

Abstract
Ras oncogenes (Hras, Kras, and Nras) are important drivers of carcinogenesis. However, tumors with Ras mutations often show loss of the corresponding wildtype (WT) allele, suggesting that proto-oncogenic forms of Ras can function as a suppressor of carcinogenesis. In vitro studies also suggest that WT Ras proteins can suppress the tumorigenic properties of alternate mutant Ras family members, but in vivo evidence for these heterologous interactions is lacking. We have investigated the genetic interactions between different combinations of mutant and WT Ras alleles in vivo using carcinogen-induced lung and skin carcinogenesis in mice with targeted deletion of different Ras family members. The major suppressor effect of WT Kras is observed only in mutant Kras-driven lung carcinogenesis, where loss of one Kras allele led to increased tumor number and size. Deletion of one Hras allele dramatically reduced the number of skin papillomas with Hras mutations, consistent with Hras as the major target of mutation in these tumors. However, skin carcinoma numbers were very similar, suggesting that WT Hras functions as a suppressor of progression from papillomas to invasive squamous carcinomas. In the skin, the Kras proto-oncogene functions cooperatively with mutant Hras to promote papilloma development, although the effect is relatively small. In contrast, the Hras proto-oncogene attenuated the activity of mutant Kras in lung carcinogenesis. Interestingly, loss of Nras increased the number of mutant Kras-induced lung tumors but decreased the number of mutant Hras-induced skin papillomas. These results show that the strongest suppressor effects of WT Ras are only seen in the context of mutation of the cognate Ras protein, and only relatively weak effects are detected on tumor development induced by mutations in alternative family members. The data also underscore the complex and context-dependent nature of interactions between proto-oncogenic and oncogenic forms of different Ras family members during tumor development.
Keywords: Ras, Hras, Kras, Nras, skin carcinogenesis, lung carcinogenesis
Introduction
Mutational activation of the RAS family of genes is one of the most common oncogenic events in cancer, occurring in ~30% of human solid tumors. Studies in the mouse have demonstrated that expression of mutant Ras results in tumor development in a range of tissues, underscoring the potency of Ras oncogenes as drivers of tumorigenesis. There is, however, a high degree of specificity with respect to which RAS gene family member is mutated in different tumor types1. While mutations in KRAS are common in lung, colon and pancreatic cancers, those in NRAS predominate in melanoma, and HRAS mutations are commonly seen in bladder, head and neck, and skin cancers 1. Strong tissue-specific mutation patterns are also seen in mouse models of cancer. Chemically induced tumors in mouse lung and skin show complete specificity for mutations involving Kras and Hras, respectively 2, 3. Differences in the regulation of expression play an important role in determining Ras mutation specificity, as insertion of Hras into the Kras locus in Hras knock-in mice demonstrated the capability of Hras to induce lung tumors in vivo, in spite of the complete specificity for Kras mutations in lung tumors from WT mice 4, 5.
RAS genes encode small GTPases that cycle between active (GTP-bound) and inactive (GDP-bound) states in response to extracellular cues. In their active conformations RAS proteins engage and activate effectors that include RAF, phosphatidylinositol 3-kinase (PI3K) and Ral guanine-dissociation stimulator (RalGDS), to regulate diverse cellular functions including cell growth, proliferation, and differentiation. RAS activating mutations found in tumors occur predominantly at codons 12, 13 and 61, and have historically been viewed as functionally dominant because they render RAS proteins constitutively active. However, the status of the WT RAS allele may also play a role in tumors carrying mutant RAS genes. Studies in the mouse showed that skin tumors initiated by somatic activating mutations in Hras are frequently accompanied by an increase in copy number of the mutant allele or loss of the WT allele 6. Furthermore, these genomic events contribute to the progression of squamous carcinomas to more invasive tumors 7. Mutant KRAS alleles are also often expressed at higher levels compared to the WT allele in human lung tumors, likely as a result of preferential amplification of the mutant copy of the gene 8. We and others have shown that copy number alteration involving the Kras locus on chromosome 6 is the earliest and most common somatic genetic event in mouse lung tumors initiated by oncogenic Kras 9, 10. Finally, genetic and in vitro functional studies have shown that WT Kras can functionally suppress the oncogenic activity of mutant Kras through mechanisms that remain to be elucidated 4, 11, 12. Therefore, the imbalance in favor of mutant Ras alleles in tumors is compatible with the requirement of tumor cells to overcome the suppressor effects of the respective WT Ras.
Members of the Ras family of genes share extensive sequence identity with one another, and are ubiquitously expressed, albeit at varying levels. In addition to the respective WT counterpart, the oncogenic activity of one mutant Ras protein may be further modulated by other members of this gene family. Data from in vitro studies suggest complex interactions among Ras oncogenes and proto-oncogenes 13, but these studies involved ectopic overexpression of Ras alleles and relied on reporter systems as functional readouts. Therefore, it is not clear to what extent these interactions actually contribute to the cancer phenotype. To address this question, we took advantage of mouse models of lung and skin cancers to study the in vivo effects of Ras proto-oncogenes on mutant Ras-driven carcinogenesis. We found that the same Ras proto-oncogene could have positive or negative effects on mutant Ras-driven carcinogenesis, depending on the tumor type and/or the mutant Ras oncogene. These findings suggest that the interactions between Ras oncogenes and proto-oncogenes during carcinogenesis are complex and context-dependent.
Results
Chemical carcinogenesis has been widely used to study tumorigenesis and to identify important genetic determinants of this process. Mice treated with a single dose of urethane by intra-peritoneal injection develop multiple lung tumors, the majority of which contain an activating mutation at codon 61 of the Kras gene 4, 12. On the other hand, topical application of a single dose of 7,12-dimethylbenz(a)anthracene (DMBA) to dorsal skin followed by promotion with 12-O-tetradecanoylphorbol-13-acetate (TPA) results in the development of skin tumors, the majority of which harbor a codon 61 activating mutation in the Hras gene 2. We have used these chemical carcinogenesis models to assess the effects of targeted deletions of Ras gene family members on lung and skin tumor development. All studies were conducted in the FVB/N strain of mice, and on this background, Hras, Kras and Nras are highly expressed in both skin and lung (Supplementary Table 1). While levels of Nras are similar in both tissues, Kras is more highly expressed in the lung and Hras in the skin. These differences in expression levels could potentially account, at least in part, for the specificity of Ras mutations in tumors of these tissues.
Deletion of Kras enhances lung but attenuates skin tum,or development
Homozygous deletion of Kras results in embryonic lethality, but mice with one functional copy of the Kras gene are viable and have no apparent developmental defects 14. As a surrogate for a conventional Kras null allele, we used the LSL-KrasG12D allele, which we backcrossed into the FVB/N genetic background. The LSL-KrasG12D allele contains a transcriptional termination STOP element which renders the allele nonfunctional until removed by Cre recombinase 15, 16. We found Kras levels in mice heterozygous for the LSL-KrasG12D (KrasLSL/WT) allele reduced by approximately 2-fold, both in RNA (Figure 1a) and protein (Figure 1b), compared to WT animals. While we cannot completely rule out the possibility of some level of transcriptional leakage, we noted that animals containing the LSL-KrasG12D allele did not develop any lung tumor, or tumors in other tissues, without administration of adeno-Cre.
Figure 1.
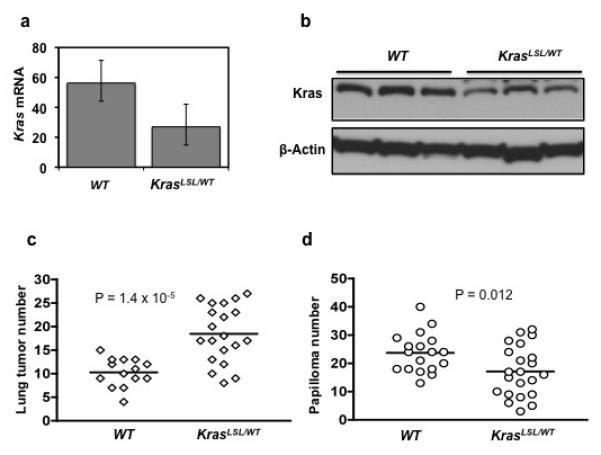
Lung and skin carcinogenesis in mice with one or two functional copies of Kras. The LSL-KrasG12D allele contains a transcriptional STOP element, and is used as a surrogate Kras knockout allele. Kras expression is reduced approximately 2-fold in lungs of KrasLSL/WT mice compared to WT mice, both at (a) RNA and (b) protein levels. Error bars indicate s.d. (c) Lung tumor number at 20 weeks after treatment with urethane. (d) Skin papilloma number at 20 weeks after initiation with DMBA. Data points correspond to tumor number of individual mice of the indicated genotypes. Horizontal line indicates the average tumor number for each genotype. Statistics were calculated using the Wilcoxon-Mann-Whitney test.
Kras is the major mutational target in carcinogen-induced lung tumors 4, 12, but KrasLSL/WT mice containing only one functional Kras allele developed almost 2-fold more lung tumors (18.4 ± 6.0, mean ± s.d.) than WT littermates (10.3 ± 3.0) with two functional Kras alleles (P = 1.4 × 10−5) (Figure 1c). We also observed that tumors from KrasLSL/WT animals were larger in size (data not shown). As expected, more than 95% of lung tumors from mice of both genotypes contained Kras codon 61 activating mutations (Supplementary Table 2). These codon 61 mutations must occur on the functional Kras allele in tumors from heterozygous mice, given that the LSL-KrasG12D allele is nonfunctional. These observations in the LSL-KrasG12D model are in concordance with previous studies using conventional Kras null alleles 11, emphasizing the suppressor function of WT Kras during lung carcinogenesis.
While WT Kras can suppress the activity of mutant Kras, it is not known whether WT Kras exerts a similar effect on cancers driven by mutant forms of other Ras gene family members. To address this question we treated KrasLSL/WT and WT mice with DMBA/TPA to induce skin tumor development. Skin papillomas that developed in both groups of mice contained the codon 61 activating mutation in Hras, as expected (Supplementary Table 3). There was however a ~30% reduction in number of papillomas in KrasLSL/WT mice compared to WT littermates (17.1 ± 8.9 vs. 23.7 ± 6.9, P = 0.012) (Figure 1d). These data suggest that in the skin Kras functions cooperatively with mutant Hras to drive the formation of papillomas.
Deletion of Hras suppresses skin but potentiates lung tumor development
Hras is the major target of mutation in skin tumors induced by DMBA/TPA treatment 2. A previous study used mice of a mixed genetic background (129/Sv, C57BL/6, and DBA/2) to study the effect of Hras deletion on skin tumor development 17. Although C57BL/6 is highly resistant to DMBA/TPA skin carcinogenesis and developed only a modest number of papillomas in WT mice (~16 tumors/mouse), it was found that HrasKO/KO animals developed significantly fewer tumors (~3 tumors/mouse) 17. Because genetic background can affect tumor development as well as pattern of genetic alterations in tumors 10, 18-20, we backcrossed the HrasKO allele into the FVB/N background for more than 15 generations. FVB/N mice are highly susceptible to the development of epithelial tumors, and particularly skin tumors 21. In this genetic background, the difference in papilloma number between WT and HrasKO/KO littermates was much more dramatic (Figure 2a). Whereas WT mice developed between 18-41 papillomas (29.3 ± 6.0), HrasKO/KO mice developed almost 30-fold fewer papillomas (1.3 ± 1.3, P = 1.3 × 10−19), compared to the ~6-fold difference in the previous study, and with more than 65% of animals having only one or no tumors. Mice heterozygous for Hras (HrasKO/WT) showed skin tumor numbers (15.3 ± 5.4) intermediate to those of WT and HrasKO/KO animals, indicating a clear gene-dosage effect on skin papilloma development.
Figure 2.
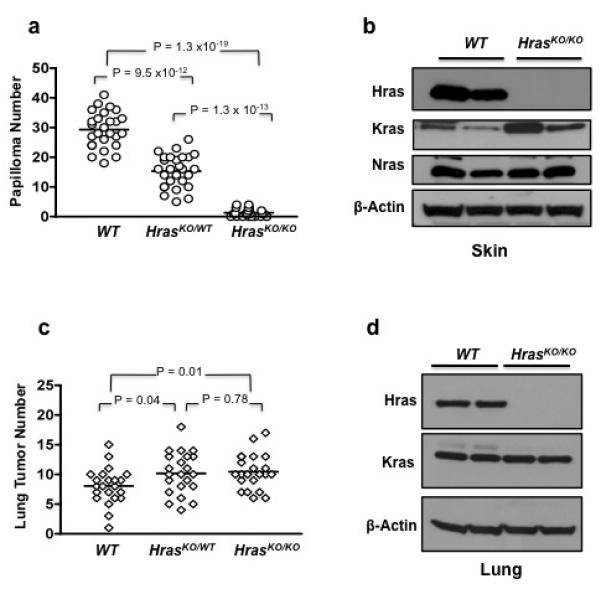
Papilloma and lung tumor development in mice with targeted deletion of Hras. (a) Skin papilloma numbers at 20 weeks after initiation with DMBA. (b) The level of Kras protein is elevated in the skin of HrasKO/KO mice compared to WT animals. (c) Lung tumor numbers at 20 weeks after treatment with urethane. (d) In the lung, there is no difference in level of Kras protein between HrasKO/KO and WT mice.
As expected, papillomas from WT mice had a 100% incidence of Hras mutations at codon 61 (Supplementary Table 3). The majority of papillomas (8 of 10) from HrasKO/WT mice also contained the codon 61 activating Hras mutation. In contrast, of the 6 papillomas from HrasKO/KO mice that were available for analysis, 5 (83%) had activating mutations in Kras, at codons 13 or 61. We found levels of Kras protein, but not Nras protein, to be higher in the skin of HrasKO/KO mice compared to WT animals (Figure 2b), possibly contributing to the preference for Kras mutations in papillomas from HrasKO/KO mice. These data demonstrate that while oncogenic Kras can functionally replace mutant Hras during skin carcinogenesis, the preference is clearly in favor of Hras in mice with functional Hras alleles.
In the lung, mutational activation of Kras occurs in the majority of tumors induced by urethane 4, 12. To determine whether Hras levels influence lung tumor development, we treated WT, HrasKO/WT, and HrasKO/KO littermates with urethane and determined their lung tumor number after 20 weeks. Mice of all three genotypes developed multiple lung tumors, and molecular analyses showed that Kras mutations are common in these tumors and occur at similar frequencies in all three genotype groups (Supplementary Table 2). However, we observed a relatively modest but statistically significant increase in lung tumor number in HrasKO/WT (10.2 ± 3.6, P = 0.04) and HrasKO/KO (10.5 ± 3.0, P = 0.01) mice compared to WT (8.0 ± 3.0) animals (Figure 2c). Unlike in the skin, we did not detect a change in the level of Kras protein in the lungs of HrasKO/KO mice that could potentially have accounted for the increase in lung tumor number (Figure 2d). Nevertheless, these data suggest that Hras negatively modulates the oncogenic activity of mutant Kras during lung carcinogenesis.
Hras heterozygosity increases progression rate of papillomas to squamous carcinomas
Genomic imbalances favoring the mutated Ras allele are observed in lung and skin tumors, suggesting that the WT counterpart of the mutant Ras protein functions as a suppressor of carcinogenesis in both tissues. Heterozygosity at the Kras locus indeed resulted in a significant increase in number of carcinogen-induced lung tumors (Figure 1c). In contrast, mice heterozygous for Hras developed fewer papillomas than WT mice (Figure 2a). Because genomic imbalances at the Hras locus are common in invasive squamous carcinomas 6, 7, we speculated that these genetic events might be contributing to skin tumor progression. Papillomas are thought to be precursors to invasive carcinomas 22, and previous studies have used the ratio of carcinomas to papillomas to measure the rate of malignant progression 23. We therefore monitored HrasKO/WT and WT mice up to one year for the occurrence of carcinomas. Although HrasKO/WT mice developed almost 2-fold fewer papillomas than WT mice, we found the incidence and time of onset of carcinomas to be very similar between both groups (P = 0.18, Kaplan-Meier analysis). In addition, carcinomas from both WT and HrasKO/WT mice have the activating mutation at codon 61 of Hras (Supplementary Table 4). However, Hras KO/WT mice had a significantly higher rate of malignant progression compared to WT mice (Table 1, P = 0.046, Fisher’s exact test). These data suggest that WT Hras functions as a suppressor of skin tumor progression rather than initiation, and that its loss, either through genetic manipulation of the mouse germline or somatic genetic alterations in tumor cells, promotes the conversion of benign skin papillomas to invasive squamous carcinomas.
Table 1.
Hras heterozygosity increases rate of malignant progression of skin tumors.
| Genotype | No. of Mice | Cumulative No. of Papillomas |
Cumulative No. of Carcinomas |
Conversion Ratea |
|---|---|---|---|---|
| HrasWT/WT | 24 | 700 | 29 | 4.1% |
| HrasKO/WT | 26 | 393 | 28 | 7.1% |
Ratio of cumulative number of carcinomas (at 52 weeks) to cumulative number of papillomas (at 20 weeks).
Opposing effects of Nras deletion on lung and skin tumor development
Genetic studies in mice have shown that the Nras proto-oncogene can suppress the malignant phenotype of thymic lymphomas driven by its oncogenic counterpart 24. However, it is not known whether Nras exerts similar effects on the development of tumors driven by mutant Kras or mutant Hras. To address these questions, we subjected NrasWT/WT, NrasKO/WT, and NrasKO/KO littermates to the urethane protocol to induce lung tumors, and to the DMBA/TPA protocol to induce skin tumors.
Urethane induced lung tumors from NrasWT/WT, NrasKO/WT, and NrasKO/KO mice all contained activating mutation at codon 61 of Kras (Supplementary Table 2). However, we observed an increase in lung tumor number in both NrasKO/WT (11.1 ± 2.9, P = 0.015) and NrasKO/KO (12.5 ± 5.0, P = 0.008) mice compared to NrasWT/WT mice (8.4 ± 3.3) (Figure 3a). NrasKO/KO developed more tumors than NrasKO/WT, but the difference was not statistically significant. These data suggest that the Nras proto-oncogene attenuates the oncogenic activity of mutant Kras during lung tumor development.
Figure 3.
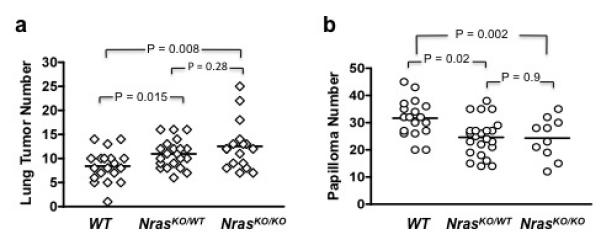
Lung tumor and papilloma development in mice with targeted deletion of Nras. (a) Lung tumor number at 20 weeks after IP injection of urethane. (b) Skin papilloma number at 20 weeks after initiation with DMBA.
The status of Nras had no effect on the frequency of Hras mutations in papillomas, as all tumors analyzed contained the codon 61 Hras mutation (Supplementary Table 3). However, both NrasKO/WT (24.6 ± 6.9, P = 0.002) and NrasKO/KO (24.3 ± 7.5, P = 0.02) and mice developed fewer papillomas compared to WT mice (31.7 ± 6.8) (Figure 3b). While Nras has a negative role in lung carcinogenesis, these data show that in the skin it has a positive effect on mutant Hras-induced carcinogenesis.
Discussion
We have investigated the genetic interactions between proto-oncogenic and oncogenic forms of Ras family members during the process of tumor development in the lung and the skin. Several studies have noted that WT alleles of various members of the Ras family are lost in tumors carrying mutations of the same Ras family member. The observation that Ras genes appear to be co-expressed, albeit at different levels, in mouse and human tissues raised the possibility that WT proteins of one isoform may suppress mutant Ras activity driven by a different isoform. Previous studies aimed at addressing this question were carried out using transfection assays with resultant expression of Ras proteins at non-physiological levels 13. We therefore initiated this study to investigate interactions between the different Ras family members in an in vivo context. The use of chemical carcinogenesis to induce tumor formation in the mouse mimics the effects of carcinogen exposure in human cancer development and also recapitulates the specificity of Ras mutations observed in human cancers. Our data show that the major effect of loss of WT Kras is seen only in lung tumors driven by mutant Kras, and only marginal effects are seen due to altered gene dosage of other Ras family members.
Mice with only one functional copy of Kras developed more and larger lung tumors than WT littermates, consistent with the notion that the remaining WT copy of Kras is a potent suppressor of lung tumor formation and progression 11. This is compatible with the observation that human and murine lung tumors with Kras mutations frequently have copy number alterations involving the Kras locus 8-10, likely as a mechanism to shift the balance in favor of mutant Kras. The molecular mechanism that underlies the suppressor function of WT Kras is not known, but could potentially involve competition for common downstream effectors 11. Alternatively, signaling through WT Kras could occur independently of mutant Kras in the lung to trigger cellular functions that have evolved as protective mechanisms against oncogenic conditions. We recently showed that mutational activation of Kras4A, the minor isoform of Kras, is necessary for lung tumor development, and that this isoform is also responsible for the suppressor function of WT Kras 4. Detailed functional analyses of Kras4A could provide important mechanistic insights into the oncogenic and suppressor functions of Kras.
We found papilloma numbers to directly correlate with the number of functional Hras alleles, compatible with Hras as the target of mutation in these tumors. As previously noted 17, animals that completely lack Hras developed few skin papillomas with the majority having activating mutations in Kras, indicating that mutational activation of Kras can induce papilloma formation in vivo. These observations agree with the fact that Kras mutations occur in a broad range of tumor types in different tissues, perhaps due to the unique function of Kras in stem cell expansion 25. In the skin, the preference for Hras mutations in papillomas may be attributed, at least in part, to the higher levels of Hras compared to Kras in this tissue. Alternatively, the preference for Hras mutations may be due to the particular signaling networks involving these two Ras proteins in skin. In HrasKO/KO animals, levels of Kras are upregulated in the skin, and this could potentially render signaling through Kras more conducive for skin carcinogenesis. Interestingly, mice carrying the KrasLA2 allele, which undergoes spontaneous somatic recombination that results in oncogenic activation of Kras, develop skin papillomas but only on a mixed genetic background and not on the FVB/N inbred strain 26, suggesting that genetic background may also contribute to the specificity of Ras mutations.
Hras and Kras are major targets of mutation in skin and lung tumors respectively, but targeted deletions of these genes showed opposite effects on tumor number in the respective tissues. In the lung, the suppressor function of WT Kras is a major determinant of lung tumor formation, as targeted deletion of one Kras allele resulted in an increase in tumor number. This is in agreement with genetic studies showing that the balance between levels of mutant and WT Kras regulates lung cancer susceptibility 12. On the other hand, deletion of one Hras allele resulted in a decrease in number of papillomas despite the fact that Hras also undergoes loss of WT allele or gain of mutant allele during skin tumor development 6, 7. However, HrasKO/WT mice demonstrated a significantly higher rate of malignant progression from papillomas to carcinomas, suggesting that WT Hras functions as a suppressor of skin tumor progression rather than skin tumor initiation. This is consistent with the high frequency of genomic imbalance at the Hras locus in invasive squamous carcinomas with Hras mutations 7. Furthermore, the effect on skin tumor progression appeared to be specific to WT Hras, as reduced Nras or Kras level had no effect on this process (data not shown).
We also found that Ras proto-oncogenes are capable of influencing tumor formation driven by mutant forms of other Ras family members, albeit on a modest level. These results are summarized in Figure 4. Deletion of Hras or Nras increased the number of mutant Kras-driven lung tumors, suggesting that Hras and Nras attenuate the ability of mutant Kras to promote lung carcinogenesis. In contrast, deletion of Kras or Nras reduced the number of mutant Hras-driven skin papillomas, suggesting that Kras and Nras signaling function cooperatively with oncogenic Hras during skin carcinogenesis. These findings, particularly those involving Nras, demonstrate that the effects of Ras proto-oncogenes on mutant Ras-driven carcinogenesis are context-dependent. It is not clear whether this is due to differences in oncogene function (Hras vs. Kras) or tissue-specific signaling networks (skin vs. lung). One possible explanation for the data from our skin carcinogenesis studies is that signaling through WT Kras and WT Nras has additive contributions to the oncogenic activity of mutant Hras. However, in vitro studies have shown that expression of Nras has little to no effect on the activity of mutant Hras 13, suggesting that the interactions between Nras and mutant Hras, as well as other Ras proto-oncogene and oncogene pairs, may have a non-cell autonomous mechanistic basis. For example, deletion of Ras proto-oncogenes could affect aspects of tissue physiology that are relevant to carcinogenesis, such as the inflammatory response, which has been shown to play an important role in mutant Ras-driven cancer development 27. Additive effects of Kras and Nras deletion on vascular development and haemaotpoiesis during embryogenesis 14 may also reflect complex non cell-autonomous roles in tumorigenesis. Future work incorporating in vitro and in vivo studies are necessary in order to elucidate the mechanisms that underlie the interactions between proto-oncogenic and oncogenic forms of Ras family members, and to understand their contributions to cancer development.
Figure 4.
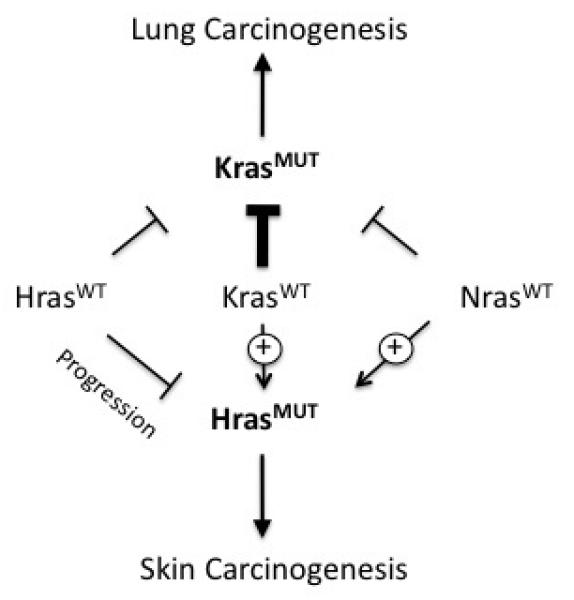
Genetic interactions between Ras proto-oncogenes and oncogenes during lung and skin carcinogenesis. Thickness of the lines indicates the strength of interactions. The Kras proto-oncogene is a strong suppressor of lung tumor development driven by oncogenic Kras. Both Nras and Hras proto-oncogenes also suppress mutant Kras-driven lung carcinogenesis, but the effects were relatively modest. In the skin, Nras and Kras proto-oncogenes had positive effects on the development of mutant Hras-driven papillomas. The Hras proto-oncogene also function as a suppressor of skin carcinogenesis, but at the level of progression rather than initiation.
Materials and Methods
Animals
The HrasKO, NrasKO, and LSL-KrasG12D alleles have been backcrossed into the FVB/N background over multiple generations to minimize the effects of genetic heterogeneity on tumor development. For the HrasKO and NrasKO alleles, heterozygous animals were bred to generate littermates of all three possible genotypes. Because LSL-KrasG12D is a non-functional allele until activated by Cre recombinase, it can effectively be used as a functional knockout allele of Kras. LSL-KrasG12D heterozygous mice were bred with FVB/N animals to generate WT and heterozygous mice.
Lung Carcinogenesis
Male mice were treated with a single dose of urethane (in PBS; at 1g/kg body mass) by intraperitoneal injection at 5-8 weeks of age. Animals were sacrificed twenty weeks after the injection, and lungs were collected, fixed overnight in formalin, and stored in 70% ethanol. Lung tumor numbers were counted under a dissecting microscope.
Skin Chemical Carcinogenesis
Skin tumor development was initiated with a single dose of DMBA on the dorsal skin at 8 weeks of age, followed by biweekly promotion with TPA for twenty weeks as previously described 2. Because male animals often fight, causing skin wounding, only female mice were used in this assay. The number of skin tumors that developed on individual animals was counted at different time points during the course of the study.
RNA and Protein Analysis
Frozen skin and lung tissues were ground up in liquid nitrogen and processed for RNA and protein as previously described 28. Gene expression was measured on the Affymetrix M430 2.0 platform, and by real-time PCR using Mm00517491_m1 (Kras2) and Mm00607939_s1 (β-Actin) assays on demand from Applied Biosystems. Ras proteins were detected using antibodies against Hras (C-20), Kras (F234), and Nras (C-20), purchased from Santa Cruz Biotechnology.
Mutational Analysis
Tumor tissues were incubated overnight at 55 °C with proteinase K, and DNA was purified by phenol/chloroform extraction. The status of Hras codon 61 was determined using a digestion assay as previously described 29. Kras mutations were identified by direct DNA sequencing.
Supplementary Material
Acknowledgements
We are grateful to Dr. Motoya Katsuki and Dr. Hiroki Nagase for the HrasKO mice, and Dr. Tyler Jacks for the LSL-KrasG12D mice. This work was supported by NCI grants CA111834-01 and CA84244 to AB. PMKW is supported NIH Training Grant T32 GM007175 and a National Science Foundation Graduate Research Fellowship. MDT acknowledges the support from the Nan Tucker McEvoy Research Fund in Thoracic Oncology. AB acknowledges support from the Barbara Bass Bakar Chair in Cancer Genetics.
Financial Support: This work was supported by CA111834-01 and CA84244 to AB
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://nature.com.onc)
References
- 1.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 2.Balmain A, Pragnell IB. Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature. 1983;303:72–74. doi: 10.1038/303072a0. [DOI] [PubMed] [Google Scholar]
- 3.You M, Candrian U, Maronpot RR, Stoner GD, Anderson MW. Activation of the Ki-ras protooncogene in spontaneously occurring and chemically induced lung tumors of the strain A mouse. Proc Natl Acad Sci U S A. 1989;86:3070–3074. doi: 10.1073/pnas.86.9.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.To MD, Wong CE, Karnezis AN, Del Rosario R, Di Lauro R, Balmain A. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat Genet. 2008;40:1240–1244. doi: 10.1038/ng.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manenti G, Trincucci G, Pettinicchio A, Amendola E, Scarfo M, Dragani TA. Cis-acting genomic elements of the Pas1 locus control Kras mutability in lung tumors. Oncogene. 2008;27:5753–5758. doi: 10.1038/onc.2008.194. [DOI] [PubMed] [Google Scholar]
- 6.Bremner R, Balmain A. Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell. 1990;61:407–417. doi: 10.1016/0092-8674(90)90523-h. [DOI] [PubMed] [Google Scholar]
- 7.Buchmann A, Ruggeri B, Klein-Szanto AJ, Balmain A. Progression of squamous carcinoma cells to spindle carcinomas of mouse skin is associated with an imbalance of H-ras alleles on chromosome 7. Cancer Res. 1991;51:4097–4101. [PubMed] [Google Scholar]
- 8.Modrek B, Ge L, Pandita A, Lin E, Mohan S, Yue P, et al. Oncogenic activating mutations are associated with local copy gain. Mol Cancer Res. 2009;7:1244–1252. doi: 10.1158/1541-7786.MCR-08-0532. [DOI] [PubMed] [Google Scholar]
- 9.Sweet-Cordero A, Tseng GC, You H, Douglass M, Huey B, Albertson D, et al. Comparison of gene expression and DNA copy number changes in a murine model of lung cancer. Genes Chromosomes Cancer. 2006;45:338–348. doi: 10.1002/gcc.20296. [DOI] [PubMed] [Google Scholar]
- 10.To MD, Quigley DA, Mao JH, Del Rosario R, Hsu J, Hodgson G, et al. Progressive genomic instability in the FVB/Kras(LA2) mouse model of lung cancer. Mol Cancer Res. 2011;9:1339–1345. doi: 10.1158/1541-7786.MCR-11-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Wang Y, Vikis HG, Johnson L, Liu G, Li J, et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat Genet. 2001;29:25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
- 12.To MD, Perez-Losada J, Mao JH, Hsu J, Jacks T, Balmain A. A functional switch from lung cancer resistance to susceptibility at the Pas1 locus in Kras2LA2 mice. Nat Genet. 2006;38:926–930. doi: 10.1038/ng1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz R, Lue J, Mathews J, Yoon A, Ahn D, Garcia-Espana A, et al. Inhibition of Ras oncogenic activity by Ras protooncogenes. Int J Cancer. 2005;113:241–248. doi: 10.1002/ijc.20563. [DOI] [PubMed] [Google Scholar]
- 14.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 17.Ise K, Nakamura K, Nakao K, Shimizu S, Harada H, Ichise T, et al. Targeted deletion of the H-ras gene decreases tumor formation in mouse skin carcinogenesis. Oncogene. 2000;19:2951–2956. doi: 10.1038/sj.onc.1203600. [DOI] [PubMed] [Google Scholar]
- 18.Nagase H, Bryson S, Cordell H, Kemp CJ, Fee F, Balmain A. Distinct genetic loci control development of benign and malignant skin tumours in mice. Nat Genet. 1995;10:424–429. doi: 10.1038/ng0895-424. [DOI] [PubMed] [Google Scholar]
- 19.Hager JH, Hodgson JG, Fridlyand J, Hariono S, Gray JW, Hanahan D. Oncogene expression and genetic background influence the frequency of DNA copy number abnormalities in mouse pancreatic islet cell carcinomas. Cancer Res. 2004;64:2406–2410. doi: 10.1158/0008-5472.can-03-3522. [DOI] [PubMed] [Google Scholar]
- 20.Dworkin AM, Ridd K, Bautista D, Allain DC, Iwenofu OH, Roy R, et al. Germline variation controls the architecture of somatic alterations in tumors. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hennings H, Glick AB, Lowry DT, Krsmanovic LS, Sly LM, Yuspa SH. FVB/N mice: an inbred strain sensitive to the chemical induction of squamous cell carcinomas in the skin. Carcinogenesis. 1993;14:2353–2358. doi: 10.1093/carcin/14.11.2353. [DOI] [PubMed] [Google Scholar]
- 22.Perez-Losada J, Balmain A. Stem-cell hierarchy in skin cancer. Nat Rev Cancer. 2003;3:434–443. doi: 10.1038/nrc1095. [DOI] [PubMed] [Google Scholar]
- 23.Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, et al. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531–542. doi: 10.1016/s0092-8674(00)80127-0. [DOI] [PubMed] [Google Scholar]
- 24.Diaz R, Ahn D, Lopez-Barcons L, Malumbres M, Perez de Castro I, Lue J, et al. The N-ras proto-oncogene can suppress the malignant phenotype in the presence or absence of its oncogene. Cancer Res. 2002;62:4514–4518. [PubMed] [Google Scholar]
- 25.Quinlan MP, Settleman J. Explaining the preponderance of Kras mutations in human cancer: An isoform-specific function in stem cell expansion. Cell Cycle. 2008;7:1332–1335. doi: 10.4161/cc.7.10.5927. [DOI] [PubMed] [Google Scholar]
- 26.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 27.Lee KE, Bar-Sagi D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell. 2010;18:448–458. doi: 10.1016/j.ccr.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao JH, To MD, Perez-Losada J, Wu D, Del Rosario R, Balmain A. Mutually exclusive mutations of the Pten and ras pathways in skin tumor progression. Genes Dev. 2004;18:1800–1805. doi: 10.1101/gad.1213804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagase H, Mao JH, Balmain A. Allele-specific Hras mutations and genetic alterations at tumor susceptibility loci in skin carcinomas from interspecific hybrid mice. Cancer Res. 2003;63:4849–4853. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.