

SUMMARY
Ribonuclease H1 is a conserved enzyme that cleaves the RNA strand of RNA•DNA heteroduplexes, and has important functions in the nuclear and mitochondrial compartments. The therapeutic action of antisense oligodeoxynucleotides involves the recruitment of RNase H1 to cleave disease-relevant RNA targets. Recombinant human(Hs)-RNase H1 was purified from a bacterial expression host, and conditions were identified that provided optimal oligonucleotide-directed RNA cleavage in vitro. Hs-RNase H1 exhibits optimal catalytic activity in pH 7.5 HEPES buffer, and a salt (KCl) concentration of ~100–150 mM. Mg2+ best supports Hs-RNase H1, with an optimal concentration of 10 mM, but at higher concentrations inhibits enzyme activity. Mn2+ and Co2+ also support catalytic activity, while Ni2+ and Zn2+ exhibit only modest activities as cofactors. The optimized assay was used to show that an antisense oligonucleotide, added in substoichiometric amounts to initiate RNA cleavage, supports up to thirty rounds of reaction in 30 minutes. Mutation to alanine of the conserved histidine at position 264 causes a ~100-fold decrease in kcat under multiple-turnover conditions, but does not alter the Km. Under single-turnover conditions, the H264A mutant exhibits a 12-fold higher exponential time constant for substrate cleavage. The defective activity of the H264A mutant is not rescued in either assay condition by higher Mg2+ concentrations. These data implicate the H264 side chain in phosphodiester hydrolysis as well as in product release, and are consistent with a proposed model in which the H264 side chain interacts with a divalent metal ion to support catalysis.
Keywords: Ribonuclease H1, Antisense oligonucleotides, Metal-dependent phosphodiester hydrolysis, Conserved histidine function, RNA-DNA heteroduplex
Introduction
The ribonucleases H constitute a family of divalent metal ion-dependent endoribonucleases that cleave the RNA strand of RNA•DNA heteroduplexes [1–3]. Based on structural and biochemical features the eukaryotic enzymes are classified either as RNase H1 or RNase H2. RNase H2 is a nuclear-localized, heterotrimeric enzyme that participates in DNA replication and repair, while RNase H1 is present in the mitochondrial and nuclear compartments, and functions as a monomer in vitro [1,3]. RNase H1 is essential for mitochondrial DNA replication [4], with the nuclear functions yet to be fully established, but likely include specific aspects of DNA replication, recombination, repair, and transcription [1,3]. The RNase H1 polypeptide is comprised of an N-terminal hybrid-binding domain (HBD) [5,6] that is joined by a connecting domain (CD) to the catalytic domain (RNase H domain, or RNHD) [7,8]. The HBD confers binding affinity and processivity of action, wherein engagement of substrate by the HBD allows cleavage of multiple RNA phosphodiesters by the RNHD, prior to HBD disengagement of the processed hybrid [5]. The catalytic chemistry involves metal ion activation of a water nucleophile that creates 5′-phosphomonoester, 3′-hydroxyl product termini [9]. Structural and enzymatic studies have implicated two closely-positioned Mg2+ ions as core components of the catalytic site. One of the metals (MgA) binds the water nucleophile, which is predicted on the basis of computational studies to donate a proton to a nonbridging oxygen of the scissile bond during nucleophilic attack [10–12], while the second metal (MgB) facilitates the departure of the 3′-oxygen in alkoxide form [12]. The 3′-alkoxide anion is predicted to accept the proton from the nonbridging oxygen (see above) [11] or from a protein side chain [12]. The two metal ions are predicted to cooperatively stabilize a phosphorane intermediate [10] in an overall pathway involving inversion of configuration at phosphorus. The side chains of a set of highly conserved carboxylic acids bind the two metal ions. However, the involvement of other amino acid residues, including a highly conserved histidine (vide infra), as well as the involvement of additional metal ions or metal ion binding sites have yet to be fully understood.
Antisense oligonucleotides with 2′-deoxyribose backbones can recruit RNase H1 to cleave disease-relevant RNA targets in vivo [13–15]. The efficacy of action depends in part upon the ability of the oligonucleotide to support multiple rounds of RNA cleavage. To accomplish this the oligonucleotide must exhibit chemical stability and nuclease resistance; have a strong affinity and selectivity for the target RNA sequence; support efficient RNA cleavage by RNase H1; and allow facile release of the RNA products to allow its engagement with new substrate [13,14]. A better understanding of the Hs-RNase H1 catalytic mechanism, in particular with respect to additional components that participate in, and perhaps also regulate, the hydrolytic and product release steps, can inform the design of next-generation antisense oligonucleotides with optimal pharmacologic action. A specific histidine was shown to be important for the catalytic activity of the RNase H of HIV reverse transcriptase [16,17]. The histidine (H264 in Hs-RNase H1) is highly conserved, and therefore may play a conserved role in the reaction pathway. How the histidine may participate in Hs-RNase H1 action has not been experimentally examined. Using purified Hs-RNase H1 in an optimized in vitro assay of oligonucleotide-directed RNA cleavage, we provide evidence for the involvement of H264 in two steps of the Hs-RNase H1 catalytic mechanism.
Results and Discussion
The mature nuclear/mitochondrial form of Hs-RNase H1 was purified from an overexpressing bacterial strain using affinity chromatography (see Materials and Methods). The yield was ~3 mg per liter of bacterial culture, with a purity of >90% as estimated by SDS-PAGE. RNA cleavage assays employed a 21 nt RNA whose sequence corresponds to a segment (nt 324–345) within the human c-myb mRNA [18]. The complementary 21 nt oligodeoxynucleotide (denoted as MOH1 in [18]) does not carry sugar or phosphodiester modifications, as it has been shown that phosphorothioate groups - which enhance nuclease resistance and improve antisense oligonucleotide action in vivo [14] – also can inhibit RNase H1 cleavage of the corresponding hybrids in vitro [19,20]. The stable products of Hs-RNase H1 cleavage of the heteroduplex containing a 5′-32P-labeled RNA strand are identified in Supplemental Figure S1. Also, the presence of the N-terminal (His)6-tag does not significantly affect the kinetics or pattern of Hs-RNase H1 cleavage of the heteroduplex (Supplemental Fig. S2). Thus, the experiments used the (His)6-tagged form of the enzyme, which for convenience is referred to as Hs-RNase H1.
Salt and pH dependence of Hs-RNase H1 catalytic activity
To identify optimal in vitro reaction conditions we assessed Hs-RNase H1 catalytic activity as a function of several experimental parameters. The fraction of RNA cleaved was measured as a function of salt (KCl) concentration (at pH 7.5 and a Mg2+ concentration of 10 mM), under conditions of excess substrate (preformed heteroduplex). Figures 1A and 1B show that the ability of Hs-RNase H1 to cleave substrate is enhanced with increasing KCl concentration, with a maximum observed at ~100–150 mM. The stable products of cleavage are not significantly altered by changes in salt concentration, and the decrease in activity at salt concentrations >150 mM may reflect salt competition for substrate binding. Catalytic activity also was assessed between pH 5.5 and 11.5, in buffer containing 150 mM KCl and 10 mM Mg2+. Figures 1C and 1D show that maximal activity is achieved at pH 7.5. Based on these findings, 150 mM KCl and a pH of 7.5 were included as standard reaction conditions.
Figure 1.
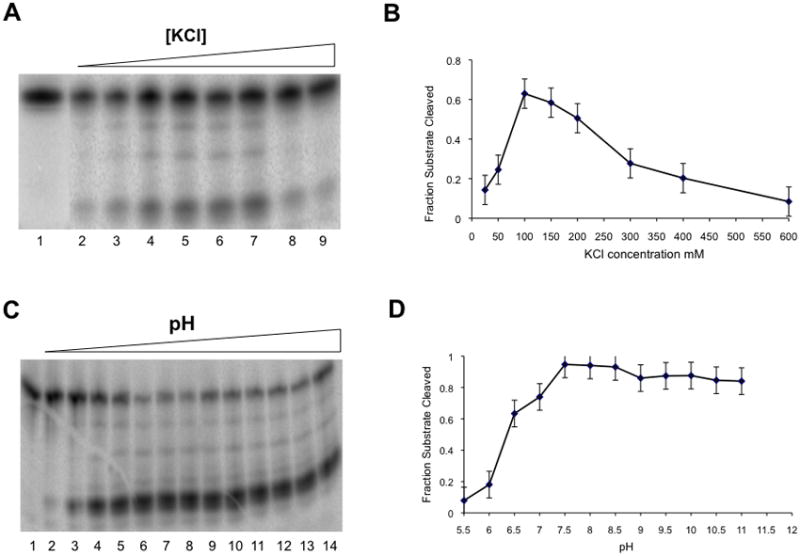
Salt and pH dependence of Hs-RNase H1. The action of Hs-RNase H1 on the 21 bp hybrid substrate was measured as a function of KCl concentration (panels A,B) or pH (panels C,D) in reactions performed at 30°C in buffer containing 10 mM MgCl2, and 10 mM DTT (see Materials and Methods). Both analyses used 30 nM Hs-RNase H1 and 100 nM 5′-32P-labeled (preformed) hybrid. For the KCl concentration dependence experiment, 20 mM Tris-HCl (pH 7.5) was used as buffer. In panel A, lanes 2–9 show reactions at KCl concentrations of 25, 50, 100, 150, 200, 300, 400 and 600 mM, respectively. Lane 1 is a control reaction that lacked Mg2+. Panel B graphically displays the fraction of substrate cleaved as a function of KCl concentration. The maximum error is shown for each point. C. pH dependence. Lanes 2–14 show reactions at pH values of 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, 8.5, 9.0, 9.5, 10.0, 10.5, 11.0, and 11.5, respectively. The KCl concentration was 150 mM. Tris buffer was used at each pH value. Here, the use of Tris rather than HEPES for pH titration at fixed Mg2+ concentration allowed the 5.5–11.5 pH range to be achieved. Panel D displays the fraction of substrate cleaved as a function of pH. Experiments were carried out in duplicate, and the average values are shown with the associated maximum error.
Divalent metal ion dependence of Hs-RNase H1 catalytic activity
Structural studies of Hs-RNase H1 and Bacillus halodurans (Bh) RNase H1 identified a two metal ion catalytic mechanism, with Mg2+ as the presumptive physiologically relevant cofactor [7–9]. It was shown that Hs-RNase H1 either with [20] or without [21] the mitochondrial targeting sequence exhibits maximal activity at either ~1 mM Mg2+ or ~1 μM Mn2+, but with the latter metal supporting a lower level of activity. The isolated RNHD of Hs-RNase H1 can cleave substrate in 10 mM Mn2+ [8], and it also has been shown that Thermotoga maritima RNase H1 activity is supported by Mn2+ and Mg2+, but with negligible activity provided by Ni2+, Co2+, Zn2+, or Ca2+ [22]. We examined the ability of Hs-RNase H1 to cleave substrate as a function of the concentration of the above-mentioned metal ions. The use of HEPES buffer in these experiments avoided the problem of metal ion chelation in solution (see Materials and Methods). Figure 2 shows that Mg2+ ion best supports Hs-RNase H1 activity, with maximum activity achieved at 10 mM, but with progressive inhibition observed at higher concentrations. Based on this result, 10 mM Mg2+ was used in the optimized assay system, which also used HEPES as the buffer species. In addition, ~5 mM Mn2+ or ~10 mM Co2+ support activity, with only minor inhibition observed at higher concentrations. Ni2+ and Zn2+ also support Hs-RNase H1, but to significantly lesser extents (Fig. 2 inset), while Ca2+ is inactive over the concentration range examined (data not shown). We conclude that Hs-RNase H1 prefers Mg2+ as cofactor, but that Mn2+ and Co2+ also support significant levels of activity.
Figure 2.

Divalent metal ion dependence of Hs-RNase H1. Cleavage assays used Hs-RNase H1 (30 nM) and 32P-labeled (preformed) RNA•DNA hybrid (100 nM) at 30°C in buffer consisting of 150 mM KCl, 20 mM HEPES (pH 7.5), and the specified concentration of divalent metal ion in the form of the chloride salt. The fraction of RNA cleaved was plotted as a function of divalent metal ion concentration. Experiments were performed in duplicate and the average values are shown along with the associated maximum errors. Symbols are: Mg2+ (solid diamonds); Mn2+ (solid squares); and Co2+ (solid triangles). The inset diagram shows Zn2+ (open triangles) and Ni2+ (open squares).
Substoichiometric amounts of oligodeoxynucleotide support multiple rounds of RNA cleavage
We next assessed whether the 21 nt oligodeoxynucleotide could direct RNA cleavage in vitro in a manner reflective of antisense oligonucleotide action in vivo. For this experiment substoichiometric amounts of the oligonucleotide were added to initiate cleavage of an excess amount of RNA, and in the presence of a relatively low concentration of Hs-RNase H1. Under these conditions, efficient action of the oligonucleotide would require multiple rounds of RNA binding, recruitment of RNase H1, achievement of catalysis, dissociation of the RNA products, and binding of the oligonucleotide to new substrate. The extent of reaction was determined by measuring the amount of intact substrate remaining as a function of time. Thus, the assay measures the initial (primary) cleavage event, irrespective of the precise site. The number of rounds of RNA cleavage at each RNA:DNA ratio is provided in Table 1, which shows that the oligonucleotide is capable of directing up to ~30 rounds of RNA cleavage at an RNA:DNA ratio of 100. The limit to the reaction at longer incubation times, as indicated by the failure to achieve full cleavage of the RNA, probably reflects a loss of enzyme activity, as addition of fresh Hs-RNase H1 allowed additional cleavage of substrate (N.A. and A.W.N., data not shown). We conclude that this assay recapitulates the proposed steps of antisense oligonucleotide action in vivo, and thus provides a stringent test of oligonucleotide efficacy. We also examined whether RNase H1 could be functionally “programmed” with an oligonucleotide, as can occur with RISC assembly [23]. A variety of protocols involving incubation of the oligonucleotide with Hs-RNase H1, prior to addition of RNA, revealed no significant enhancement of the rate of RNA cleavage (N.A. and A.W.N., data not shown). These results indicate that an antisense oligonucleotide does not function on its own as a guide strand for RNase H1 cleavage of RNA, and is consistent with structural studies showing that the HBD and the RNHD specifically and selectively recognize the RNA•DNA heteroduplex [6–8].
Table 1.
Sustained action of substoichiometric amounts of an oligonucleotide in directing RNA cleavage by Hs-RNase H1.
See Materials and Methods and Results & Discussion for experimental procedures. The turnover number (column 3) is defined as the number of rounds of RNA cleavage in 30 minutes, and is determined by multiplying the RNA:DNA ratio (column 1) by the fraction of RNA cleaved (column 2).
| RNA:DNA ratio | Fraction of heteroduplex cleaved in 30 min. | Turnover Number |
|---|---|---|
| 10 | 0.90 | 9 |
| 50 | 0.56 | 28 |
| 100 | 0.29 | 29 |
Histidine 264 is important for Hs-RNase H1 catalytic activity
Structural studies of Hs-RNase H1 and Bh-RNase H1 [7,8], as well as computational modeling of the Bh-RNase H1 catalytic site [24] implicate the conserved residue H264 (or E188 in Bh-RNase H1) in the catalytic pathway. The Hs-RNase H1 H264A mutant was purified and its catalytic behavior assessed. Under conditions of substrate excess the H264A mutant is defective in its ability to cleave RNA (compare Fig. 3B with Fig. 3A). Comparison of the kinetic behavior reveals a 100-fold reduction in kcat, but essentially no change in Km (Table 2). To assess the source(s) of the kcat reduction, a time course for (preformed) heteroduplex cleavage was performed under single-turnover (enzyme excess) conditions (Fig. 3D). Here, the rate of disappearance of the substrate is determined by the rate of the hydrolytic step without contribution from events associated with product release. The exponential time constant for disappearance of the heteroduplex in the presence of the H264A mutant (6.9 ± 0.4 min) is ~12-fold greater than the value obtained with wild-type enzyme (0.6 ± 0.2 min). These data indicate an involvement of H264 in the chemical step, and also implicate H264 in product release, since the 12-fold difference in the time constants only partially accounts for the 100-fold difference in the steady-state kcat values. In either reaction condition, the impaired activity of the H264A mutant is not rescued by increasing the Mg2+ concentration to 80 mM (N.A. and A.W.N., data not shown). H264A mutant activity also is inhibited to a greater extent than WT enzyme by higher Mg2+ concentrations (Supplemental Fig. S3; see also below).
Figure 3.
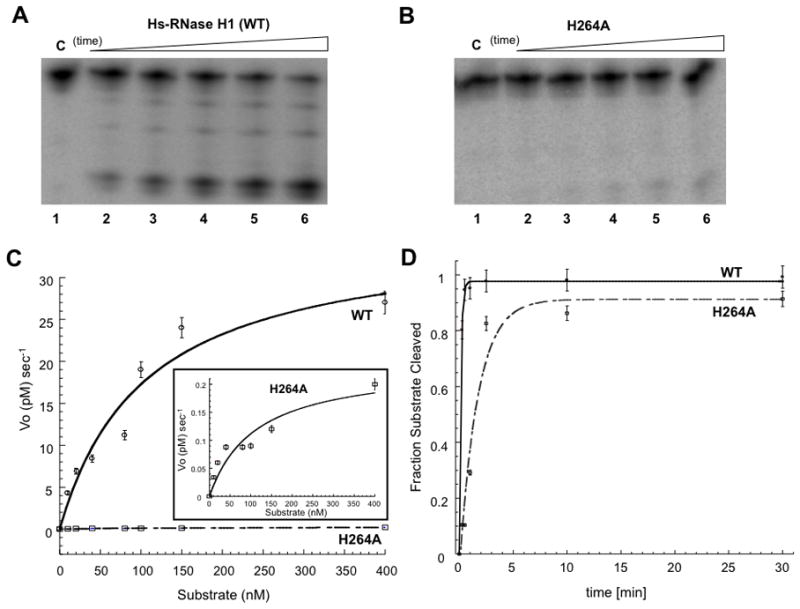
Defective catalytic activity of the H264A mutant. Panels A and B display phosphorimages of time course assays of hybrid substrate cleavage by Hs-RNase H1 and the H264A mutant, respectively, under steady-state (substrate excess) conditions. The assay used 150 nM RNA•DNA (preformed) hybrid and 10 nM enzyme. Reactions were initiated by adding Mg2+ (10 mM final concentration) followed by incubation at 30°C in a buffer containing 150 mM KCl, 25 mM HEPES (pH 7.5), and 10 mM DTT. Aliquots were combined with excess EDTA at specified times and analyzed by electrophoresis in a 15% denaturing polyacrylamide gel as described in Materials and Methods. In panel A, lanes 1–6 display 0, 1, 2.5, 5, 10, and 30 min time points, respectively, and in panel B, lanes 1–6 display 0, 2.5, 5, 10, 30, and 60 min time points, respectively. C. Michaelis-Menten steady-state analysis. The initial rate (Vo) was obtained as a function of substrate concentration. A best-fit curve to a Michaelis-Menten scheme is shown, with each point representing the average of two experiments. The inset has an expanded vertical axis, to better display the substrate concentration dependence of initial rate of cleavage by the H264A mutant. D. Single-turnover kinetic analysis. 5′-32P labeled RNA•DNA (preformed) hybrid (5 nM) was reacted at 30°C with Hs-RNase H1 or H264A mutant (50 nM) in buffer containing 150 mM KCl, 10 mM MgCl2, 25 mM HEPES (pH 7.5), and 10 mM DTT. Reactions were initiated by adding MgCl2, and aliquots quenched with excess EDTA at the specified times, followed by analysis by denaturing PAGE (see Materials and Methods). Lanes 1–6 display 0, 0.25, 0.5, 1, 2.5, and 5 min reaction times for Hs-RNase H1, and lanes 7–12 represent the same time points for the H264A mutant. The graph displays the fraction of RNA cleaved as a function of time. Shown are the best-fit curves to a single exponential equation, using Kaleidagraph software. The exponential time constant is 0.6 ± 0.2 min for Hs-RNase H1, and 6.9 ± 0.4 min for the H264A mutant.
Table 2.
Kinetic parameters of Hs-RNase H1 and the H264A mutant.
See Materials and Methods, and Results & Discussion for the determination of the kinetic values and best-fit error estimates. Reported values are from reactions performed at 30°C.
| kcat (sec−1) | Km (nM) | kcat/Km (M−1 sec−1) | Vmax (pM sec−1) | |
|---|---|---|---|---|
| Hs-RNase H1 | 3.5 ± 0.5 | 97 ± 21 | 3.6 × 107 | 35 ± 5 |
| H264A | 0.025 ± 0.004 | 106 ± 34 | 2.4 × 105 | 0.25 ± 0.04 |
A proposed dual functional role for the H264 side chain
An efficient disengagement of the product 5′-phosphomonoester from the catalytic site would allow the RNHD to cleave additional phosphodiesters in a processive manner while the HBD remains bound to the heteroduplex. Based on crystallographic and modeling data it was proposed that product release from the catalytic site is facilitated through relief of a steric clash between the H264 side chain and the 5′-phosphomonoester [8]. Removal of the imidazole side chain would be expected to stabilize product binding and inhibit turnover under steady-state conditions. The kinetic data presented here are consistent with the proposed model, as based on the reduced reactivity of the Bh-RNase H1 E118A mutant [7] (the E188 side chain is proposed to be functionally equivalent to the H264 side chain [9]). How H264 also may function in the hydrolytic step is suggested by a molecular dynamics simulation analysis of Bh-RNase H1 [24] that identified an additional Mg2+ ion (MgC) that interacts with the E188 side chain, and which is near MgA. MgC is proposed to provide a local electrostatic environment optimal for phosphodiester hydrolysis, perhaps through optimal positioning of the water nucleophile [24]. A primary function of H264 therefore would be to establish binding site C and may be a predominant energetic determinant of metal binding, since high Mg2+ concentrations do not rescue H264A mutant activity. The inhibition of Hs-RNase H1 at higher Mg2+ concentrations is consistent with the computational analysis, which shows that the increased solvent charge density perturbs the position of MgC, which in turn impairs catalysis [24]. Interestingly, the H264A mutant is more sensitive than wild-type enzyme to inhibition by high Mg2+ concentrations (Supplemental Fig. 3). Perhaps the absence of the imidazole side chain renders the catalytic site more accessible to solvent Mg2+ ion, thereby enhancing the inhibitory effect of elevated concentrations. Additional enzymological and computational studies are needed to more precisely define the role of Mg2+ ion and the conserved histidine in the catalytic cycle, and product release in particular. The flexibility of the peptide loop containing H264 is a conserved feature of RNase H family members [8,25–27]. It was proposed that the product 5′-phosphomonoester group initially engages with the Mg2+ bound to site A, then with Mg2+ at site C, in the process of product disengagement from the catalytic site [9]. Perhaps this proposed movement is enabled by H264 loop flexibility. Finally, comparative insight is provided by studies of Escherichia coli (Ec) RNase HI, which lacks the HBD, but otherwise is structurally similar to the RNHD of RNase H1. Similar to the Hs-RNase H1 H264A mutation, the Ec-RNase HI H124A mutation causes a ~50-fold reduction in kcat. In contrast, there is a ~4-fold increase in Km [28]. However, a similar effect of the H264A mutation on RNHD engagement of substrate cannot be ruled out, since the N-terminal HBD of RNase H1 is the major determinant of substrate binding [5,6], and as such is expected to dominate the Km.
Summary
An in vitro assay involving purified recombinant human RNase H1 supports multiple cycles of oligonucleotide-directed RNA cleavage, and provides a stringent test of the ability of an antisense oligonucleotide to function in a sustained fashion. The efficiency of antisense oligonucleotide action in vivo is expected to depend upon the catalytic behavior of RNase H1, which relies on H264 (and perhaps an associated Mg2+ ion) in the chemical and product release steps. The in vitro assay system supports up to 30 rounds of substrate cleavage in 30 minutes. The rate-limiting step is not known, but does not appear to involve an event in the enzyme-substrate complex. It is possible that dissociation of cleaved RNA fragments from the antisense oligonucleotide may dictate the overall turnover rate. Finally, whereas the gene silencing action of an antisense oligonucleotide requires only a single RNA phosphodiester to be cleaved, the processivity of RNase H1 action may be important for the optimal metabolism of cellular heteroduplex structures.
Materials and Methods
Water was deionized and distilled. Chemicals and reagents were molecular biology grade and were purchased from Sigma-Aldrich (St. Louis, MO) or from Fisher Scientific (Chicago, IL). Dialysis tubing (Spectra-Por CE, 10,000 MWCO) was purchased from Fisher Scientific. CoCl2, ZnCl2, NiCl2, and standardized 1 M aqueous solutions of MgCl2, MnCl2 and CaCl2 were obtained from Sigma-Aldrich. Ni2+-NTA chromatography resin and restriction grade thrombin were purchased from Novagen (Madison, WI). His-Trap (1 ml) HP Ni-NTA chromatography columns for ÄKTA FPLC were purchased from GE Healthcare Life Sciences (Piscataway, NJ). Protein assay kits and protein standards were purchased from Bio-Rad Laboratories (Hercules, CA). Amicon spin concentrators were purchased from Millipore (Billerica, MA). [γ-32P]ATP (3000 Ci/mmol) was purchased from Perkin-Elmer (Boston, MA). T4 polynucleotide kinase was purchased from New England Biolabs (Beverly, MA). Oligodeoxynucleotides were synthesized by Life Technologies (Carlsbad, CA), and the deprotected forms purified by denaturing gel electrophoresis then stored at −80°C in TE buffer (pH 8.0).
Overexpression and purification of Hs-RNase H1 and Hs-RNase H1 H264A were accomplished as follows. The H264A mutation was introduced by QuikChange site-directed mutagenesis (Agilent Technologies, Santa Clara, CA), and the mutation confirmed by sequencing. A single colony of E. coli BL21(DE3)[pLysS] (Novagen), freshly transformed with the recombinant pET-15b plasmid containing the cloned cDNA for Hs-RNase H1 lacking the mitochondrial targeting sequence [29], was directly inoculated into 1 liter of LB broth containing 50 μg/ml ampicillin, and grown with aeration at 30°C. When the OD (600 nm) reached ~0.5, IPTG was added (1 mM final concentration) and incubation continued at 30°C for 3 hr. Cells were harvested by centrifugation and were resuspended in Buffer A (300 mM NaCl, 5 mM imidazole, 25 mM HEPES (pH 7), 1 mM EDTA, 2 mM 2-mercaptoethanol, and 10% glycerol). Cells were disrupted by repeated bursts of sonication with intermittent cooling on ice. The sonicated solution was centrifuged (10,000 rpm in an SS34 rotor for 30 min), and the supernatant loaded on a His-Trap HP Ni-NTA column, equilibrated with Buffer A, and attached to an ÄKTA FPLC Explorer System. The column was subjected to a linear gradient of imidazole (0–1 M) in Buffer A. His-tagged Hs-RNase H1 eluted between 350 and 400 mM imidazole, and was dialyzed for 3 hr against Buffer A + 200 mM imidazole, followed by dialysis for 3 hr against Buffer A + 100 mM imidazole, and dialysis overnight against Buffer A. The purity of the protein was >90%, as estimated by 15% SDS-PAGE.
Preparation of substrate
A 21 bp RNA-DNA hybrid served as a substrate for Hs-RNase H1. The RNA, 5′-GAAAUACGGUCCGAAACGUUG-3′, corresponds to a sequence within the coding sequence of human c-Myb mRNA [18] and was obtained from ThermoFisher Scientific (Waltham, MA). The sequence of the complementary oligodeoxynucleotide is 5′-CAACGTTTCGGACCGTATTTC-3′. RNA (200 pmol) was 5′-32P-labeled by incubation (37°C, 1 hr) with T4 polynucleotide kinase (10 units), [γ-32P]ATP (3 pmol), and unlabeled ATP (290 pmol) in a 30μl volume. Labeled RNA was subjected to denaturing PAGE, extracted from excised gel slices, and ethanol precipitated. The purified RNA was resuspended in RNA storage buffer (Ambion) and stored at −20°C. Hybrid substrates were prepared in 50μl reactions containing 1μM 32P-labeled RNA and a two-fold molar excess of the complementary oligodeoxynucleotide in annealing buffer (50 mM KCl, 20 mM HEPES (pH 7.5), 1 mM EDTA). Reactions were heated at 90°C for 5 min, cooled to 37°C, then placed on ice.
Substrate cleavage assays
Assays were performed using a buffer consisting of 150 mM KCl, 10 mM MgCl2, 20 or 25 mM HEPES (pH 7.5), and 10 mM DTT. HEPES was chosen as the buffer since it exhibits negligible interactions with divalent metal ions [30,31]. In this regard, the use of Tris buffers reduced the activity of Hs-RNase H1, relative to that obtained in HEPES-based buffers, and also caused perturbation of the metal ion (Mg2+, Mn2+) concentration dependence of enzyme activity, presumably reflecting the metal-chelating property of Tris [32] (N.A. and A.W.N, unpublished observations). For determination of kinetic parameters, preformed 32P-labeled RNA•DNA hybrid (see above), ranging in concentration from 1–250 nM, was incubated at 30°C with 40 nM Hs-RNase H1. Reactions were initiated by adding Mg2+, followed by incubation at 30°C. Aliquots were removed at the indicated times and quenched by adding an equal volume of stop mix (95% deionized formamide, 20 mM EDTA). Samples were heated at 90°C for 2 min then electrophoresed in a 15% polyacrylamide gel containing 7M urea and TBE buffer. Reactions were visualized by phosphorimaging (Typhoon 9400 system) and quantified using ImageQuant software (v5.0). Under conditions of substrate excess, the fraction of substrate cleaved was calculated by determining the total radioactivity in each gel lane as well as the specific amount of uncut RNA. Initial rates were determined under conditions in which the percentage of substrate cleaved was <20%. The fraction of substrate cleaved determined as a function of time provided the initial velocity of cleavage (Vo). Vo was measured as a function of substrate concentration, and the Vmax and Km values determined from a best-fit curve to a Michaelis-Menten kinetic scheme using Kaleidagraph software. The kcat values were determined from the Vmax values and the total enzyme concentration. Under single-turnover conditions, the fraction of substrate cleaved as a function of reaction time was fit to a single-exponential decay equation.
Supplementary Material
Supplemental Figure 1. Stable products of Hs-RNase H1 cleavage of the 21 bp RNA-DNA heteroduplex substrate.
Supplemental Figure 2. Comparison of heteroduplex substrate cleavage by Hs-RNase H1, with or without an N-terminal (His)6-tag.
Supplemental Figure 3. Comparison of the Mg2+ titration behavior of Hs-RNase H1 and the H264A mutant.
Acknowledgments
We thank Susana Cerritelli and Robert Crouch for providing the recombinant pET-15b plasmid, containing the cDNA encoding the mature nuclear/mitochondrial form of Hs-RNase H1. The authors also acknowledge original suggestions and encouragement from Alan M. Gewirtz (in memorium), and Rhonda H. Nicholson for comments on the manuscript. This research was supported in part by the NIH (RO1 GM56772).
Abbreviations
- HBD
Hybrid binding domain
- RNHD
RNase H domain
- CD
Connecting Domain
- Hs
Human (Homo sapiens)
References
- 1.Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tadokoro T, Kanaya S. Ribonuclease H: Molecular diversities, substrate binding domains, and catalytic mechanism of the prokaryotic enzymes. FEBS J. 2009;276:1482–1493. doi: 10.1111/j.1742-4658.2009.06907.x. [DOI] [PubMed] [Google Scholar]
- 3.Hollis T, Shaban NM. Structure and function of RNase H enzymes. In: Nicholson AW, editor. Ribonucleases, Nucleic Acids and Molecular Biology. Vol. 26. Springer-Verlag; Berlin Heidelberg: 2011. pp. 299–317. [Google Scholar]
- 4.Cerritelli SM, Frolova EG, Feng C, Grinberg A, Love PE, Crouch RJ. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol Cell. 2003;11:807–815. doi: 10.1016/s1097-2765(03)00088-1. [DOI] [PubMed] [Google Scholar]
- 5.Gaidamakov SA, Gorshkova II, Schuck P, Steinbach PJ, Yamada H, Crouch RJ, Cerritelli SM. Eukaryotic RNases H1 act processively by interactions through the duplex RNA-binding domain. Nucleic Acids Res. 2005;33:2166–2175. doi: 10.1093/nar/gki510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nowotny M, Cerritelli SM, Ghirlando R, Gaidamakov SA, Crouch RJ, Wang Y. Specific recognition of RNA/DNA hybrid and enhancement of human RNase H1 activity by HBD. EMBO J. 2008;27:1172–1181. doi: 10.1038/emboj.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structure of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 8.Nowotny M, Gaidamakov SA, Ghirlando R, Cerritelli SM, Crouch RJ, Yang W. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol Cell. 2007;28:264–276. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 9.Nowotny M, Yang W. Stepwise analysis of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006;25:1924–1933. doi: 10.1038/sj.emboj.7601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Vivo M, Dal Peraro M, Klein ML. Phosphodiester cleavage in ribonuclease H occurs via an associative two-metal-aided catalytic mechanism. J Am Chem Soc. 2008;130:10955–10962. doi: 10.1021/ja8005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elsässer B, Fels B. Atomistic details of the associative phosphodiester cleavage in human ribonuclease H. Phys Chem Chem Phys. 2010;12:11081–11088. doi: 10.1039/c001097a. [DOI] [PubMed] [Google Scholar]
- 12.Rosta E, Nowotny M, Yang W, Hummer G. Catalytic mechanism of RNA backbone cleavage by ribonuclease H from quantum mechanics/molecular mechanics simulations. J Am Chem Soc. 2011;133:8934–8941. doi: 10.1021/ja200173a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crooke ST. Antisense Strategies. Curr Mol Medicine. 2004;4:465–487. doi: 10.2174/1566524043360375. [DOI] [PubMed] [Google Scholar]
- 14.Bennet CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Ann Rev Pharm Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 15.Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem. 2004;279:17181–17189. doi: 10.1074/jbc.M311683200. [DOI] [PubMed] [Google Scholar]
- 16.Schatz O, Cromme FV, Grüninger-Leitz F, Le Grice SFJ. Point mutations in conserved amino acid residues within the C-terminal domain of HIV-1 reverse transcriptase specifically repress RNase H function. FEBS Lett. 1989;257:311–314. doi: 10.1016/0014-5793(89)81559-5. [DOI] [PubMed] [Google Scholar]
- 17.Tisdale M, Schulze T, Larder BA, Moelling K. Mutations within the RNase H domain of human immunodeficiency virus type 1 reverse transcriptase abolish virus infectivity. J Gen Virol. 1991;72:59–66. doi: 10.1099/0022-1317-72-1-59. [DOI] [PubMed] [Google Scholar]
- 18.Kalota A, Karabon L, Swider CR, Viazovkina E, Elzagheid M, Damha MJ, Gewirtz AM. 2′-Deoxy-2′-fluoro-β-D-arabinonucleic acid (2′F-ANA) modified oligonucleotides (ON) effect highly efficient, and persistent, gene silencing. Nucl Acids Res. 2006;34:451–461. doi: 10.1093/nar/gkj455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao W, Han F, Storm C, Egan W, Cheng YC. Phosphorothioate oligonucleotides are inhibitors of human DNA polymerases and RNase H: implications for antisense technology. Mol Pharmacol. 1992;41:223–229. [PubMed] [Google Scholar]
- 20.Wu H, Lima WF, Crooke ST. Properties of cloned and expressed human RNase H1. J Biol Chem. 1999;274:28270–28278. doi: 10.1074/jbc.274.40.28270. [DOI] [PubMed] [Google Scholar]
- 21.Tadokoro T, Chon H, Koga Y, Takano K, Kanaya S. Identification of the gene encoding a type 1 RNase H with an N-terminal double-stranded RNA binding domain from a psychrotrophic bacterium. FEBS J. 2007;274:3715–3727. doi: 10.1111/j.1742-4658.2007.05903.x. [DOI] [PubMed] [Google Scholar]
- 22.Jongruja N, You D, Kanaya E, Koga Y, Takano K, Kanaya S. The N-terminal hybrid binding domain of RNase HI from Thermotoga maritima is important for substrate binding and Mg2+-dependent activity. FEBS J. 2010;277:4474–4489. doi: 10.1111/j.1742-4658.2010.07834.x. [DOI] [PubMed] [Google Scholar]
- 23.Kawamata T, Tomari Y. Making RISC. Trends Biochem Sci. 2010;35:368–376. doi: 10.1016/j.tibs.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 24.Ho M, De Vivo M, Dal Perro M, Klein ML. Understanding the effect of magnesium ion concentration on the catalytic activity of ribonuclease H through computation: does a third metal binding site modulate endonuclease catalysis? J Am Chem Soc. 2010;132:13702–13712. doi: 10.1021/ja102933y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang W, Hendrickson WA, Crouch RJ, Satow Y. Structure of ribonuclease H phased at 2 Å resolution by MAD analysis of the selenomethionyl protein. Science. 1990;249:1398–1405. doi: 10.1126/science.2169648. [DOI] [PubMed] [Google Scholar]
- 26.Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Ikehara M, Matsuzaki T, Morikawa K. Three-dimensional structure of ribonuclease H from E. coli. Nature. 1990;347:306–309. doi: 10.1038/347306a0. [DOI] [PubMed] [Google Scholar]
- 27.Davies JF, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA. Crystal structure of the RNase H domain of HIV-1 reverse transcriptase. Science. 1991;252:88–95. doi: 10.1126/science.1707186. [DOI] [PubMed] [Google Scholar]
- 28.Kanaya S, Kohara A, Miura Y, Sekiguchi A, Iwai S, Inoue H, Ohtsuka E, Ikehara M. Identification of the amino acid residues involved in the active site of Escherichia coli ribonuclease H by site-directed mutagenesis. J Biol Chem. 1990;265:4615–4621. [PubMed] [Google Scholar]
- 29.Cerritelli SM, Crouch RJ. Cloning, expression and mapping of ribonucleases H of human and mouse related to bacterial RNase HI. Genomics. 1998;53:300–307. doi: 10.1006/geno.1998.5497. [DOI] [PubMed] [Google Scholar]
- 30.Blanchard JS. Buffers for enzymes. Methods Enzymol. 1984;104:404–414. doi: 10.1016/s0076-6879(84)04107-0. [DOI] [PubMed] [Google Scholar]
- 31.Perrin DD, Dempsey B. Buffers for pH and Metal Ion Control. Chapman & Hall; London: 1974. [Google Scholar]
- 32.Fischer BE, Häring UK, Tribolet R, Sigel H. Metal ion/buffer interactions. Stability of binary and ternary complexes containing 2-amino-2(hydroxymethyl)-1,3-propanediol (Tris) and adenosine 5′-triphosphate (ATP) Eur J Biochem. 94:523–530. doi: 10.1111/j.1432-1033.1979.tb12921.x. (19790) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Stable products of Hs-RNase H1 cleavage of the 21 bp RNA-DNA heteroduplex substrate.
Supplemental Figure 2. Comparison of heteroduplex substrate cleavage by Hs-RNase H1, with or without an N-terminal (His)6-tag.
Supplemental Figure 3. Comparison of the Mg2+ titration behavior of Hs-RNase H1 and the H264A mutant.