

Abstract
The addition of mass spectrometry (MS) analysis to the hydrogen exchange (HX) proteolytic fragmentation experiment extends powerful HX methodology to the study of large biologically important proteins. A persistent problem is the degradation of HX information due to back exchange of deuterium label during the fragmentation-separation process needed to prepare samples for MS measurement. This paper reports a systematic analysis of the factors that influence back exchange (solution pH, ionic strength, desolvation temperature, LC column interaction, flow rates, system volume). The many peptides exhibit a range of back exchange due to intrinsic amino acid HX rate differences. Accordingly, large back exchange leads to large variability in D-recovery from one residue to another as well as one peptide to another that cannot be corrected for by reference to any single peptide-level measurement. The usual effort to limit back exchange by limiting LC time provides little gain. Shortening the LC elution gradient by two-fold only reduced back-exchange by ~2 % (from ~30% to 28%), while sacrificing S/N and peptide count. An unexpected dependence of back exchange on ionic strength as well as pH suggests a strategy in which solution conditions are changed during sample preparation. Higher salt should be used in the first stage of sample preparation (proteolysis and trapping) and lower salt (< 20 mM) and pH in the second stage before electrospray injection. Adjustment of these and other factors together with recent advances in peptide fragment detection yields hundreds of peptide fragments with D-label recovery of 90 ± 5%.
Introduction
The naturally occurring exchange of protein amide hydrogens with the hydrogens in water depends on and therefore can provide detailed information about protein structure, biophysical properties and functional behavior, in principle resolved to the amino acid level. This powerful capability has been very widely exploited in HX NMR studies but routine NMR analysis is limited to relatively small, highly soluble proteins that are available in quantity and labeled with stable isotopes. Hydrogen exchange investigations of larger and biologically more interesting protein systems can be achieved by a proteolytic fragmentation method [1] followed by mass spectrometry analysis [2–5]. In this method, protein samples taken from an H-D exchange experiment are proteolytically fragmented and separated in preparation for MS analysis to determine the quantity and position of carried D-label at a fragment-resolved level. The comparison of high quality data for very many overlapping fragments promises to provide HX information at the amino acid-resolved level [6–8]. This is the capability that has made the HX NMR method so valuable. A problem is that some D-label is variably lost during sample preparation due to back exchange in the H2O solutions used. The different residues in any given peptide fragment unavoidably lose D-label at different rates [9], and this residue-level variability cannot be reconstructed and corrected for when one has only fragment-level data. The problem can only be minimized by reducing the level of back exchange.
A related HX MS method uses electron transfer or capture dissociation to produce a series of protein fragments that differ by only one terminal residue[3,10–20]. Comparison of results for overlapping fragments can then resolve the position and quantity of carried D-label at the individual residue level by simple subtraction. This method can use direct whole molecule sample injection and thus minimizes the sample preparation steps that allow back exchange. However, it seems likely that analysis of large proteins will still rely on submolecular protein fragments prepared as just described. In this case back exchange will continue to be a problem.
Because back exchange quickly degrades HX MS analysis, it continues to receive a great deal of attention [8,12,21–29]. The typical level of D-label recovery reported in the fragment separation literature is about 70% (30% back exchange). Higher reported values generally depend on results for only one or a few peptides. However we find that different peptide fragments experience a wide range of back exchange values. Among other implications, any computational correction for back exchange using reference peptides will be flawed. This is true even for direct measurement of back exchange in the peptide of interest since different amide sites will be labeled in experimental and reference situations. We systematically studied the conditions that determine back exchange including pH, ionic strength, ion transfer tube temperature, the interaction of peptides with reverse phase columns, and the time consumed at each stage of sample preparation. The optimization of these variables reduces back exchange by a factor of two to three.
Experimental Section
Maltose binding protein from E. coli (370 amino acids), was expressed and purified as previously described [30]. For full deuteration 10 μM MBP was incubated in D2O with 2 M D6-GdmCl at pD 9 and 45°C for 30 minutes and then refolded by dilution into fresh D2O. In D-recovery experiments, the fully deuterated protein sample was diluted into H2O at the quench condition (minimal HX rate as specified below) and injected into the online temperature controlled system described before [31] to produce, separate, and analyze many peptide fragments. The ExMS program [32] was used to identify each peptide and determine its centroid mass. Subtraction of the centroid mass of the all-H peptide yields the number of deuterons still carried by each peptide. Fractional D-recovery was calculated for each peptide fragment as peptide-bound deuterium recovered divided by the total number of exchangeable amide sites (peptide amino acids minus proline and minus 2 to account for the free N-terminal amino group and the rapidly lost D on the second residue; see main text. It was also assumed that deuterons on exchangeable side chains are lost rapidly during sample preparation, as has been shown in earlier calibrations of structurally unprotected HX rates [9,33,34].
Experiments used a ThermoScientific LTQ-Orbitrap XL mass spectrometer in positive ion mode over the m/z window 200–1500. Samples are injected into a home-built system contained in thermoelectrically cooled chamber for online digestion, buffer exchange, and LC separation (see Fig. 1 in Mayne et al. [31]). The protein sample (0.3 μM MBP) flows first through an immobilized pepsin column and then directly onto a small C4 trap column where the injection buffer salts and denaturant are washed away. Isocratic flow proceeds until a volume equivalent to ~1.5 times the initial injection volume has flowed over the column as we find this ensures complete buffer exchange. Peptides are eluted by acetonitrile gradient through a 0.3 mm × 5 cm C18 analytical column, directly to the electrospray source. Additional information pertaining to our online system and instrument parameters can be found in previous work which details our methodology to produce very many overlapping peptides[31]. Chromatographic gradients were shaped to yield roughly equal numbers of peptides per unit time as described in supplemental text (Fig. 6 in Online Resource 1). Aqueous running buffers contained 0.1% formic acid adjusted to the desired pH and ionic strength by the addition of TFA and NH4OH. The organic running buffer contained 0.1% formic acid in acetonitrile.
Results & Discussion
In the typical HX MS fragment separation analysis, an experimental protein is exposed to H-D exchange for a period of time. Each amide hydrogen exchanges at its own rate determined by solvent conditions, its intrinsic chemical rate, and protecting structure, modified by the experimental variable being studied. To measure the extent of D-labeling, protein samples are taken and prepared for MS analysis by quenching into a minimum HX rate condition (low pH and temperature). The protein unfolds but HX is greatly slowed, allowing a short time for sample preparation without excessive loss of D-label. In the present experiments, the protein was proteolytically fragmented (immobilized pepsin column), the peptide fragments were caught on a trap column, washed and buffer exchanged, roughly separated by fast reverse phase chromatography, and then injected by ESI into the spectrometer to determine the mass of each fragment and thus the amount of carried D. These sample preparation steps were performed in an online flow system described before [31].
To study the effect of various preparatory conditions on back exchange, we used maltose binding protein (MBP, 370 residues) that had been fully deuterated by exchange in D2O. We measured the recovery of D-label for each of many MBP peptide fragments after passage through the entire analysis. The difference between the known fully deuterated mass of each peptide and the mass experimentally recovered directly measures back-exchange. Although our methods [31] found ~200 MBP fragments (pepsin proteolysis alone), we used for each experimental series only the peptides that were observed in all experiments in order to ensure unbiased comparisons. Identification and analysis of these many peptides used SEQUEST (ThermoScientific Bioworks 3.3.1) and the ExMS program [32].
pH and Ionic Strength
Fig. 1A shows the expected dependence of HX rate on pH for a hypothetical peptide with all amino acids, calculated from standard reference values [9,33]. The minimum HX rate is expected to be reached at pH 2.5. Accordingly, the quench and running buffers in fragment-separation experiments have always been prepared near this condition [1]. To test this expectation we performed a series of D-recovery experiments over a range of experimental pH values (Fig. 1B). Single peptide values are often used as a back exchange reference in the literature. In fact, different peptides display a wide range of D-label recoveries. This can be expected since amide HX rate varies with amino acid type and nearest neighbors [9,33]. Unexpectedly however, the minimum rate with significantly reduced back exchange was reached at pH 2.25 (Fig. 1C).
Fig 1.

Dependence of HX rates on pH and ionic strength at 0 °C. A. Expected pH dependence for the hypothetical peptide GGVALISTDENQRHKCMFTW. The rate at any pH is the sum of the H3O+ ion catalyzed reaction (red) and the OH− ion catalyzed reaction (blue), each of which varies by 10-fold per pH unit. The additional pH-independent contribution due to water catalysis is shown with hatch marks. The averaged fragment-level HX rate constant shown is taken as the geometric mean (log averaged) of the 20 amides, each of which exchange with somewhat different rate constants. B. Cumulative population distribution pH series. C. Slices taken across B at given population percentiles. D. Effect of ionic strength at pH 2.5.
Testing showed that the shift in the pH of minimum rate depends on ionic strength. When ionic strength is 20 mM or higher, HX rate is a minimum at pH 2.5 and matches expected values (Fig. 1D). The earlier HX rate calibrations [9,33] were done in high salt (0.5 M KCl) purposely to shield against extraneous charge effects. However, MS analysis requires electrospray solutions with low salt where, we find, the pH of minimum HX rate is significantly shifted. A similar shift was noted before [35]. The amide group acts like it has a small net positive partial charge which, at low ionic shielding, favors the OH− -catalyzed reaction and disfavors H3O+, shifting the pH-rate curve to the left.
The present results show how these different requirements for minimizing back exchange rate can be satisfied. Experimental HX samples normally contain significant salt and after quench may have added GdmCl (0.5 to 2 M) to promote protein unfolding and improve digestion in the proteolysis step. Therefore, in this first stage of sample preparation we use quench buffer at pH 2.5. The sample is then caught on a trap column, washed, eluted with an acetonitrile gradient through the LC step, and injected online into the mass spectrometer. These latter steps should use low salt, desirable for ESI MS, and the lower pH. We use wash and elution buffers with 0.1% formic acid adjusted to pH 2.25 with TFA. Solution pH values were measured and adjusted in the pertinent solutions at room temperature and then used at 0° C.
Desolvation Temperature
The details of ion source depend on instrument design. In our spectrometer, after nebulization at the electrospray needle, droplets of solution are pulled by a pressure differential through a heated capillary (~200 °C), which speeds solvent evaporation and ionization. As exchange rates in solution depend sharply on temperature (~3-fold per 10 °C) [9,33], sample heating in the capillary might greatly promote back-exchange.
We measured back exchange as a function of capillary temperature. Cumulative recovery distributions are in Fig. 2. The results show a broad maximum in D-recovery when capillary temperature is set between 100 and 200 °C, with declining recovery at higher and lower temperature. We did not observe a difference in recovery between charge states of the same peptide as reported before [29], apparently due to instrumental differences. Results for given peptides with different charge state agreed in these and our other experiments to < 0.1 D.
Fig 2.
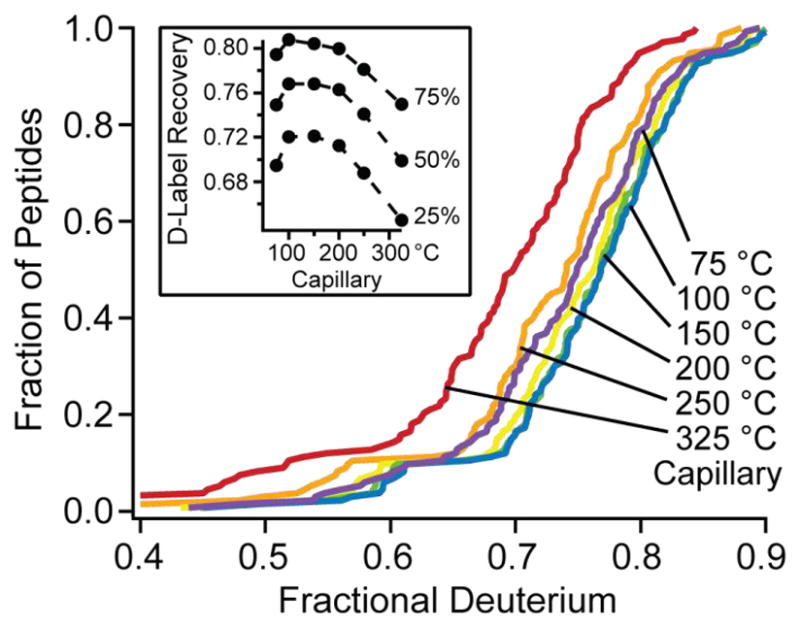
The dependence of D-label recovery on transfer tube temperature.
Interestingly, the 75 °C data shows distinctly reduced recovery. Less efficient evaporation at 75 °C could lead to increased time at temperature above 0 °C in the liquid phase before solvent evaporation, leading to increased back-exchange. Given these results, we adopted a capillary temperature setting of 100 °C.
Time on the LC column
To study the HX behavior of peptides bound to the C18 media of reverse phase columns, we compared HX rates of column-bound peptides with rates expected from earlier calibrations in free solution. Fully deuterated MBP samples were placed into quench conditions in H2O, injected into the online flow system, digested, and washed onto the trap column (5 min elapsed time). Peptides were held on the column for an additional experimental delay time between 0 and 45 min, then eluted from the trap column, through the analytical LC column, and into the mass spectrometer (3 to 18 minutes additional time).
Fig. 3 shows cumulative recovery distributions across the time series, and compares these results with the expected time-dependent loss of D-label in free solution, calculated by summing for each peptide’s individual amides. We assumed that D-label on side chains [9] and the N-terminal amino group is lost too rapidly to measure (expected rate > 10 s−1), and similarly for the amide on the second residue. The accelerated rate for the second residue is due to the absence of an amide group on the prior residue (10-fold in rate), and it is promoted by another 10-fold by the fixed positive charge on the neighboring N-terminal amino group, especially at the low salt concentration used here. This effect is contained in the older literature on HX of peptide models (see Table 1 in Molday et al. [34]) and has been directly measured more recently [36].
Fig 3.

Exchange on the column. A. Observed D-recovery for all peptides across the delay time series. The inset places the variable delay time during sample preparation. B. Recovery for the various peptides after a 20 minute delay on the trap column. C – E. Observed (data points) and theoretical (dashed lines) D-label recovery. C. Some peptides with normal recovery. D. A peptide with large slowing on the column due to structure formation and two component peptides with normal recovery. E. Some histidine-containing peptides with accelerated early loss.
A comparison between observed and expected D-recovery from the 20 minute delay experiment is shown for the whole peptide population in Fig. 3B and for a number of individual peptides across the delay series in Fig. 3C to 3E. During the sample preparation time including proteolysis and column interaction, most of the peptides exchange as expected. A few are much slower. Large retardation with HX slowing up to 20-fold while bound to the column matrix was seen for 11 overlapping peptides between the C-terminal residues 340 to 370. Fig. 3D shows one of these and two shorter component peptides which exchange as expected. Interestingly, in native MBP this segment adopts a helix-turn-helix motif and docks with a hydrophobic interface on the C-domain (see Fig. 7 in Online Resource 1). Evidently this peptide and some subfragments are induced to form mildly stable H-bonded structure, perhaps aided by hydrophobic interaction with the hydrocarbon chains of the reverse phase column. The slowing factor decreases systematically as either (helix) segment is cut back. Similar but more modest slowing, up to 4-fold, was seen for sets of peptides derived from several other protein segments (116–149, 169–194, 283–301, 312–330), apparently due to tentative helix formation. The tendency of an apolar environment to promote helix formation is well known; unsatisfied H-bonding is energetically expensive.
Some peptides show a small but noticeable negative offset between the expected and observed number of D atoms at the earliest time point, indicating additional D-loss. This included all of the 15 peptides that contain one of the three MBP histidine residues, suggesting some (acid) catalysis of nearby residues by the imidazolium side chain. However, we have not seen indications of this phenomenon with histidine-containing peptides in some other proteins.
Sample preparation time
Most previous attempts to minimize back exchange focus on minimizing the time that samples spend on the reverse phase column, with modest improvement. We find that time reduction accomplished by shortening the acetonitrile elution gradient (15, 10 or 5 minutes) produces surprisingly small gains (Fig. 4). The reason appears to be that early eluting peptides experience almost no time reduction while later eluting peptides tend to have a slower intrinsic HX rate [9] (more large apolar side chains [9], more time in higher acetonitrile) so that increased exposure time has less than the expected effect on back exchange. For example, whereas the amides of polar residues lose label at the rate of 1% to 2% per minute, the large apolar residues do so ~4 times more slowly [9]. Experimental evidence for this view comes from the fact that we find no correlation between the level of D-recovery and column elution time. In these experiments, total back exchange time varied between 10 and 20 minutes.
Fig 4.
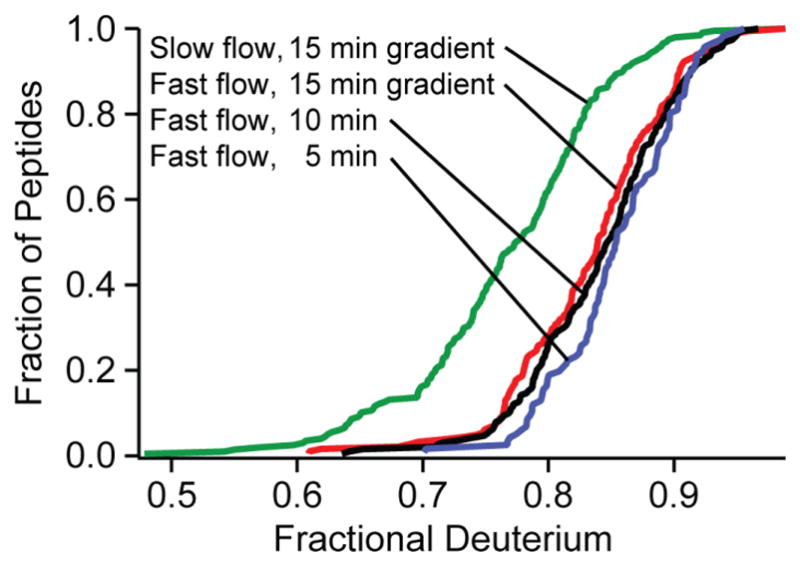
Minimizing preparation time by increasing flow rates (termed the fast condition, see text) increases D-recovery from the green to the colored distributions, which also show the effect of sharper elution gradients.
Fig 4 Change in sample exposure time. Sharper elution gradients provide little reduction in back exchange and sacrifice peptide fragment yield. Minimizing preparation time by increasing flow rates (termed the fast flow condition, see text) increased recovery levels from the green to the colored distributions.
In fact, the reduction of column retention time proved counter-productive. In order to obtain ultimate HX resolution at the amino acid level, it will be necessary to obtain a large number of sequentially overlapping peptides and multiple residue coverage. Chromatographic crowding became a problem in the 5 minute gradient resulting in 40% fewer useful peptides. Gradient shaping used to equalize peptide density through the chromatogram reduces but does not overcome this problem (described in supplemental text and Fig 6 in Online Resource 1). In addition, peak sharpening may reduce the number of MS scans per peptide and therefore S/N.
We more broadly reduced the time required to navigate the free volume in our flow system by increasing overall system flow rates. An increase in flow rates (300 μl/min during digestion, 450 μl/min for buffer exchange, 10 μl/min during peptide elution) reduced overall sample preparation time by 4.3 minutes. These flow rates maintained pressures below 2000 psi as recommended for POROS media (protease column). The increased D-recovery illustrated in Fig. 4 is consistent with the expected back exchange loss rate of about 1 to 2% per minute on average of carried D-label at the pH minimum and 0 °C [9].
Other considerations
Are these results for maltose binding protein results typical for proteins in general? Each test shown here used ~90 peptides, and they vary over a wide range in size, amino acid content, hydrophobicity, etc. It seems unlikely that sets of peptides from other proteins will behave differently. In agreement, we have now used our previous sample processing conditions and the improved conditions described here in ongoing experiments with other proteins (cytochrome c, staphylococcal nuclease, ribonuclease H, apolipoprotein A-I, Hsp104). The gain in D-recovery was comparable in all cases.
When is back exchange important? For HX MS experiments in which one attempts to define epitopic or ligand binding sites, one may be satisfied with crude peptide-level changes. These are less dependent on back exchange. Back exchange becomes most important when reaching for the amino acid level of resolution that has made the HX NMR experiment so powerful for protein studies. Recent progress using ECD and ETD to strive for site resolution minimizes the back exchange problem. In this case the whole protein can be injected directly into the mass spectrometer, avoiding the fragment separation analysis. However, a major advantage of the fragment separation analysis is the ability to study much larger and biologically more important proteins than HX NMR can accomplish. This goal probably exceeds the capability of direct ECD/ETD methods. In order to study large proteins by these methods, it seems likely that the fragment separation approach will be required as an initial step, resurrecting the back exchange problem.
The ability of the HX MS method to achieve high structural resolution depends on obtaining high quality data for many overlapping peptide fragments. We previously described methods for obtaining [31] and efficiently analyzing [32] hundreds of useful protein fragments with data accuracy to ~0.1 D. The present work shows that attention to the various factors that determine back exchange can increase D-recovery into the range 75 to 95%, as summarized in Fig. 5. These capabilities taken together give the investigator freedom to choose among different options. For example, if the effort to reach single amino acid resolution requires exceptionally low back exchange, the sacrifice of the lower half of the peptide population shown in Fig. 4 would still retain a very large number of peptides with high data quality, i.e. with D-recovery in the range of 90 ± 5%.
Fig. 5. Summary of gains in D-label recovery.
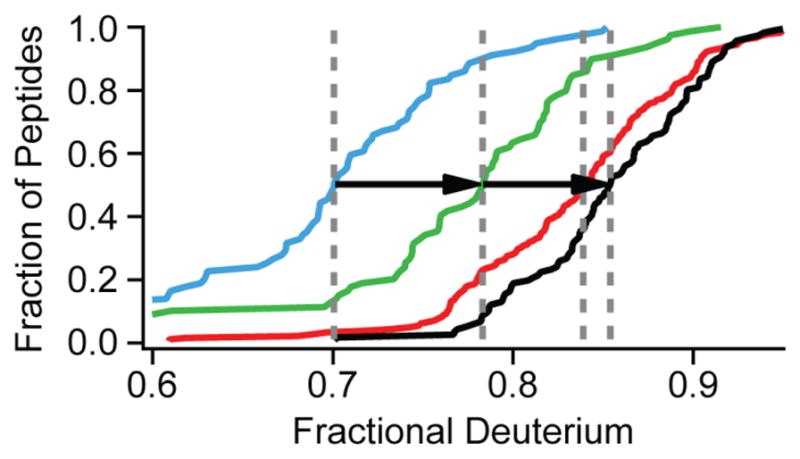
Arrows show the improvement made by manipulating sample ionic strength at pH 2.5 (blue to green) and increasing system flow rates during the prep/wash (green to red) and elution (red to black).
Conclusions
A systematic study of the factors that influence back exchange in the typical HX MS setup reveals a number of surprises and shows how the back exchange problem can be minimized. We find that different peptides exhibit a range of back exchange levels. Among other implications, this situation negates the use of any one or a small number of peptides as a reference marker for the degree of back exchange or its correction by computation. This must be done peptide by peptide and even then is imperfect since different amide sites will be detected in experimental and reference situations.
Results show that there is no single best back exchange condition; it varies with ionic strength. The first stage of sample preparation, involving proteolysis and sample trapping, is best performed at pH 2.5 and 0°C in high ionic strength, often with substantial GdmCl. The trapped peptides should then be washed and passed through the analytical HPLC column in pH 2.25 solution at low ionic strength. The common approach of trying to limit chromatographic time (reduced column size; shorter elution gradient) yields limited gains and is potentially counter-productive in respect to the yield of useful peptides. Sample exposure time can be more simply minimized by using high flow rates to rapidly clear system free volume. Putting aside the previously unexpected ionic strength effect, the loss of D-label through the sample preparation time proceeds closely as predicted from previous amide HX rate calibrations [9,33] although peptide-column interaction can have some unexpected effects such as structure formation. The combination of previously described methods for producing [31] and analyzing [32] many peptide fragments together with the ability to largely negate deleterious back exchange moves toward the goal of obtaining ultimate amino acid structural resolution for HX MS analysis.
Supplementary Material
Acknowledgments
This work was supported by research grants from the NIH (RO1 GM031847), the NSF (MCB1020649), and the Mathers Charitable Foundation, and a structural biology predoctoral training grant from the NIH (GM08275).
Footnotes
Conflict of interest. The authors declare no conflict of interest.
Bibliography
- 1.Englander JJ, Rogero JR, Englander SW. Protein hydrogen exchange studied by the fragment separation method. Anal Biochem. 1985;147:234–244. doi: 10.1016/0003-2697(85)90033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 4.Marcsisin SR, Engen JR. Hydrogen exchange mass spectrometry: What is it and what can it tell us? Anal Bioanal Chem. 2010;397:967–972. doi: 10.1007/s00216-010-3556-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsutsui Y, Wintrode PL. Hydrogen/deuterium, exchange-mass spectrometry: A powerful tool for probing protein structure, dynamics and interactions. Curr Med Chem. 2007;14:2344–2358. doi: 10.2174/092986707781745596. [DOI] [PubMed] [Google Scholar]
- 6.Althaus E, Canzar S, Ehrler C, Emmett MR, Karrenbauer A, et al. Computing H/D-exchange rates of single residues from data of proteolytic fragments. BMC Bioinf. 2010;11:424. doi: 10.1186/1471-2105-11-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pascal BD, Chalmers MJ, Busby SA, Griffin PR. HD Desktop: An integrated platform for the analysis and visualization of H/D exchange data. J Am Soc Mass Spectrom. 2009;20:601–610. doi: 10.1016/j.jasms.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z, Zhang A, Xiao G. Improved protein hydrogen/deuterium exchange mass spectrometry platform with fully automated data processing. Anal Chem. 2012;84:4942–4949. doi: 10.1021/ac300535r. [DOI] [PubMed] [Google Scholar]
- 9.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hagman C, Tsybin YO, Hakansson P. Solution-phase deuterium/hydrogen exchange at a specific residue using nozzle-skimmer and electron capture dissociation mass spectrometry. Rapid Commun Mass Spectrom. 2006;20:661–665. doi: 10.1002/rcm.2339. [DOI] [PubMed] [Google Scholar]
- 11.Huang RYC, Garai K, Frieden C, Gross ML. Hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry determine the interface and dynamics of apolipoprotein e oligomerization. Biochemistry. 2011;50:9273–9282. doi: 10.1021/bi2010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rand KD, Lund FW, Amon S, Jørgensen TJD. Investigation of amide hydrogen back-exchange in Asp and His repeats measured by hydrogen (1H/2H) exchange mass spectrometry. Int J Mass Spectrom. 2011;302:110–115. [Google Scholar]
- 13.Rand KD, Pringle SD, Morris M, Engen JR, Brown JM. ETD in a traveling wave ion guide at tuned Z-spray ion source conditions allows for site-specific hydrogen/deuterium exchange measurements. J Am Soc Mass Spectrom. 2011;22:1784–1793. doi: 10.1007/s13361-011-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zehl M, Rand KD, Jensen ON, Jorgensen TJD. Electron transfer dissociation facilitates the measurement of deuterium incorporation into selectively labeled peptides with single residue resolution. J Am Chem Soc. 2008;130:17453–17459. doi: 10.1021/ja805573h. [DOI] [PubMed] [Google Scholar]
- 15.Abzalimov RR, Kaplan DA, Easterling ML, Kaltashov IA. Protein conformations can be probed in top-down HDX MS experiments utilizing electron transfer dissociation of protein ions without hydrogen scrambling. J Am Soc Mass Spectrom. 2009;20:1514–1517. doi: 10.1016/j.jasms.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan J, Han J, Borchers CH, Konermann L. Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein. J Am Chem Soc. 2009;131:12801–12808. doi: 10.1021/ja904379w. [DOI] [PubMed] [Google Scholar]
- 17.Pan J, Han J, Borchers CH, Konermann L. Characterizing short-lived protein folding intermediates by top-down hydrogen exchange mass spectrometry. Anal Chem. 2010;82:8591–8597. doi: 10.1021/ac101679j. [DOI] [PubMed] [Google Scholar]
- 18.Pan JX, Han J, Borchers CH, Konermann L. Conformer-specific hydrogen exchange analysis of a beta(1–42) oligomers by top-down electron capture dissociation mass spectrometry. Anal Chem. 2011;83:5386–5393. doi: 10.1021/ac200906v. [DOI] [PubMed] [Google Scholar]
- 19.Rand KD, Adams CM, Zubarev RA, Jorgensen TJD. Electron capture dissociation proceeds with a low degree of intramolecular migration of peptide amide hydrogens. J Am Chem Soc. 2008;130:1341–1349. doi: 10.1021/ja076448i. [DOI] [PubMed] [Google Scholar]
- 20.Rand KD, Zehl M, Jensen ON, Jorgensen TJD. Protein hydrogen exchange measured at single-residue resolution by electron transfer dissociation mass spectrometry. Anal Chem. 2009;81:5577–5584. doi: 10.1021/ac9008447. [DOI] [PubMed] [Google Scholar]
- 21.Hotchko M, Anand GS, Komives EA, Ten Eyck LF. Automated extraction of backbone deuteration levels from amide H/2H mass spectrometry experiments. Protein Sci. 2006;15:583–601. doi: 10.1110/ps.051774906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Engen JR, Hobbins WB. Ultra performance liquid chromatography (UPLC) further improves hydrogen/deuterium exchange mass spectrometry. J Am Soc Mass Spectrom. 2006;17:163–167. doi: 10.1016/j.jasms.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 23.Wu Y, Kaveti S, Engen JR. Extensive deuterium back-exchange in certain immobilized pepsin columns used for H/D exchange mass spectrometry. Anal Chem. 2006;78:1719–1723. doi: 10.1021/ac0518497. [DOI] [PubMed] [Google Scholar]
- 24.Kipping M, Schierhorn A. Improving hydrogen/deuterium exchange mass spectrometry by reduction of the back-exchange effect. J Mass Spectrom. 2003;38:271–276. doi: 10.1002/jms.437. [DOI] [PubMed] [Google Scholar]
- 25.Emmett MR, Kazazic S, Marshall AG, Chen W, Shi SD, et al. Supercritical fluid chromatography reduction of hydrogen/deuterium back exchange in solution-phase hydrogen/deuterium exchange with mass spectrometric analysis. Anal Chem. 2006;78:7058–7060. doi: 10.1021/ac060693n. [DOI] [PubMed] [Google Scholar]
- 26.Zhang HM, Bou-Assaf G, Emmett M, Marshall A. Fast reversed-phase liquid chromatography to reduce back exchange and increase throughput in H/D exchange monitored by FT-ICR mass spectrometry. J Am Soc Mass Spectrom. 2009;20:520–524. doi: 10.1016/j.jasms.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keppel TR, Jacques ME, Young RW, Ratzlaff KL, Weis DD. An efficient and inexpensive refrigerated LC system for H/D exchange mass spectrometry. J Am Soc Mass Spectrom. 2011;22:1472–1476. doi: 10.1007/s13361-011-0152-6. [DOI] [PubMed] [Google Scholar]
- 28.Valeja S, Emmett M, Marshall A. Polar aprotic modifiers for chromatographic separation and back-exchange reduction for protein hydrogen/deuterium exchange monitored by fourier transform ion cyclotron resonance mass spectrometry. J Am Soc Mass Spectrom. 2012:1–9. doi: 10.1007/s13361-011-0329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coales SJ, Tomasso JC, Hamuro Y. Effects of electrospray capillary temperature on amide hydrogen exchange. Rapid Commun Mass Spectrom. 2008;22:1367–1371. doi: 10.1002/rcm.3512. [DOI] [PubMed] [Google Scholar]
- 30.Gardner KH, Zhang X, Gehring K, Kay LE. Solution NMR studies of a 42 kDa escherichia coli maltose binding protein/beta-cyclodextrin complex: Chemical shift assignments and analysis. J Am Chem Soc. 1998;120:11738–11748. [Google Scholar]
- 31.Mayne L, Kan ZY, Sevugan Chetty P, Ricciuti A, Walters B, et al. Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J Am Soc Mass Spectrom. 2011:1–8. doi: 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kan ZY, Mayne L, Chetty PS, Englander SW. ExMS: Data analysis for HX-MS experiments. J Am Soc Mass Spectrom. 2011;22:1906–1915. doi: 10.1007/s13361-011-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Connelly GP, Bai Y, Jeng MF, Englander SW. Isotope effects in peptide group hydrogen exchange. Proteins. 1993;17:87–92. doi: 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
- 34.Molday RS, Englander SW, Kallen RG. Primary structure effects on peptide group hydrogen exchange. Biochemistry. 1972;11:150–158. doi: 10.1021/bi00752a003. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Z. Thesis. Purdue University; United States -- Indiana: 1995. Protein hydrogen exchange determined by mass spectrometry: A new tool for probing protein high-order structure and structural changes; p. 224. [Google Scholar]
- 36.Skinner JJ, Limb WK, Bédard S, Black BE, Englander SW. Protein hydrogen exchange: Testing current models. Protein Sci. 2012 doi: 10.1002/pro.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.