to afford the hydrobromide salt (49 mg, 94.2%); mp 126-127 °C (dec.). 1H NMR: (300 MHz, d6-DMSO): δ 9.05 (s, 1H), 8.16 (s, 1H), 7.69 (bs, 3H), 6.58 (AB spin system, 2H), 3.46-3.30 (m, 1H), 3.00-2.87 (m, 1H), 2.86-2.75 (m, 2H), 2.11-1.98 (m, 2H), 1.84-1.62 (m, 5H), 1.40-0.98 (m, 8H). MS (ESI): [M+H]+ = 276. HRMS (ESI): [M+H]+ calcd for C17H26NO2: 276.1964, found: 276.1966.
Cis-5-(aminomethyl)-7-adamantyl-5,6,7,8-tetrahydronaphthalene-1,2-diol hydrobromide, 3c
In the same manner as 3b above, 33c (56 mg, 0.143 mmol) was transformed into 3c (53 mg, 92.0%) as a light brown powder; mp 188 °C (dec.). 1H NMR: (300MHz, CD3OD): δ 6.62 (AB spin system, 2H), 3.47-3.39 (m, 1H), 3.09-3.00 (m, 2H), 2.98-2.88 (m, 1H), 2.29-2.11 (m, 2H), 2.00 (bs, 3H), 1.81-1.59 (m, 12H), 1.28-1.16 (m, 1H), 1.09 (q, 1H, J = 12.1 Hz). MS (ESI): [M+H]+ = 328. HRMS (ESI): [M+H]+ calcd for C21H30NO2: 328.2277, found: 328.2275.
Cis-5-(aminomethyl)-7-propyl-5,6,7,8-tetrahydronaphthalene-1,2-diol hydrobromide, 3e
In the same manner as 3b above, 33e (70 mg, 0.234 mmol) was deprotected to yield the title compound (71 mg, 95.3%); mp 175 °C (dec.). 1H NMR: (300 MHz, d6-DMSO): δ 9.06 (bs, 1H), 8.16 (bs, 1H), 7.71 (bs, 3H), 6.58 (AB spin system, 2H), 3.48-3.35 (m, 1H), 3.04-2.71 (m, 3H), 2.09-1.85 (m, 2H), 1.59-1.30 (m, 5H), 1.05-0.87 (m, 4H). MS (ESI): [M+H]+ = 236. HRMS (ESI): [M+H]+ calcd for C14H22NO2: 236.1651, found: 236.1650.
Pharmacology
Materials
[3H]Spiperone (95 Ci/mmol) and [3H]SCH-23390 (81 Ci/mmol) were purchased from Amersham Biosciences (Piscataway, NJ). Butaclamol, SCH-23390, ketanserin, and most other reagents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO).
Competition Binding Experiments
Fresh porcine striatal tissue was obtained from the Purdue Butcher Block and prepared as previously described.[55] In brief, the striatal tissue was homogenized using a potter-type homogenizer, suspended in homogenization buffer (20 mM Hepes, 0.32 M sucrose, pH 7.4), and centrifuged at 1,000g for 10 min at 4 °C. The pellet (P1) was discarded, and the supernatant was centrifuged at 30,000g for 10 min at 4 °C. The resulting pellet (P2) was resuspended in 50 mM Tris buffer (pH 7.4) by briefly using a Kinematica homogenizer, followed by centrifuging at 30,000g for 30 min at 4 °C. This pellet was resuspended again in 50 mM Tris buffer, dispensed into 1.0 mL aliquots, and centrifuged again at 13,000g for 10 min at 4 °C. A BCA protein assay was used to quantify the final protein concentration in each pellet. The supernatant was removed, and the pellets were frozen at −80 °C until use.
The radioligand binding assays were performed as previously described,[56] with minor modifications. The pellets were resuspended (1 mg/mL) in receptor binding buffer (50 mM Hepes, 4 mM MgCl2, pH 7.4), and 75 μg of protein was used per assay tube. Receptor isotherms were performed with [3H]SCH-23390 and [3H]spiperone to determine Bmax and Kd values for D1-like and D2-like receptor sites, respectively (760 fmol/mg and 0.44 nM for [3H]SCH-23390; 250 fmol/mg and 0.075 nM for [3H]spiperone). All D2-like binding assays were performed with 50 nM ketanserin to block 5-HT2A binding sites. Nonspecific binding was defined with 5 μM butaclamol. Drug dilutions for competition binding assays were made in receptor binding buffer and added to assay tubes containing 75 μg of protein and either 1 nM [3H]SCH-23390 or 0.15 nM [3H]spiperone. All binding experiments were incubated at 37 °C for 30 min and were terminated by harvesting with ice-cold wash buffer (10 mM Tris, 0.9% NaCl) using a 96-well Packard Filtermate cell harvester. After the samples were dried, 30 μL of Packard Microscint O was added to each well. Radioactivity was counted with a Packard Topcount scintillation counter.
Computational Chemistry
Methods
All renderings were performed in PyMol.[57] Trajectories were viewed using VMD (Visual Molecular Dynamics).[58] The crystal structure of the β2 adrenergic receptor co-crystallized with the inverse agonist carazolol was downloaded from the RCSB Protein Data Bank (pdb file #2rh1).[59] The fused T4 lysozyme, acetamide group, 1,4-butanediol molecules, dodecaethylene glycol molecules, maltose molecules, and sulfate ions were removed, leaving only the palmitoyl group attached to C341 in the protein, cholesterol molecules, ligand (carazolol), and waters with two or more hydrogen bond contacts with the protein. The N187E mutation was reversed in silico using the mutation feature of PyMol. Acetyl and N-methylamide caps, as well as non-standard residue hydrogen atoms were added in PyMol; the rest were added using the pdb2gmx module of GROMACS (GROningen MAchine for Chemical Simulations).[60] The orientation of Asn, Gln, and His residues, as well as the protonation state of acidic and basic residues, was visually inspected and no modifications were deemed necessary. Ligand, cholesterol, and palmitoyl cysteine parameters were generated using the antechamber program, part of the AmberTools 1.4 package,[61, 62] based on an ab initio HF/6-31G* optimization[63] performed on Gaussian03[64] and subsequent resp (restrained electrostatic potential) fitting.[65, 66]
Membrane simulations
The prepared receptor system was merged into a pre-equilibrated 85×80Å united-atom POPC (palmitoyl oleoyl phosphatidylcholine) bilayer system, solvated with 12Å of SPC waters on either side and ionized with 0.5M NaCl. All subsequent calculations were performed with GROMACS 4.0,[60] using the AMBER03 force field port[67] (from http://ffamber.cnsm.csulb.edu/) with optimized parameters for united-atom lipids (from http://www.bioinf.uni-sb.de/RB/).[68] The system was energy minimized (steepest descent algorithm), and molecular dynamics (MD) simulations were performed for 10 ns (2 fs/step) at 300K using the NPT ensemble (V-rescale thermostat;[69] Parrinello-Rahman barostat)[70] with position restraints on the protein and ligand heavy atoms, followed by unrestrained simulations for 30 ns.[71] No major changes in the protein-ligand interaction profile or overall tertiary structure of the protein were observed during the simulation.
Receptor activation
The agonist isoproterenol was built in-place from the structure of the antagonist carazolol inside the binding pocket, due to their topological similarity. The new protein-agonist system was energy minimized and MD simulations were performed with soft protein-ligand distance restraints for 5 ns. At this point, the simulations were modified to reflect experimental observations from different sources (see Supporting Information 1), including a comparison between the 3D structures of bovine rhodopsin and opsin (RCSB pdb codes 1U19 and 3CAP, respectively)[72, 73] and a computational study of the activation of the CB1 receptor.[74] 368 ns were logged under various conditions (see Supporting Information 1). Finally, the protein-ligand restraints were removed (protein-protein restraints were conserved, mainly to retain the helicity of TMs 5 and 6, compensating for the absence of IL3) and, after another 10 ns of simulation, the system was energy minimized. A detailed description of the activation process, as well as the structures of the ligands used, can be found in the Supporting Information.
Homology Models
Homology models of the D1 receptor based on the resulting agonist-bound structure of the β2AR were created with Modeller 9 version 2.[75, 76] Alignments were made manually, utilizing key conserved residues as references. Protein sequences were obtained from the Protein Information Resource[77, 78] (See Supporting Information for alignment). 1000 models were generated (including disulfide bridges between C96-C186 and C298-C307), and the model with the lowest internal score was inspected for helix conservation, loop conformations, and key residue alignment. Extracellular loop 3 (EL3) was refined with Modeller, taking the best out of 1000 structures ranked by internal score. Any necessary torsional modifications were carried out in order to preserve relevant motifs (important hydrogen bond, aromatic, and salt bridge interactions). The molecule was prepared in a manner similar to the β2AR, and embedded in the same membrane system as the original template.
The dopamine D1 receptor agonist doxanthrine (DOX) was manually docked into the receptor binding pocket by achieving the best possible overlap between the catechol and amine moieties. After inspecting the system for bad contacts from the insertion of the protein into the membrane, the system was energy minimized. MD simulations were performed using position restraints on the protein and ligand for the first 10 ns, and then distance restraints between the protein and the ligand (see Supporting Information), as well as within the protein (corresponding to those used in the β2AR), for the next 10 ns of simulation. The protein-ligand restraints were removed for the following 20 ns of simulation, during which, after some initial shifts, the protein-ligand interaction profile remained largely unchanged. Energy minimization provided a receptor structure that was used for docking studies.
Docking
Docking of compounds 1c, 2c, and 3c (constructed and energy minimized in vacuum using SYBYL 8.1 with the MMFF94s force field)[79] in the receptor binding site was performed using the GOLD program (Genetic Optimization for Ligand Docking) version 3.2.[80, 81] Ten residues in the binding cavity were allowed to rotate during the docking process (See Supporting Information for full conditions). A distance constraint was used to preserve the known salt bridge between D103 and the ligand ammonium moiety, and a water molecule present in the vicinity of S199 and N292 was included. 100 docking orientations were calculated and the best five were inspected. If these were in good agreement with each other the docking pose with the best GOLD score was taken as the result for the docking run. The protein side chain torsions were modified according to the GOLD output as appropriate using SYBYL, and then the previous ligand was replaced by the docked structure in the system coordinate file via the text editor. The system was then energy minimized and MD simulations were performed without any protein-ligand restraints until convergence was achieved, typically 16-22 ns. After deeming the system converged (see Supporting Information), energy minimization was performed again and the output structures were used for evaluation.
Supplementary Material
Acknowledgements
This work was funded by NIH grants MH42705 (DEN), GM085604 (MAL), MH60397 (VJW), and an endowed AFPE fellowship (LAB). The authors would like to thank Abbott Laboratories for their very generous gift of the isochroman compounds. We also would like to thank Dow Hurst and Dr. Patricia Reggio for providing structures of their in silico activated cannabinoid CB1 receptor, and Dr. Ronald Dror for a valuable discussion regarding in silico GPCR activation. Much of the work presented here is from the PhD theses of LAB and UL.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.
References
- [1].Tarazi FI. SQU Journal for Scientific Research: Medical Sciences. 2001;3(2):93–104. [PMC free article] [PubMed] [Google Scholar]
- [2].Zhang A, Neumeyer JL, Baldessarini RJ. Chemical Reviews. 2007;107(1):274–302. doi: 10.1021/cr050263h. [DOI] [PubMed] [Google Scholar]
- [3].Odowd BF. Journal of Neurochemistry. 1993;60(3):804–816. doi: 10.1111/j.1471-4159.1993.tb03224.x. [DOI] [PubMed] [Google Scholar]
- [4].Kebabian JW, Calne DB. Nature. 1979;277(5692):93–96. doi: 10.1038/277093a0. [DOI] [PubMed] [Google Scholar]
- [5].Kalani MYS, Vaidehi N, Hall SE, Trabanino RJ, Freddolino PL, Kalani MA, Floriano WB, Kam VWT, Goddard WA. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(11):3815–3820. doi: 10.1073/pnas.0400100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shi L, Javitch JA. Annu.Rev.Pharmacol.Toxicol. 2002;42:437–467. doi: 10.1146/annurev.pharmtox.42.091101.144224. [DOI] [PubMed] [Google Scholar]
- [7].Tomic M, Seeman P, George SR, O’Dowd BF. Biochem.Biophys.Res.Commun. 1993;191(3):1020–1027. doi: 10.1006/bbrc.1993.1319. [DOI] [PubMed] [Google Scholar]
- [8].Cueva JP. Purdue University. 2008 [Google Scholar]
- [9].Huang X, Lawler CP, Lewis MM, Nichols DE, Mailman RB. Int.Rev.Neurobiol. 2001;48:65–139. doi: 10.1016/s0074-7742(01)48014-7. [DOI] [PubMed] [Google Scholar]
- [10].Neve KA, Neve RL. The Dopamine Receptors. Humana Press Inc.; Totowa, N.J.: 1996. [Google Scholar]
- [11].Kaiser C, Dandridge PA, Garvey E, Hahn RA, Sarau HM, Setler PE, Bass LS, Clardy J. J.Med.Chem. 1982;25(6):697–703. doi: 10.1021/jm00348a017. [DOI] [PubMed] [Google Scholar]
- [12].Jacob JN, Nichols DE, Kohli JD, Glock D. J.Med.Chem. 1981;24(8):1013–1015. doi: 10.1021/jm00140a021. [DOI] [PubMed] [Google Scholar]
- [13].Brewster WK, Nichols DE, Riggs RM, Mottola DM, Lovenberg TW, Lewis MH, Mailman RB. J.Med.Chem. 1990;33(6):1756–1764. doi: 10.1021/jm00168a034. [DOI] [PubMed] [Google Scholar]
- [14].DeNinno MP, Schoenleber R, Perner RJ, Lijewski L, Asin KE, Britton DR, MacKenzie R, Kebabian JW. J.Med.Chem. 1991;34(8):2561–2569. doi: 10.1021/jm00112a034. [DOI] [PubMed] [Google Scholar]
- [15].Michaelides MR, Schoenleber R, Thomas S, Yamamoto DM, Britton DR, MacKenzie R, Kebabian JW. J.Med.Chem. 1991;34(10):2946–2953. doi: 10.1021/jm00114a002. [DOI] [PubMed] [Google Scholar]
- [16].Estacio SG, Cabral d. C. P., Cabral BJC, Minas d. P. M. E., Simoes JAM. J. Phys. Chem. A. 2004;108(49):10834–10843. [Google Scholar]
- [17].Varfolomeev MA, Abaidullina DI, Solomonov BN, Verevkin SP, Emel’yanenko VN. J. Phys. Chem. B. 2010;114(49):16503–16516. doi: 10.1021/jp108459r. [DOI] [PubMed] [Google Scholar]
- [18].Boduszek B, Uher M. Synthetic Communications. 2000;30(10):1749–1754. [Google Scholar]
- [19].Xu S, Xu SP, Li LM. Yao Xue.Xue.Bao. 2002;37(2):113–116. [PubMed] [Google Scholar]
- [20].Liu HJ, Shia KS, Shang X, Zhu BY. Tetrahedron. 1999;55(13):3803–3830. [Google Scholar]
- [21].Pine SH, Pettit RJ, Geib GD, Cruz SG, Gallego CH, Tijerina T, Pine RD. Journal of Organic Chemistry. 1985;50(8):1212–1216. [Google Scholar]
- [22].Kraus GA, Frazier KA, Roth BD, Taschner MJ, Neuenschwander K. Journal of Organic Chemistry. 1981;46(11):2417–2419. [Google Scholar]
- [23].Doodeman R, Rutjes FPJT, Hiemstra H. Tetrahedron Letters. 2000;41(31):5979–5983. [Google Scholar]
- [24].Ku YK, Grieme T, Raje P, Sharma P, Morton HE, Rozema M, King SA. Journal of Organic Chemistry. 2003;68(8):3238–3240. doi: 10.1021/jo0268613. [DOI] [PubMed] [Google Scholar]
- [25].Cohen N, Schaer B, Saucy G, Borer R, Todaro L, Chiu AM. Journal of Organic Chemistry. 1989;54(14):3282–3292. [Google Scholar]
- [26].Miki Y, Hachiken H, Noguchi K, Ohta M, Nakano A, Takahashi K, Takemura S. Chem.Pharm.Bull.(Tokyo) 1990;38(12):3257–3260. doi: 10.1248/cpb.38.3257. [DOI] [PubMed] [Google Scholar]
- [27].Ku YY, Grieme T, Raje P, Sharma P, King SA, Morton HE. Journal of the American Chemical Society. 2002;124(16):4282–4286. doi: 10.1021/ja0171198. [DOI] [PubMed] [Google Scholar]
- [28].Bernotas RC, Cube RV. Synthetic Communications. 1990;20(8):1209–1212. [Google Scholar]
- [29].Brown JM. Angewandte Chemie-International Edition in English. 1987;26(3):190–203. [Google Scholar]
- [30].Ghosh AK, Krishnan K. Tetrahedron Letters. 1998;39(9):947–948. doi: 10.1016/S0040-4039(97)10666-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sajiki H. Tetrahedron Letters. 1995;36(20):3465–3468. [Google Scholar]
- [32].Sajiki H, Hirota K. Tetrahedron. 1998;54(46):13981–13996. [Google Scholar]
- [33].Kasal A, Chodounska H, Szczepek WJ. Tetrahedron Letters. 1996;37(34):6221–6222. [Google Scholar]
- [34].Schoenleber RW, Kebabian JW, Martin YC, DeNinno MP, Perner RJ, Stout DM, Hsaio CN, DiDomenico S, Jr., DeBernardis JF, Basha FZ, Meyer MD, De B, Erlich PP, Campbell JR, Morton HE, Lijewski L. Dopamine Agonists. 1996 [Google Scholar]
- [35].DeBernardis JF, Kerkman DJ, Winn M, Bush EN, Arendsen DL, McClellan WJ, Kyncl JJ, Basha FZ. J.Med.Chem. 1985;28(10):1398–1404. doi: 10.1021/jm00148a005. [DOI] [PubMed] [Google Scholar]
- [36].Stevens DR, Till CP, Whiting DA. Journal of the Chemical Society-Perkin Transactions. 1992;1(2):185–190. [Google Scholar]
- [37].DeBernardis JF, Kyncl JJ, Basha FZ, Arendsen DL, Martin YC, Winn M, Kerkman DJ. J.Med.Chem. 1986;29(4):463–467. doi: 10.1021/jm00154a006. [DOI] [PubMed] [Google Scholar]
- [38].Meyer MD, Hancock AA, Tietje K, Sippy KB, Prasad R, Stout DM, Arendsen DL, Donner BG, Carroll WA. J.Med.Chem. 1997;40(7):1049–1062. doi: 10.1021/jm960723m. [DOI] [PubMed] [Google Scholar]
- [39].Adachi K, Taniguchi N. Bulletin of the Chemical Society of Japan. 1982;55(5):1655–1656. [Google Scholar]
- [40].Kasturi TR, Sivaramakrishnan R. Indian Journal of Chemistry. 1975;13(7):648–651. [Google Scholar]
- [41].Sperber N, Papa D, Schwenk E. Journal of the American Chemical Society. 1948;70(9):3091–3094. doi: 10.1021/ja01189a076. [DOI] [PubMed] [Google Scholar]
- [42].Jin J, Weinreb SM. Journal of the American Chemical Society. 1997;119(25):5773–5784. [Google Scholar]
- [43].Leete E, Friedman AR. Journal of the American Chemical Society. 1964;86(6):1224–1226. [Google Scholar]
- [44].Bielmeier SR, Lewis MM, Nichols DE, Tropsha A, Mailman RB. Rational design of selective drugs for D1 and D5 dopamine receptors, in Society for Neuroscience. Vol. 809.5. New Orleans, LA: 2000. [Google Scholar]
- [45].Lewis MM, Hoffman B, Henage LG, Miller DW, Nicholas RA, Nichols DE, Tropsha A, Mailman RB. Society for Neuroscience. Vol. 489.4. Miami, FL: 1999. Mutation of the D1A Dopamine Receptor Reveals Specific Residies Involved in Agonist Recognition and Receptor Activation. [Google Scholar]
- [46].Bonner LA, Chemel BR, Watts VJ, Nichols DE. Bioorg Med Chem. 2010;18(18):6763–6770. doi: 10.1016/j.bmc.2010.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nichols DE. The Dopamine Receptors. Humana Press; New York: 2010. [Google Scholar]
- [48].Ahmed B, Khan SA, Alam T. Pharmazie. 2003;58(3):173–176. [PubMed] [Google Scholar]
- [49].Sakai T, Chotaro K. Journal of the Pharmaceutical Society of Japan. 1935;55:691–704. [Google Scholar]
- [50].Perkin WH, Robinson R. Journal of the Chemical Society, Transactions. 1914;105:2376–2392. [Google Scholar]
- [51].Beetz T, Meuleman DG, Wieringa JH. J.Med.Chem. 1982;25(6):714–719. doi: 10.1021/jm00348a020. [DOI] [PubMed] [Google Scholar]
- [52].Elmore NF, King TJ. Journal of the Chemical Society. 1961 Oct;:4425–4428. [Google Scholar]
- [53].Wamser CC, Wagner WR. Journal of the American Chemical Society. 1981;103(24):7232–7234. [Google Scholar]
- [54].Barton DHR, Chern CY, Jaszberenyi JC. Australian Journal of Chemistry. 1995;48(2):407–425. [Google Scholar]
- [55].Hulme EC. Receptor-Ligand Interactions: A Practical Approach. IRL Press at Oxford University Press; Oxford, U.K.: 1992. [Google Scholar]
- [56].Watts VJ, Lawler CP, Gilmore JH, Southerland SB, Nichols DE, Mailman RB. Eur.J.Pharmacol. 1993;242(2):165–172. doi: 10.1016/0014-2999(93)90076-t. [DOI] [PubMed] [Google Scholar]
- [57].The PyMOL Molecular Graphics System. Schrîdinger, LLC: 2006. [Google Scholar]
- [58].Humphrey W, Dalke A, Schulten K. Journal of Molecular Graphics. 1996;14(1):33. doi: 10.1016/0263-7855(96)00018-5. &. [DOI] [PubMed] [Google Scholar]
- [59].Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hess B, Kutzner C, van der Spoel D, Lindahl E. Journal of Chemical Theory and Computation. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- [61].Wang J, Wang W, Kollman PA, Case DA. J.Mol.Graph.Model. 2006;25(2):247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- [62].Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J.Comput.Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- [63].Kuyper LF, Hunter RN, Ashton D, Merz KM, Kollman PA. Journal of Physical Chemistry. 1991;95(17):6661–6666. [Google Scholar]
- [64].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Revision E. 01 Gaussian,Inc.; 2007. [Google Scholar]
- [65].Bayly CI, Cieplak P, Cornell WD, Kollman PA. Journal of Physical Chemistry. 1993;97(40):10269–10280. [Google Scholar]
- [66].Wang JM, Cieplak P, Kollman PA. Journal of Computational Chemistry. 2000;21(12):1049–1074. [Google Scholar]
- [67].Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. J.Comput.Chem. 2003;24(16):1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- [68].Berger O, Edholm O, Jahnig F. Biophysical Journal. 1997;72(5):2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bussi G, Donadio D, Parrinello M. Journal of Chemical Physics. 2007;126(1) doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- [70].Parrinello M, Rahman A. Journal of Applied Physics. 1981;52(12):7182–7190. [Google Scholar]
- [71].Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. Journal of Computational Chemistry. 1997;18(12):1463–1472. [Google Scholar]
- [72].Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J.Mol.Biol. 2004;342(2):571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- [73].Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Nature. 2008;454(7201):183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- [74].Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, Gawrisch K, Pitman MC, Reggio PH. J.Biol.Chem. 285(23):17954–17964. doi: 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Fiser A, Do RK, Sali A. Protein Sci. 2000;9(9):1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sali A, Blundell TL. J.Mol.Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- [77].Wu CH, Huang HZ, Arminski L, Castro-Alvear J, Chen YX, Hu ZZ, Ledley RS, Lewis KC, Mewes HW, Orcutt BC, Suzek BE, Tsugita A, Vinayaka CR, Yeh LSL, Zhang J, Barker WC. Nucleic Acids Research. 2002;30(1):35–37. doi: 10.1093/nar/30.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wu CH, Yeh LSL, Huang HZ, Arminski L, Castro-Alvear J, Chen YX, Hu ZZ, Kourtesis P, Ledley RS, Suzek BE, Vinayaka CR, Zhang J, Barker WC. Nucleic Acids Research. 2003;31(1):345–347. doi: 10.1093/nar/gkg040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].SYBYL 8.1, Tripos International. 2009.
- [80].Jones G, Willett P, Glen RC. Journal of Molecular Biology. 1995;245(1):43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- [81].Jones G, Willett P, Glen RC, Leach AR, Taylor R. Journal of Molecular Biology. 1997;267(3):727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.