

Summary
The DNA repair enzyme TDP2 resolves 5′-phosphotyrosyl-DNA adducts, and is responsible for resistance to anti-cancer drugs that target covalent topoisomerase-DNA complexes. TDP2 also participates in key signaling pathways during development and tumorigenesis, and cleaves a protein-RNA linkage during picornavirus replication. The crystal structure of zebrafish TDP2 bound to DNA reveals a deep and narrow basic groove that selectively accommodates the 5′-end of single-stranded DNA in a stretched conformation. The crystal structure of the full-length C. elegans TDP2 shows that this groove can also accommodate an acidic peptide stretch in vitro, with Glu and Asp sidechains occupying the DNA backbone phosphate binding sites. This extensive molecular mimicry suggests a potential mechanism for auto-regulation and how TDP2 may interact with phosphorylated proteins in signaling. Our study provides a framework to interrogate functions of TDP2 and develop inhibitors for chemotherapeutic and antiviral applications.
Topoisomerases release torsional stress or resolve catenation problems in DNA by cleaving DNA strands and resealing them after changing their topological states1–3. During the topoisomerase-catalyzed reactions, a transiently formed cleavage complex, in which the topoisomerase is covalently bound to DNA through a phosphotyrosine linkage, coordinates the cleavage and re-ligation steps4 (Fig. 1). Failed topoisomerase reactions lead to a persisting DNA strand break with the enzyme stuck to the cleaved DNA end, and are implicated in genome instability and carcinogenesis5,6. Several classes of anticancer drugs target the cleavage complex of topoisomerases to induce the formation of such cytotoxic DNA lesions1,7,8. The recently identified DNA repair enzyme TDP2 is capable of hydrolyzing a 5′-phosphotyrosine linkage9–11, the bond formed by most types of topoisomerases including type-II and type-IA1–4. As expected from its enzymatic activity (Fig. 1), TDP2 plays a critical role in cellular resistance to topoisomerase II-induced DNA damage and has been linked to chemotherapy resistance against etoposide12, an anti-cancer drug that inhibits type-II topoisomerases8.
Figure 1. Repair of topoisomerase II cleavage complexes by TDP2.

(a, b) Topoisomerase II (TOP2) functions as a homodimer, where each monomer cleaves one strand of a double-stranded DNA by forming a covalent 5′-phosphotyrosyl bond. The resulting cleavage complex (b) allows the passage of another duplex DNA through the break, thereby enabling DNA relaxation, catenation-decatenation, and knotting-unknotting1. Under normal conditions, religation of the cleaved DNA strands is highly efficient and most of TOP2 is non-covalently bound to DNA as in (a). In the presence of anticancer drugs such as etoposide, mitoxantrone, doxorubicin, and daunorubicin, or food flavonoids or DNA damage or oxidative lesions, the cleavage complex accumulates and needs to be removed for DNA repair1. (c) Prior to TDP2 activity, the cleavage complex needs to be denatured or proteolyzed to expose the DNA-5′-phosphotyrosyl bond. (d) TDP2 releases the TOP2 polypeptide and leaves the 5′-phosphate end. (e) The DNA break may be repaired by direct ligation after annealing of the two ends with the 4-base pair stagger, or through double-strand break (DSB) repair mechanisms.
While TDP2 complements the activity of the extensively studied TDP1 that is specific for the 3′-phosphotyrosine bond formed by the type-IB topoisomerases13–15, the two classes of TDP enzymes are mechanistically distinct from one another, and the substrate recognition mechanisms of TDP2 remain unknown. Moreover, TDP2 is a multi-functional protein also known as TTRAP16,17 or EAPII18–20 and acts in signal-transduction and transcription regulation, but it is unknown how TDP2 serves several seemingly unrelated roles in the cell. Beside its normal cellular roles, TDP2 also plays a role in the replication cycle of picornaviruses as a co-opted host factor that resolves the tyrosyl-RNA linkage formed between viral RNA genome and the primer protein (VPg)21. Thus, identification of specific TDP2 inhibitors may lead to novel therapeutics to treat cancer and various diseases caused by picornavirus infections. Here we performed structural and biochemical studies of TDP2 from two different organisms, in order to better understand TDP2 functions and gain insights into its multifunctionality.
RESULTS
TDP2 fold and active site structure
We determined crystal structures of the full-length TDP2 from Danio rerio and Caenorhabditis elegans (hereinafter referred to as zTDP2 and cTDP2) at 1.66Å and 2.35Å resolution, respectively (Table 1). The zTDP2 structure was determined with one of the two molecules in the asymmetric unit in complex with DNA revealing the mechanism of substrate recognition (Fig. 2). In the zTDP2 crystals, the N-terminal 110-120 residues preceding the catalytic domain are poorly ordered and were not modeled in the electron density map. The cTDP2 structure, on the other hand, reveals the overall architecture of the full-length TDP2 molecule without DNA (Fig. 3).
Table 1.
Data collection and refinement statistics
| zTDP2+DNA | zTDP2+DNA+Mg2+ | cTDP2 (Se) | cTDP2 (native) | cTDP2ΔN107 | |
|---|---|---|---|---|---|
|
| |||||
| 4F1H | 4FPV | 4F1I | 4GEW | 4FVA | |
| Data collection | |||||
| Space group | P212121 | P212121 | P41212 | P41212 | P21 |
| Cell dimensions | |||||
| a, b, c (Å) | 57.9, 95.1, 104.7 | 58.1, 95.0, 104.5 | 100.7, 100.7, 121.4 | 101.3, 101.3, 120.9 | 52.6, 104.4, 84.7 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 91.0, 90 |
| Wavelength (Å) | 0.979 | 0.979 | 0.979 | 0.979 | 0.979 |
| Resolution (Å) | 50 - 1.66 (1.71 - 1.66)a | 50 - 1.73 (1.81 - 1.73) | 50 - 2.50 (2.59 - 2.50) | 50 - 2.35 (2.39 - 2.35) | 50 - 1.95 (1.98 - 1.95) |
| Rsym | 0.068 (0.57) | 0.045 (0.51) | 0.126 (0.82) | 0.085 (0. 69) | 0.066 (0.56) |
| I/σI | 29.9 (2.4) | 26.9 (1.7) | 8.3 (2.1) | 24.1 (1.8) | 14.2 (1.8) |
| Completeness (%) | 100.0 (99.8) | 96.5 (95.3) | 100.0 (100.0) | 100.0 (99.7) | 99.1 (97.9) |
| Redundancy | 6.7 (4.5) | 6.7 (4.5) | 3.8 (3.6) | 12.3 (10.3) | 3.8 (3.5) |
| Refinement | |||||
| Resolution (Å) | 50–1.66 | 50–1.73 | 50–2.50 | 50–2.35 | 50–2.07 |
| No. reflections | 67732 | 59083 | 22218 | 26841 | 55529 |
| Rwork/Rfree | 0.16/0.19 | 0.17/0.20 | 0.18/0.22 | 0.17/0.21 | 0.17/0.22 |
| No. atoms | |||||
| Protein | 4187 | 4192 | 2626 | 2600 | 7896 |
| DNA | 109 | 105 | - | - | - |
| Ligand/ion | 25 | 22 | 48 | 54 | 20 |
| Water | 586 | 496 | 142 | 137 | 646 |
| B-factors | |||||
| Protein | 23.6 | 25.4 | 62.2 | 71.4 | 32.9 |
| DNA | 39.6 | 51.0 | - | - | - |
| Water | 37.9 | 38.3 | 62.1 | 70.9 | 41.8 |
| Ligand/ion | 54.6 | 45.8 | 84.0 | 90.0 | 43.7 |
| R.m.s deviations | |||||
| Bond lengths (Å) | 0.014 | 0.008 | 0.004 | 0.004 | 0.008 |
| Bond angles (°) | 1.53 | 1.21 | 0.88 | 0.91 | 1.11 |
The data of highest resolutionshell are shown in parenthesis.
Figure 2. Structure of zTDP2, the unique mode of DNA-binding, and the active site architecture.

(a) The catalytic domain of zTDP2 (ribbons) and the bound DNA (sticks), with simulated annealing composite omit 2Fo−Fc electron density contoured at 1.0σ shown for 1.8Å from the DNA atoms. (b) The molecular surface of zTDP2 with DNA bound in a narrow groove leading to the active site. (c–e) Top, surface electrostatic potential (red:−5kT e−1 to blue:+5kT e−1) of zTDP2, DNaseI22, and APE1 (ref. 23) with bound DNA, in the same orientation as in the bottom panels. Bottom, backbone fold of each protein in ribbons, with sidechains for the conserved tetrad of catalytic residues (Asn, Glu, Asp, His) in sticks. The green arrows in (c) indicate the shallow grooves opposite the DNA-binding groove across the active site of zTDP2 potentially involved in binding topoisomerase-derived substrate peptides. (f) A wall-eye stereo pair showing the 5′-terminus of the single-stranded DNA and surrounding protein residues in the active site. The 4 sidechains shown in (c) are highlighted by red labels. The green sphere represents a divalent metal ion, whereas the red spheres represent water molecules. Hydrogen bonds or metal coordination interactions are denoted by dotted lines. (g) In vitro 5′-phosphotyrosine bond hydrolysis activity of the wild-type and E161A mutant zTDP2. ssY19 and p19 represent the 5′-phosphotyrosyl-oligonucleotide substrate and 5′-phospho-oligonucleotide product, respectively.
Figure 3. The full-length TDP2 has a modular architecture.

(a) Full-length cTDP2 molecule in the crystal, with residue numbers indicated. The extreme N-terminal 20 residues as well as the linker between the α-helical bundle and the catalytic domain are disordered (dotted lines). (b) Structural comparison between the catalytic domains of zTDP2 (yellow) and cTDP (blue). Two different views (90° rotated) are shown. (c) Molecular surface of the cTDP2 catalytic domain colored according to the electrostatic potential, with the N-terminal regions of two other molecules (yellow and green) shown in ribbons. (d) Top view, similar to that of the zTDP2-DNA complex in the top panel in Fig. 2c. The basic DNA-binding groove is occupied by the N-terminal residues of one molecule (yellow), while the active site pocket and the adjacent groove are blocked by those from another molecule (green). The surface electrostatic potential is color-scaled as in Fig. 2.
The catalytic domain of zTDP2 spans residues 120 through 369, accounting approximately for the C-terminal two thirds of the polypeptide. The protein fold consists of a double layer of β-sheets sandwiched between α-helices (Fig. 2a), resembling a family of Mg2+ or Mn2+-dependent endonucleases including DNaseI22 and the apurinic/apyrimidinic site endonuclease-1 (APE1)23 (Fig. 2c–e). The active site residues of zTDP2 are readily identifiable from this structural conservation and include Glu161 and Asp271, corresponding to the catalytically important residues Glu152 and Asp262 of the human TDP2 identified through mutation analyses9,11. These residues are positioned at the bottom of a deep pocket along with other putative catalytic residues including Asn129 and His360 (Fig. 2c,f and Supplementary Fig. 1a,b). The arrangement of the tetrad of active site residues of zTDP2 (Asn129, Glu161, Asp271, His360) resembles those for the corresponding residues of DNaseI22 (Asn7, Glu39, Asp168, His252) and human APE1 (Asn68, Glu96, Asp210, His309) (ref. 23), with r.m.s.d. for the sidechain atoms of 0.43Å and 0.48Å, respectively (Fig. 2c–e, bottom panels). The conserved active site architecture indicates that TDP2 employs a divalent metal-dependent catalytic mechanism similarly to other members of this family9,24.
Mode of single-stranded DNA binding
Despite the similar domain folds and active site arrangements, we found that zTDP2 binds DNA very differently from DNaseI or APE1 (Fig. 2c–e, top panels). Unlike DNaseI and APE1 that bind to double-stranded DNA substrates using a broad and shallow DNA-binding face22,23, TDP2 has a deep and narrow groove leading to the enzyme active site. By soaking a single-stranded DNA with 5′-phosphotyrosine modification (5′-Tyr-pTGCAGC-3′) into the zTDP2 crystal, we observed clear electron density for 5 bases of single-stranded DNA with its 5′-end anchored in the active site (Fig. 2a and Supplementary Fig. 1). The DNA bound to zTDP2 has a strikingly stretched-out conformation for the 5′-terminal bases T1 and G2 occupying the narrow DNA-binding groove, with the following C3, A4, and G5 bases stacked on one another as in a double-stranded DNA. The structure shows that at least 2 nucleotides at the 5′-terminus of a DNA substrate need to be single-stranded (unpaired) to access the active site of zTDP2 (Fig. 2b), recalling the double-nucleotide unpairing required for productive DNA binding to FEN1 (ref. 25). This substrate requirement fits well with the expected structure of a trapped topoisomerase-II reaction intermediate in which tyrosine is linked to a 4-base 5′-overhang of a double-stranded DNA8 (Fig. 1b, c), and explains the recent observation that TDP2 processes 5′-overhanged substrates much more efficiently than blunt-end substrates11. The shallow grooves on the opposite side of the DNA-binding groove across the active site appear suitable to accommodate a topoisomerase-derived peptide linked to the tyrosine backbone (indicated by green arrows in Fig. 2c, top and Fig. 3d).
zTDP2 interacts extensively with the bound single-stranded DNA, making direct or water-mediated contacts with the backbone phosphates of all 5 nucleotides from the 5′-end, as well as forming hydrogen bonds with the deoxyribose O4′ atoms of the T1, G2, and C3 positions (Fig. 4a, b). In addition, Tyr318 that forms part of a side-wall of the DNA-binding groove stacks against the G2 base, stabilizing the stretched-out single-stranded conformation (Supplementary Fig. 1c). Notably, except for the hydrogen bond that Arg321 makes with the C3 base in one of its two sidechain conformers (Fig. 4b and Supplementary Fig. 1c), the DNA interactions made by zTDP2 are all sequence-independent, as expected for TDP2’s role in repairing 5′-phosphotyrosine adducts in a sequence non-specific fashion. In contrast to the well-defined electron density for the single-stranded DNA up to the scissile phosphate bridging the 5′-end and tyrosine (Fig. 2a), density for the tyrosine moiety was missing in the 1.66Å resolution map from a crystal soaked with the phosphotyrosyl DNA substrate in the absence of added metals or EDTA (Supplementary Fig. 1a). As the observed geometry of the 5′-phosphate group in the active site was consistent with that expected for the cleaved product (Fig. 2f) and we observed electron density for a putative divalent metal ion interacting with the 5′-phosphate and Glu161, it was suspected that residual metal ions in solution supported hydrolysis of the phosphotyrosine linkage. When the zTDP2 crystal was soaked with the phosphotyrosyl DNA substrate in the presence of added Mg2+, the resulting 1.73Å resolution map was very similar to that described above except that water molecules coordinating the Mg2+ ion were better ordered (Supplementary Fig. 1b), confirming that our zTDP2-DNA structure represents a product complex after tyrosine removal. Of note, clear density for the tyrosine moiety was still not observed when the soaking was done in the presence of 25mM EDTA to inhibit the enzyme activity or with crystals grown using an inactive E161A mutant (Fig. 2g), which in fact rendered the DNA electron density much weaker in both cases. These observations imply that the intact active site with a bound metal ion is required for the phosphotyrosyl DNA substrate to bind stably. While our data do not address whether zTDP2 makes specific interactions with the moiety linked to the 5′-terminus of a DNA substrate, the limited active site pocket space likely selects against bulkier hetero groups or additional nucleotides linked to the 5′-terminus (Fig. 2b,c).
Figure 4. Similarities between the DNA and peptide binding modes of TDP2.
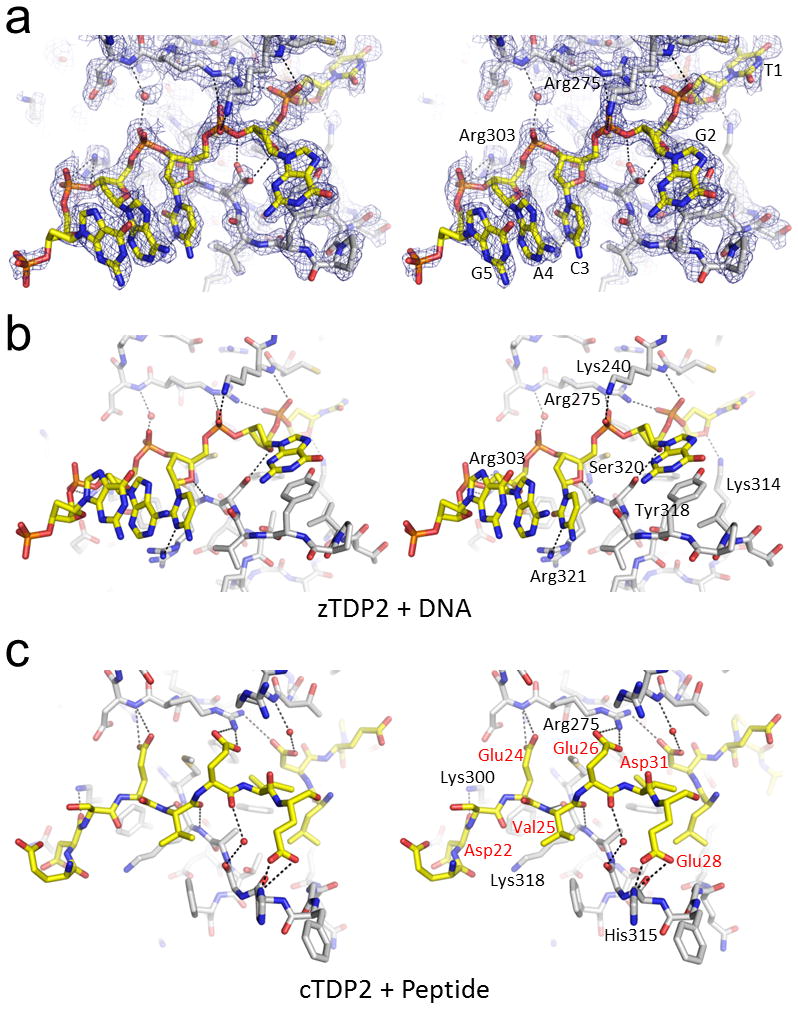
(a) Single-stranded DNA bound in the DNA-binding groove of zTDP2. The simulated annealing composite omit 2Fo−Fc electron density (1.0σ) is shown for 1.8Å from DNA or protein atoms or a water molecule involved in the protein-DNA interaction. (b) zTDP2-DNA interactions as in (a), from a slightly rotated view. Only one of the two conformers for the Ser320 sidechain with higher occupancy is shown for clarity. (c) The N-terminal acidic residues of cTDP2 bound in the DNA-binding groove. Wall-eye stereo pairs are shown for (a–c). Hydrogen bonding and salt bridge interactions are indicated by the black dotted lines.
TDP2 has a modular architecture
The crystal structure of cTDP2, unlike that of zTDP2 described above, allowed visualization of the whole TDP2 molecule. The full-length cTDP2 has a modular architecture consisting of three parts, an extended N-terminal stretch without regular secondary structure elements, a small α-helical bundle domain, and the C-terminal catalytic domain (Fig. 3a and Supplementary Fig. 2). The catalytic domain of cTDP2 resembles that of zTDP2, superimposable with an r.m.s.d. of 0.89Å over 222 Cα atoms (Fig. 3b). The catalytic domains of TDP2 consist of approximately 250 amino acid residues, in which zTDP2 and cTDP2 exhibit 32% sequence identity. The region outside the catalytic domain is not essential for the enzyme activity, as cTDP2 missing either the N-terminal 40 (cTDP2ΔN40) or 107 residues (cTDP2ΔN107) was as active as the full-length cTDP2 in an assay monitoring hydrolysis of the T5PNP substrate26 (Supplementary Fig. 3). In addition, a crystal structure of cTDP2ΔN107 refined at 2.07Å resolution showed the same conformation of the catalytic domain as observed in the full-length structure (Supplementary Fig. 4, bottom), confirming that the catalytic domain is a structurally and functionally independent entity. However, the amino acid sequence of the α-helical bundle domain is well conserved among TDP2 orthologs (Supplementary Fig. 5), suggesting structural conservation as well as functional importance of this domain.
Despite a disordered linker between the α-helical bundle and the catalytic domain (residues 98-111), the connectivity could be established unambiguously based on proximity of the broken ends within the crystal and the number of unresolved amino acid residues, and we modeled the full-length cTDP2 structure in a domain-swapped arrangement. In this domain swap, the residues N-terminal to the catalytic domain interact with the catalytic domain of two other molecules (Figs. 4c and 5a). The most N-terminal segment (residues 21-40) inserts into the DNA-binding groove of one molecule then into the active site pocket of another molecule, and the following α-helical bundle (residues 43-97) is tightly associated with the outer face of the catalytic domain via a hydrophobic interface. The interaction between the α-helical bundle and the catalytic domain buries a total of 1242Å2 of accessible surface area (Fig. 5b). Both the full-length cTDP2 and cTDP2ΔN40 were found to be at least 10 times more soluble than cTDP2ΔN107 and they showed much increased apparent hydrodynamic radii in size-exclusion chromatography than cTDP2ΔN107 (Fig. 5c), consistent with the possibility that the α-helical bundle interacts in trans with the catalytic domain and covers a hydrophobic protein surface. Further supporting the role of the N-terminal residues in cTDP2 multimerization, SAXS analyses, which can provide accurate conformations and assemblies in physiological solution conditions27,28, showed that the full-length cTDP2 is dimeric whereas cTDP2ΔN107 is monomeric in solution (Fig. 5d,e). The Rg by Guinier analysis for the full-length cTDP2 and cTDP2ΔN107 were 34 and 21 Å, respectively. The Dmax of 125 and 60 Å for the full-length and truncated cTDP2, respectively, also reflected the different oligomerization states. While the significance of TDP2 multimerization remains to be investigated, the observed interaction made by the α-helical bundle domain supports its role as a protein-protein interaction module, possibly mediating interaction of TDP2 with other proteins as well.
Figure 5. cTDP2 dimerization by domain swapping.

(a) Selected molecules within the crystal lattice are shown in different colors to illustrate the intermolecular interactions. The asymmetric unit contains one cTDP2 molecule in the full-length cTDP2 crystal, and thus all molecules are crystallographically equivalent. (b) The α-helical bundle (yellow tube) interacting with the relatively hydrophobic (white) surface of the catalytic domain. Molecular surface of the catalytic domain is colored according to the electrostatic potential scaled as in Fig. 2. (c) Size-exclusion chromatography profiles for cTDP2 and its deletion mutants. Positions of the molecular weight standards are indicated by arrows. (d) SAXS analysis shows that the N-terminal residues mediate dimer formation in full-length cTDP2 in solution. SAXS curves for the full-length cTDP2 and cTDP2ΔN107, with linear Guinier plots (inset) showing the lack of aggregation in the sample. (e) GASBOR ab inito shape predictions are consistent with a monomeric form for cTDP2ΔN107 and dimeric form for full-length cTDP2. (f) In vitro 5′-phosphotyrosine bond hydrolysis activity of the full-length and truncated cTDP2. ssY19 and p19 represent the 5′-phosphotyrosyl-oligonucleotide substrate and 5′-phospho-oligonucleotide product, respectively.
Similarities between the DNA and peptide binding modes
As a result of the extensive domain swapping, the active center of cTDP2 is occupied by peptide segments; the N-terminal residues of two separate molecules form a pseudo continuous stretch that traverses across the enzyme active site (Fig. 3c,d). On one side of the active site pocket (indicated by a green arrow in Fig. 3d), the bound peptide occupies a shallow groove possibly mimicking protein backbone interactions made by a topoisomerase-derived peptide covalently linked to the phosphotyrosyl DNA substrate. On the opposite side (indicated by a yellow arrow), the bound peptide occupies the DNA-binding groove (Fig. 4c). Surprisingly, we found strong similarities between the way in which cTDP2 binds the peptide and how zTDP2 binds DNA. In the zTDP2-DNA complex, the backbone phosphate groups of the nucleotides G2, C3, A4, and G5 interact with basic sidechains Lys240, Arg275, and Arg303 as well as mainchain amide groups on the side wall of the DNA-binding groove opposite Tyr318 (Fig. 4a,b). In the cTDP2-peptide complex, the sidechains of Asp31, Glu26, Glu24, and the mainchain carbonyl group of Asp22 take the places of the backbone phosphates of the bound DNA, interacting with basic sidechains Arg275 and Lys300 as well as the mainchain amide groups (Fig. 4c). In addition, the backbone atoms of Val25 form anti-parallel β-sheet-like hydrogen bonds to anchor the peptide stretch in the DNA-binding groove of cTDP2, where its carbonyl oxygen takes the place of the deoxyribose O4′ atom of the C3 position in the DNA bound to zTDP2. The peptide segment bound to cTDP2 makes unique contacts as well, including hydrogen bonds between His315 and Glu28. The short stretch of peptide that spans residues 21 through 28 binding in the DNA-binding groove (Fig. 4c) alone buries a total of 970Å2 of accessible surface area, highlighting the substantial contacts.
DISCUSSION
The observed single-stranded DNA mimicry by the N-terminal Asp and Glu-rich stretch of cTDP2 suggests a specific mechanism for auto-regulation, which is consistent with but distinct from other known cases of DNA mimicry29. However, we did not observe an inhibitory effect of the N-terminal residues in an assay employing either a 5′-phosphotyrosylated single-stranded DNA substrate (Fig. 5f) or the substrate mimic T5PNP26 (supplementary Fig. 3), and the particular cTDP2 N-terminal sequence is not conserved (supplementary Fig. 5). Therefore, it remains to be investigated whether the N-terminal region of TDP2 acts in auto-regulation of the enzymatic activity. On the other hand, our finding that TDP2’s DNA-binding groove can accommodate an acidic polypeptide is intriguing, particularly in the context of the alternative functions of TDP2 in signal transduction and transcription regulation. TDP2 (also known as TTRAP or EAPII) is part of various signaling cascades and interacts with multiple proteins including a TGFβ-receptor ALK4, Smad3, TNF-receptor CD40, TRAFs, and a transcription factor ETS1 (refs. 16–18). The acidic peptide-binding capability of TDP2 may play roles in such protein-protein interactions or recruitment of TDP2 to DNA-lesion sites, along with hydrophobic interactions made by the α-helical bundle domain. We hypothesize that the DNA-binding groove of TDP2 may bind to peptide stretches rich in acidic or phosphorylated residues, with the enzymatic activity perhaps involved in tyrosine dephosphorylation during cellular signaling.
In summary, our collective results revealed the structure of TDP2, mechanisms of its substrate recognition, and an unusual property of TDP2 to bind an acidic polypeptide using its DNA-binding site in vitro. Our high-resolution crystallographic study serves as a framework for designing TDP2 inhibitors useful in chemotherapeutic and antiviral applications, and facilitates further experiments to interrogate in vivo functions of this multifunctional protein.
ONLINE METHODS
Protein purification and structure determination
All proteins were expressed as SUMO-fusion from codon-optimized synthetic genes in the Escherichia coli strain BL21(DE3), and purified by nickel-affinity and size-exclusion chromatography. The His6-SUMO tag was removed by an Ulp1 treatment during purification. The purified proteins in 20mM Tris-HCl (pH7.4), 0.5M NaCl, and 10mM DTT were concentrated by ultrafiltration to 10–30 mg ml−1 (full-length zTDP2 and cTDP2) or ~3 mg ml−1 (cTDP2ΔN107) for crystallization. The full-length cTDP2 crystals were obtained by hanging drop vapor diffusion at 20°C against a well solution containing 15% polyethylene glycol (PEG) 6,000, 0.3M ammonium chloride, and 0.1M MES-NaOH (pH6.0). cTDP2ΔN107 was crystallized using a well solution containing 30% PEG 3,350 and 0.2M sodium malonate (pH5.0). The crystallization condition for zTDP2 consisted of 30% PEG 3,350 and 0.2M sodium tartrate. To obtain the zTDP2-DNA complex, zTDP2 crystals were soaked overnight with 0.5mM of an HPLC-purified 5′-phosphotyrosylated 6-base oligonucleotide before being cryo-protected and flash-cooled in liquid nitrogen. X-ray diffraction data were collected at the beamlines 24-ID-C and 24-ID-E of the Advanced Photon Source, or beamline 4.2.2 of the Advanced Light Source, and processed using HKL2000 (ref. 30). The structure of cTDP2 was determined by the selenomethionine single-wavelength anomalous dispersion (SAD) phasing using PHENIX31. 8 selenium sites were found, from which the structure factor phases were calculated with a mean figure of merit of 0.27 before density modification. The structure of zTDP2 was determined by molecular replacement with PHASER32 using the refined cTDP2 catalytic domain structure as a search model. Although the two zTDP2 molecules in the asymmetric unit have nearly identical structures (Supplementary Fig. 4, top), only one of them was found to be complexed with DNA. The DNA-binding groove of the second zTDP2 molecule is partially blocked by crystal lattice contacts, which likely prevented DNA-binding. The atomic models were built using COOT33 and refined using PHENIX. Programs from the CCP4 suite34 were used for various crystallographic calculations. The x-ray diffraction data and model refinement statistics are given in Table 1. Figures were produced using PYMOL35. Electrostatic potentials were calculated using APBS36. Buried surface areas were calculated using CNS37, and the areas reported are the total (both partners) buried surfaces.
5′-phosphotyrosine hydrolysis assay
A 19-base oligonucleotide with 5′-phosphotyrosine linkage was synthesized by the Midland Certified Reagent Company (Midland, Texas USA). Terminal deoxynucleotidyl transferase (TdT) (Invitrogen, Carlsbad, CA, USA) and [α-32P] cordycepin-5′-triphosphate (PerkinElmer, Inc. Waltham, MA, USA) were used for 3′-end labeling. 1.0 nM of the labeled DNA substrate in a 10 μl reaction volume was incubated with varying concentrations (10-fold serial dilutions) of zebrafish or C. elegans TDP2 for 30 minutes at 25 °C in 80 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol (DTT), 40 μg ml−1 bovine serum albumin, 50 mM Tris-HCl (pH 7.5) and 0.01% Tween 20. The reactions were terminated by adding an equal volume of gel loading buffer (formamide containing 5 mM EDTA). Samples were subjected to 16% denaturing PAGE. Gels were dried, exposed on PhosphorImager screens, and the bands were visualized using a Typhoon 8600 imager (GE Healthcare).
SAXS analyses
SAXS data of the full-length and ΔN107 cTDP2 were collected at the Advanced Light Source SIBYLS 12.3.1 beamline28,38. Scattering measurements were performed on 20 μl samples at 15 °C loaded into a helium-purged sample chamber, 1.5 m from the Mar165 detector. Prior to data collection, the proteins were purified by gel filtration on a 24 ml Superdex200 column equilibrated in 20 mM Tris-HCl (pH7.5), 0.5 M NaCl, 10 mM DTT. Data were collected on the gel filtration fractions. Sequential exposures (0.5, 0.5, 2, 5, and 0.5 secs) were taken at 12 keV. Both proteins showed slight radiation-induced aggregation, so initial 2 sec exposures were used for both full-length and truncated cTDP2. Data was analyzed using the ATSAS suite39.
Size-exclusion chromatography
The full-length cTDP2 and its deletion mutants at the protein concentration of 1.0 mg ml−1 were injected into the Superdex200 10/300 GL column operating with a running buffer [20mM Tris-HCl (pH7.4), 0.5M NaCl, and 10mM DTT]. The molecular weight standards used were as follows; bovine γ-globulin (158K), chicken ovalbumin (44K), and horse myoglobin (17K).
Supplementary Material
Acknowledgments
We thank the beamline staff at Sector-24 of the Advanced Photon Source (APS) and J. Nix of the Molecular Biology Consortium at the Advanced Light Source (ALS) for help in data collection, H. Hiasa, D. Grandgenett, and J. Lee for comments on the manuscript. Computer resources were provided by the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute. The work conducted at the APS NE-CAT beamlines was supported by award RR15301 from the National Center for Research Resources at NIH. Use of the APS, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Supported by NIH grants GM095558 and AI087098 (H.A.), the Center for Cancer Research (Z01 BC 006150-19), Intramural Program of the National Cancer Institute (R.G. and Y.P.), and NCI P01 CA092584 and GM046312 (J.A.T.). SAXS data were collected at the ALS SIBYLS beamline 12.3.1 supported by the IDAT DOE program DE-AC02-05CH11231 and by NIH R01GM105404.
Footnotes
Accession codes:
The atomic coordinates and the structure factors have been deposited in the Protein Data Bank under accession codes 4F1H (zTDP2 with DNA), 4FPV (zTDP2 soaked with DNA in the presence of Mg2+), 4GEW (full-length cTDP2: native crystal), 4F1I (full-length cTDP2: Selenomethionine derivative), and 4FVA (cTDP2ΔN107).
AUTHOR CONTRIBUTIONS
K.K. purified the proteins and performed the activity assay on a substrate mimic. K.K., K.S., and H.A. crystallized the proteins. K.S. collected x-ray diffraction data and determined all crystal structures. R.G. and Y.P. examined enzyme activities on the 5′-phosphotyrosyl DNA substrate. S.E.T. and J.A.T. collected and analyzed the SAXS data. H.A. wrote the manuscript. All authors contributed in editing and figure preparation.
Competing financial interests:
The authors declare no competing financial interests.
References
- 1.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 3.Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 4.Corbett KD, Berger JM. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu Rev Biophys Biomol Struct. 2004;33:95–118. doi: 10.1146/annurev.biophys.33.110502.140357. [DOI] [PubMed] [Google Scholar]
- 5.Haffner MC, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deweese JE, Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 2009;37:738–748. doi: 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nature Rev Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu CC, et al. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science. 2011;333:459–462. doi: 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- 9.Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature. 2009;461:674–678. doi: 10.1038/nature08444. [DOI] [PubMed] [Google Scholar]
- 10.Zeng Z, Cortes-Ledesma F, El Khamisy SF, Caldecott KW. TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J Biol Chem. 2011;286:403–409. doi: 10.1074/jbc.M110.181016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao R, Huang SY, Marchand C, Pommier Y. Biochemical characterization of human Tyrosyl DNA Phosphodiesterase 2 (TDP2/TTRAP): a Mg2+/Mn2+-dependent phosphodiesterase specific for the repair of topoisomerase cleavage complexes. J Biol Chem. 2012;287:30842–30852. doi: 10.1074/jbc.M112.393983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Do PM, et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012;26:830–45. doi: 10.1101/gad.181685.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang SW, et al. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci USA. 1996;93:11534–11539. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raymond AC, Burgin AB., Jr Tyrosyl-DNA phosphodiesterase (Tdp1) (3′-phosphotyrosyl DNA phosphodiesterase) Methods Enzymol. 2006;409:511–524. doi: 10.1016/S0076-6879(05)09030-0. [DOI] [PubMed] [Google Scholar]
- 15.Davies DR, Interthal H, Champoux JJ, Hol WG. Crystal structure of a transition state mimic for Tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived peptide. Chem Biol. 2003;10:139–147. doi: 10.1016/s1074-5521(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 16.Pype S, et al. TTRAP, a novel protein that associates with CD40, tumor necrosis factor (TNF) receptor-75 and TNF receptor-associated factors (TRAFs), and that inhibits nuclear factor-κB activation. J Biol Chem. 2000;275:18586–18593. doi: 10.1074/jbc.M000531200. [DOI] [PubMed] [Google Scholar]
- 17.Esguerra CV, et al. Ttrap is an essential modulator of Smad3-dependent Nodal signaling during zebrafish gastrulation and left-right axis determination. Development. 2007;134:4381–4393. doi: 10.1242/dev.000026. [DOI] [PubMed] [Google Scholar]
- 18.Pei H, et al. EAPII interacts with ETS1 and modulates its transcriptional function. Oncogene. 2003;22:2699–2709. doi: 10.1038/sj.onc.1206374. [DOI] [PubMed] [Google Scholar]
- 19.Li C, et al. Oncogenic role of EAPII in lung cancer development and its activation of the MAPK-ERK pathway. Oncogene. 2011;30:3802–3812. doi: 10.1038/onc.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li C, Sun SY, Khuri FR, Li R. Pleiotropic functions of EAPII/TTRAP/TDP2: cancer development, chemoresistance and beyond. Cell Cycle. 2011;10:3274–3283. doi: 10.4161/cc.10.19.17763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Virgen-Slane R, et al. An RNA virus hijacks an incognito function of a DNA repair enzyme. Proc Natl Acad Sci USA. 2012;109:14634–14639. doi: 10.1073/pnas.1208096109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weston SA, Lahm A, Suck D. X-ray structure of the DNase I-d(GGTATACC)2 complex at 2.3 Å resolution. J Mol Biol. 1992;226:1237–1256. doi: 10.1016/0022-2836(92)91064-v. [DOI] [PubMed] [Google Scholar]
- 23.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 24.Adhikari S, et al. Characterization of magnesium requirement of human 5′-tyrosyl DNA phosphodiesterase mediated reaction. BMC Res Notes. 2012;5:134. doi: 10.1186/1756-0500-5-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsutakawa SE, et al. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011;145:198–211. doi: 10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adhikari S, et al. Development of a novel assay for human tyrosyl DNA phosphodiesterase 2. Anal Biochem. 2011;416:112–116. doi: 10.1016/j.ab.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly Reviews in Biophysics. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 28.Hura GL, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Putnam CD, Tainer JA. Protein mimicry of DNA and pathway regulation. DNA Repair (Amst) 2005;4:1410–1420. doi: 10.1016/j.dnarep.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 32.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34.Collaborative Computational Project. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.The PyMOL Molecular Graphics System. http://www.pymol.org.
- 36.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brunger AT, et al. Crystallography & NMR System (CNS), A new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 38.Classen S, et al. Software for the high-throughput collection of SAXS data using an enhanced Blu-Ice/DCS control system. J Synchrotron Radiat. 2010;17:774–781. doi: 10.1107/S0909049510028566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. ATSAS 2.1, a program package for small-angle scattering data analysis. J Appl Crystallogr. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.