

Abstract
This study investigated whether expression of the common cystic fibrosis transmembrane conductance regulator (CFTR) mutant F508del in the apical membrane of enterocytes confers increased bicarbonate secretory capacity on the intestinal epithelium of F508del mutant mice compared to that of CFTR knockout (KO) mice. CFTR KO mice, F508del mutant mice (F508del) and wild-type (WT) littermates were bred on the FVB/N background. F508del isolated brush border membrane (BBM) contained approximately 5–10% fully glycosylated band C protein compared to WT BBM. Similarly, the forskolin (FSK)-induced, CFTR-dependent short-circuit current (ΔIsc) of F508del mucosa was approximately 5–10% of WT, whereas the HCO3− secretory response (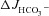 ) was almost half that of WT in both duodenum and mid-colon studied in vitro and in vivo. While WT intestine retained full FSK-induced
) was almost half that of WT in both duodenum and mid-colon studied in vitro and in vivo. While WT intestine retained full FSK-induced 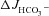 in the absence of luminal Cl−, the markedly higher
in the absence of luminal Cl−, the markedly higher 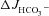 than ΔIsc in F508del intestine was dependent on the presence of luminal Cl−, and was blocked by CFTR inhibitors. The Ste20-related proline–alanine-rich kinases (SPAK/OSR1), which are downstream of the with-no-lysine (K) protein kinases (WNK), were rapidly phosphorylated by FSK in WT and F508del, but significantly more slowly in CFTR KO intestine. In conclusion, the data demonstrate that low levels of F508del membrane expression in the intestine of F508del mice significantly increased FSK-induced HCO3− secretion mediated by Cl−/HCO3− exchange. However, in WT mucosa FSK elicited strong SPAK/OSR1 phosphorylation and Cl−-independent HCO3− efflux. This suggests that therapeutic strategies which deliver F508del to the apical membrane have the potential to significantly enhance epithelial HCO3− secretion.
than ΔIsc in F508del intestine was dependent on the presence of luminal Cl−, and was blocked by CFTR inhibitors. The Ste20-related proline–alanine-rich kinases (SPAK/OSR1), which are downstream of the with-no-lysine (K) protein kinases (WNK), were rapidly phosphorylated by FSK in WT and F508del, but significantly more slowly in CFTR KO intestine. In conclusion, the data demonstrate that low levels of F508del membrane expression in the intestine of F508del mice significantly increased FSK-induced HCO3− secretion mediated by Cl−/HCO3− exchange. However, in WT mucosa FSK elicited strong SPAK/OSR1 phosphorylation and Cl−-independent HCO3− efflux. This suggests that therapeutic strategies which deliver F508del to the apical membrane have the potential to significantly enhance epithelial HCO3− secretion.
Key points
Cystic fibrosis (CF) is a lethal disease characterized by low rates of epithelial Cl− and HCO3− secretion and obstruction of the airways and gastrointestinal and reproductive organs by sticky mucus. HCO3− secretion has recently been demonstrated to be necessary for mucus hydration.
The most frequent CF mutation is F508del. This mutant protein is usually degraded in the proteasome. New therapeutic strategies have been developed which deliver F508del to the plasma membrane.
Utilizing transgenic F508del mutant and cystic fibrosis transmembrane conductance regulator (CFTR) knockout mice, apical membrane expression of F508del protein was found to be associated with enhanced stimulation of intestinal HCO3− secretion.
The predominant molecular mechanism for enhanced F508del HCO3− stimulation appeared to be the activation of a Cl− recycling pathway, with Cl− exit via membrane-resident F508del protein and Cl− entry in exchange for HCO3− by apical Cl−/HCO3− exchange. In contrast, the predominant molecular mechanism for cAMP-activated HCO3− secretion in WT intestine appears to be HCO3− exit via CFTR itself.
Introduction
Cystic fibrosis (CF) epithelia display a reduction in Cl−, HCO3− and fluid secretion and an increase in salt and fluid absorption (Greger et al. 2001; Seidler et al. 2009; Quinton, 2010). The pathophysiological correlates are sticky, viscous mucoid secretions impacting ductal structures of multiple glandular organs and the intestine, which result in progressive glandular destruction, and increased susceptibility to bacterial infections in the respiratory airways (Quinton, 2010). Recent data suggest that epithelial HCO3− secretion may be required for the release, as well as the rheological properties, of intestinal, airway and reproductive tract mucins (Garcia et al. 2009; Chen et al. 2010; Muchekehu & Quinton, 2010), and thus be of great pathophysiological relevance.
The molecular mechanisms of epithelial HCO3− secretion are complex and involve both different anion channels and Cl−/HCO3− exchangers. Likewise, the molecular correlate for defective HCO3− secretion in cystic fibrosis transmembrane conductance regulator (CFTR)-deficient or CFTR mutant epithelia is controversial, but recent data point to the involvement of various transporters in different CFTR-expressing epithelia (Steward et al. 2005; Dorwart et al. 2008; Seidler et al. 2009; Beuers et al. 2010; Quinton, 2010). While evidence has accumulated over the last two decades that the CFTR anion channel may become permeable to HCO3− under certain conditions (Poulsen et al. 1994, Clarke et al. 1998, Spiegel et al. 2003; Shcheynikov et al. 2004; Ishiguro et al. 2009; Park et al. 2010), it is also clear that anion exchangers from the Slc26 family are involved in epithelial HCO3− secretion (Steward et al. 2005; Tuo et al. 2006; Dorwart et al. 2008; Walker et al. 2009). A recent study suggests that the activation of the with no lysine (WNK) protein kinases 1 and 4–Ste20-related proline–alanine-rich kinases (SPAK/OSR1) signalling pathway in response to a decrease in intracellular Cl− concentration in pancreatic duct cells may result in a change of the anion selectivity of CFTR from Cl− to HCO3− predominance and in an inhibition of apical Cl−/HCO3− exchangers (Park et al. 2010). On the other hand, CFTR and members of the Slc26 family have been shown to structurally interact, at least in heterologous expression systems, with reciprocal activation of their individual transport functions (Ko et al. 2004). Thus, two fundamentally different potential mechanisms exist for CFTR-mediated epithelial HCO3− secretion, and both may be operative in different epithelia under distinct physiological conditions.
The most frequent mutation in CF patients, the F508del mutation, is the deletion of a phenylalanine residue at position 508 of the CFTR amino acid sequence. Early studies on the biosynthesis and localization of F508del-CFTR indicated that the mutant protein is not folded correctly and, as a result, is not delivered to the plasma membrane (Cheng et al. 1990, Gregory et al. 1991; Ward & Kopito, 1994). Premature degradation of F508del-CFTR can be partially inhibited by reducing the temperature of cultured cells or isolated tissue to 26°C (‘temperature rescue’), leading to increased F508del-CFTR membrane expression (Denning et al. 1992). But even when F508del-CFTR reaches the plasma membrane, it is prematurely degraded (Lukacs et al. 1993; Okiyoneda et al. 2010) and displays defective anion transport (Dalemans et al. 1991; Wang et al. 2000; Tang et al. 2009). However, novel drug treatment strategies have recently been developed that directly target the molecular defects in CF patients, including substances that partially prevent the degradation of mutant CFTR in the proteasome and result in improved membrane expression and increased Cl− transport by epithelial cells (Lukacs & Verkman, 2012).
We wondered if F508del-CFTR membrane expression might restore epithelial HCO3− secretion. To address this question, we utilized the F508del-CFTR mouse on the FVB/N background, in which some F508del-CFTR protein reaches the small intestinal brush border membrane (BBM), and a residual forskolin (FSK)-induced short-circuit current (Isc) is measured (French et al. 1996). We determined the amount of mature, fully glycosylated (band C) CFTR protein in the BBM of wild-type (WT) and F508del intestine, measured basal and FSK-induced HCO3− secretion in the duodenum and colon of WT, F508del and CFTR KO mice in vivo and in vitro, and assessed the transport mechanism(s) involved in F508del-augmented HCO3− secretion. Finally, we studied (i) whether SPAK/OSR1 phosphorylation may occur during agonist stimulation of intestinal anion secretion in vivo, (ii) if this pathway may be defective in the absence of CFTR expression, (iii) whether F508del-CFTR expression at the apical membrane restores WNK signalling, and (iv) if this pathway explains the differential dependence of CFTR-mediated epithelial HCO3− secretion on luminal Cl− in different experimental systems.
Methods
Animals
The CFTR KO mouse strain was originally generated in the laboratory of Martin Evans (Ratcliff et al. 1993) and the F508del-mutant strain in the laboratory of Bob Scholte (French et al. 1996). Both CFTR KO and F508del mice were congenic on the FVB/N background. Mice were bred at the animal care facility of Hannover Medical School under standard temperature and light conditions, and were allowed free access to food and water. The mice received electrolyte drinking solution containing polyethylene glycol (PEG) and high HCO3− (in mm: 40 Na2SO4, 75 NaHCO3, 10 NaCl, 10 KCl, 23 g l−1 PEG 4000), and a fibre-free diet (Altromin, C1013) to allow survival beyond weaning. Care was taken to match the mice not only as sex-matched littermates but also in terms of an equal number of weight-matched male and female pairs in each group of experiments. WT refers to the +/+ littermates of the respective strain. All experiments involving animals were approved by the Hannover Medical School Committee on investigations involving animals and an independent committee assembled by the local authorities (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit).
pH-stat titration of HCO3− secretory rates in isolated duodenal and mid-colonic mucosa
Ussing chamber experiments were performed as described previously (Seidler et al. 1997) in the open-circuit mode, with intermittent current pulses (200 μA every 60 s) to record the electrical resistance (Rt), and to calculate a nominal short-circuit current (Isc). Mice were subjected to brief CO2 narcosis prior to cervical dislocation before excising intestine for isolated mucosa preparations. The excised duodenum and mid-colon were stripped of external muscle layers and mounted in Ussing chambers (0.625 cm2 aperture). Neural activity and prostaglandin generation were blocked with tetrodotoxin (1 μm, serosal) and indomethacin (3 μm, serosal), respectively. Transepithelial Isc was calculated as μEq cm−2 tissue surface area. The serosal solution contained (in mm): 108 NaCl, 25 NaHCO3, 3 KCl, 1.3 MgSO4, 2 CaCl2, 2.25 KH2PO4, 8.9 glucose, and 10 sodium pyruvate, and was gassed with 95% O2–5% CO2 (pH 7.4). The mucosal solution (154 mm NaCl or 154 mm sodium gluconate) was gassed with 100% O2.
Surgical procedure: ‘in vivo duodenal/colonic loop experiment’
Mice were anaesthetized by spontaneous inhalation of isoflurane (Forene; Abbott Germany, Wiesbaden, Germany) and experiments performed as previously described (Sjoblom et al. 2009). In brief, a catheter was placed in the left carotid artery for continuous infusion of (in mm: 200 Na+, 100 CO32−, 5 K+ and 5 Cl−) at a rate of 0.30 ml h−1 to correct the systemic acid–base balance in isoflurane-anaesthetized mice. After ligating the pancreatic and biliary ducts, about 1 cm of the proximal duodenum was perfused for the experiments. A small polyethylene tube (PE100; polyethylene, inner diameter 1 mm) with a distal flange was advanced to the duodenal bulb, and secured by a ligature, and another PE200 (polyethylene; inner diameter 2 mm) flanged tube was secured in the distal end of the perfused segment by ligature to allow fluid drainage. A similar procedure was performed in the colon, with the input tube 2–2.5 cm distal to the caecocolonic junction and the exit tube approximately 2 cm proximal to the anus. The isolated segments with an intact blood supply were gently flushed and then perfused (Perfusor compact; Braun) at a rate of 15 ml h−1 with 154 mm NaCl. Effluents from the isolated segment were visually free of bile and blood throughout all experiments. Animals were maintained at 37°C using a heating pad controlled by a rectal thermistor probe. Mice were killed at the end of the experiment by cervical dislocation while still under anaesthesia.
Measurement of duodenal or colonic HCO3− secretion in vivo
The rate of luminal alkalinization was determined by back titration of the perfusate to pH 5.0 with 2 mm HCl by use of pH-stat equipment (PHM82 Standard pH meter, Radiometer, Copenhagen, Denmark) as described (Singh et al. 2008). HCO3− production was determined for each 10 min period and rates of luminal alkalinization are expressed as micromoles of base secreted per centimetre of intestine per hour (μmol cm−1 h−1).
Intestinal brush border membrane preparation and Western blot analysis
All processes were done at 4°C. After thoroughly rinsing with phosphate-buffered saline (PBS), intestinal segments were cut open longitudinally, the mucosa scraped with a glass slide, and mucosal scrapings suspended in 2 ml of buffer A (270 mm mannitol, 12 mm Tris, 16 mm Hepes, 1 mm EGTA, 1.006 mm CaCl2, pH 7.4, 40 μg ml−1 phenylmethylsulfonyl fluoride (PMSF), 20 μg ml−1 leupeptin, 20 μg ml−1 pepstatin A, 20 μg ml−1 antipain, 1 mm dithiothreitol (DTT), 4 mm benzamidine) and homogenized gently by thorough pottering (at least 20 up and down strokes with a rotating potter (Potter S, Braun Melsungen, Germany), 1100 U min−1 rotating speed). The volume was made up to 12 ml with buffer A and the homogenate was centrifuged at 2000 g for 10 min. The supernatant was collected and 1 m MgCl2 added to a final concentration of 10 mm with 15 min of mixing before centrifuging at 3000 g for 15 min. Then, the supernatant was centrifuged at 30,000 g for 30 min. The pellet was suspended in buffer B (270 mm mannitol, 12 mm Tris, 16 mm Hepes, pH 7.4, 40 μg ml−1 PMSF, 20 μg ml−1 leupeptin, 20 μg ml−1 pepstatin A, 20 μg ml−1 antipain, 1 mm DTT, 4 mm benzamidine) and the MgCl2 precipitation and centrifugation steps repeated. The supernatant was centrifuged at 30,000 g for 30 min. The pellet was resuspended in TN buffer (50 mm Tris-Cl, 150 mm NaCl, pH 7.4, 40 μg ml−1 PMSF, 20 μg ml−1 leupeptin, 20 μg ml−1 pepstatin A, 20 μg ml−1 antipain, 1 mm DTT, 4 mm benzamidine) and homogenized using a 25G needle to produce the brush border membrane (BBM) fraction used for SDS-PAGE. One hundred micrograms of BBM was denatured in sample buffer at 65°C for 10 min and resolved on a 10% SDS-polyacrylamide gel, transferred to PVDF membrane (GE Healthcare Life Sciences). Blots were blocked with 5% skimmed milk and first incubated with rat anti-CFTR (3G11 kindly provided by the CFTR Consortium; 1:500, http://cftrfolding.org/, generated against the first nucleotide binding domain) or rabbit anti-β-actin antibody (Abcam, Cambridge, MA, USA;1:10,000) overnight at 4°C followed by horseradish peroxidase (HRP)-conjugated anti-rat antibody (KPL; 1:5,000, KPL Inc. Gaithersburg, Maryland, USA) or anti-rabbit antibody (KPL; 1:10,000) for 1 h at room temperature. Blots were developed using ECL (GE Healthcare Life Sciences).
Assessment of SPAK phosphorylation in vivo
In vivo perfusion was performed as described above. Mice in the control groups were luminally perfused with isotonic saline for 40 min. The experimental groups of mice were perfused with saline for 20 min, followed by either isotonic sodium gluconate or saline containing 10 μm FSK. After killing, the perfused segment was excised, and the mucosa scraped, homogenized, lysed in SDS buffer, heated at 95°C for 5 min, centrifuged at 15,000 rpm for 15 min to remove cellular debris, and stored at −80°C until further use. Western analysis was performed as described above, using rabbit anti-SPAK (Cell Signaling Technology, Inc., Danvers, MA, USA, no. 2281; 1:500), rabbit anti-β-actin antibody (Abcam; 1:10,000) and rabbit anti-pSPAK/anti-pOSR1 (1:5000).
Reagents
Tetrodotoxin was purchased from Biotrend Chemicals AG (Wangen, Switzerland), forskolin from Alexis Biochemicals (Lörrach, Germany), and other reagents from Sigma-Aldrich (Deisenhofen, Germany), or the suppliers given in the text. GlyH-101 was a kind gift of Alan Verkman (University of California San Francisco).
Statistics
All results were expressed as the mean ± SEM. ‘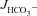 ’ and ‘Isc’ represent the mean value of basal HCO3− secretion and Isc, respectively, in either luminal NaCl or sodium gluconate in the 3–5 min before the administration of FSK. ‘
’ and ‘Isc’ represent the mean value of basal HCO3− secretion and Isc, respectively, in either luminal NaCl or sodium gluconate in the 3–5 min before the administration of FSK. ‘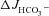 ’ and ‘ΔIsc’ represent, for each experiment, the maximal response after the secretagogue averaged over 3–5 min minus the basal value (i.e. the net stimulated murine HCO3− secretion or Isc). Because the time course of stimulation may differ from one experiment to the next, and because secretory rates may decline after a maximal response, the maximal value of the averaged time courses may differ from the calculated
’ and ‘ΔIsc’ represent, for each experiment, the maximal response after the secretagogue averaged over 3–5 min minus the basal value (i.e. the net stimulated murine HCO3− secretion or Isc). Because the time course of stimulation may differ from one experiment to the next, and because secretory rates may decline after a maximal response, the maximal value of the averaged time courses may differ from the calculated 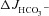 or the ΔIsc values in the bar graphs. Data were analysed by Student's t test for paired samples or by ANOVA with post hoc analysis for multiple comparisons. P < 0.05 was considered to denote statistical significance.
or the ΔIsc values in the bar graphs. Data were analysed by Student's t test for paired samples or by ANOVA with post hoc analysis for multiple comparisons. P < 0.05 was considered to denote statistical significance.
Results
Western blot analysis of CFTR protein
Western blot analysis of highly purified murine small intestinal BBM revealed that the amount of mature fully glycosylated band C protein in F508del BBM was approximately 5–10% of that found in WT BBM (Fig. 1A); no band C was detected in CFTR KO BBM (Fig. 1B). Figure 1 displays representative Western blots of pooled BBM from three mice in each group. The experiment was repeated several times with similar results. However, due to weak expression of mature fully glycosylated F508del protein, maximal loading of the gel with protein (100 μg) was necessary for its detection, which caused the bands for the loading controls (β-actin, villin) to become saturated even with very short exposure times. Precise quantification and comparison of band C between WT and F508del BBM was therefore not attempted.
Figure 1. Western blot analysis of isolated brush border membranes (BBM) for CFTR expression.
A, analysis of highly enriched small intestinal BBM from WT and F508del littermates, probed with an anti-CFTR antibody. The F508del BBM showed ∼7% of the band C concentration seen in the WT (left). Exact quantitation was not possible because maximal loading of gels with protein (100 μg) was necessary to make the faint band visible; this amount of protein caused the β-actin band to saturate. Interestingly, there is also some band B present in the BBM. This may represent alternative trafficking of CFTR to the BBM. B, in CFTR KO mice, no band B or band C was detected. Pooled BBM from 3 different mice was used for each lane. In each lane 100 μg protein was loaded.
Basal HCO3− secretion ( 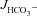 ), HCO3− secretory response to forskolin (FSK) (
), HCO3− secretory response to forskolin (FSK) ( 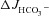 ), basal Isc, FSK-induced ΔIsc, and tissue resistance in F508del, CFTR KO and WT duodenal mucosa in vitro
), basal Isc, FSK-induced ΔIsc, and tissue resistance in F508del, CFTR KO and WT duodenal mucosa in vitro
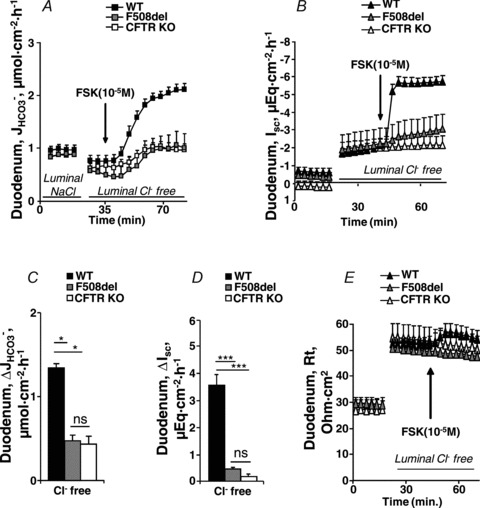
The basal HCO3− secretory rate (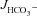 ) was not significantly different in isolated duodenal mucosa from WT and F508del, but was reduced in CFTR KO mucosa (in μmol cm−2 h−1: WT: 1.41 ± 0.12, F508del: 1.17 ± 0.28 and CFTR KO: 1.00 ± 0.22, P < 0.05, Fig. 2A). In contrast, the FSK-stimulated HCO3− secretory response (
) was not significantly different in isolated duodenal mucosa from WT and F508del, but was reduced in CFTR KO mucosa (in μmol cm−2 h−1: WT: 1.41 ± 0.12, F508del: 1.17 ± 0.28 and CFTR KO: 1.00 ± 0.22, P < 0.05, Fig. 2A). In contrast, the FSK-stimulated HCO3− secretory response (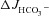 ) was significantly higher in F508del duodenum than in CFTR KO duodenum, but significantly lower than that in WT mucosa (Fig. 2A and C).
) was significantly higher in F508del duodenum than in CFTR KO duodenum, but significantly lower than that in WT mucosa (Fig. 2A and C).
Figure 2. Basal and FSK-stimulated 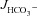 , Isc and resistance (Rt) in duodenal mucosa.
, Isc and resistance (Rt) in duodenal mucosa.
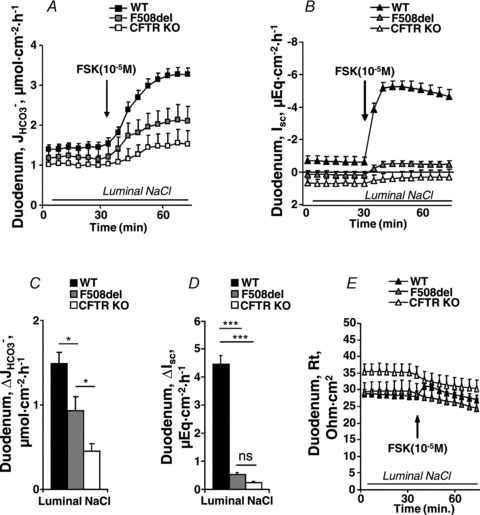
Time course of 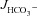 (A), Isc (B) and Rt (E) in duodenal mucosa. C and D, FSK (10−5
m)) -induced
(A), Isc (B) and Rt (E) in duodenal mucosa. C and D, FSK (10−5
m)) -induced 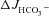 and ΔIsc in duodenal mucosa of the three different genotypes. FSK-induced ΔIsc was highly significantly different between WT and the two CF genotypes, as well as different between F508del and CFTR KO duodenum. See text for further information about basal Isc. ***P < 0.001, **P < 0.01, *P < 0.05; ns, no statistical difference; n= 5–11.
and ΔIsc in duodenal mucosa of the three different genotypes. FSK-induced ΔIsc was highly significantly different between WT and the two CF genotypes, as well as different between F508del and CFTR KO duodenum. See text for further information about basal Isc. ***P < 0.001, **P < 0.01, *P < 0.05; ns, no statistical difference; n= 5–11.
Calculated basal Isc was less negative (even slightly positive) in F508del and CFTR KO duodenum compared to WT (in μEq cm−2: F508del: 0.15 ± 0.27 and CFTR KO: 0.68 ± 0.27 vs. WT: −0.65 ± 0.32, P < 0.05, Fig. 2B), indicating a virtual lack of CFTR-mediated anion secretion in the basal state in the former epithelia, but substantial spontaneous CFTR activity in WT mucosa, despite chemical denervation and inhibition of endogenous prostaglandin synthesis. Furthermore an FSK-induced Isc response (ΔIsc) was not significantly different from zero in CFTR KO duodenum, whereas there was a small ΔIsc in F508del duodenal mucosa and a much greater one in WT mucosa (Fig. 2B and D). Tissue resistance Rt was highest in CFTR KO duodenal mucosa in the basal state (Fig. 2E), whereas a transient FSK-induced increase in Rt (which has been attributed to a collapse of the lateral spaces during secretion (Gawenis et al. 2004) was observed only in WT mucosa.
Because the F508del mutant has a gating defect in addition to reduced membrane stability (Dalemans et al. 1991), we added the CFTR potentiator genistein (10 μm) together with FSK, to isolated duodenal mucosa of F508del mutant and WT littermates (Yang et al. 2003, Yu et al. 2011). The subsequent FSK+genistein-induced 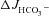 was higher than the FSK-induced
was higher than the FSK-induced 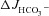 by an absolute rate of 0.40 ± 0.08 μEq cm−2 (ns), while the FSK+genistein-induced
by an absolute rate of 0.40 ± 0.08 μEq cm−2 (ns), while the FSK+genistein-induced 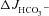 in the WT mucosa was enhanced to a similar degree (Suppl. Fig. 1). Given the fact that the effects of genistein and FSK on F508del-induced ΔIsc were not more pronounced than on WT ΔIsc, and that genistein may act through multiple mechanisms to activate CFTR (Tuo et al. 2009, 2011), this line of experiments was not pursued further. However, the experiments show that a combination of agonists stimulate HCO3− secretion in F508del intestinal mucosa even further than a maximal concentration of a cAMP agonist.
in the WT mucosa was enhanced to a similar degree (Suppl. Fig. 1). Given the fact that the effects of genistein and FSK on F508del-induced ΔIsc were not more pronounced than on WT ΔIsc, and that genistein may act through multiple mechanisms to activate CFTR (Tuo et al. 2009, 2011), this line of experiments was not pursued further. However, the experiments show that a combination of agonists stimulate HCO3− secretion in F508del intestinal mucosa even further than a maximal concentration of a cAMP agonist.
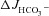 , ΔIsc and tissue resistance in F508del, CFTR KO and WT mid-colonic mucosa in vitro
, ΔIsc and tissue resistance in F508del, CFTR KO and WT mid-colonic mucosa in vitro
The next question to address was whether a HCO3− secretory response to FSK was also seen in the colon of F508del mice. Murine colon displays very low 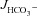 in the proximal segment, due to high NHE3 and low DRA (Slc26a3) BBM expression (Talbot & Lytle, 2010; Xiao et al. 2012), but high
in the proximal segment, due to high NHE3 and low DRA (Slc26a3) BBM expression (Talbot & Lytle, 2010; Xiao et al. 2012), but high 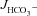 in the mid- and distal colon (Xiao et al. 2012). We therefore used the mid-colon (starting 2 cm after the caecocolonic junction and ending approximately 2 cm proximal to the anus) for our studies.
in the mid- and distal colon (Xiao et al. 2012). We therefore used the mid-colon (starting 2 cm after the caecocolonic junction and ending approximately 2 cm proximal to the anus) for our studies.
Figure 3A shows the high basal 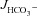 of mid-colonic mucosa for all three genotypes (in μmol cm−2 h−1: WT: 4.45 ± 0.19, F508del: 4.35 ± 0.36 and CFTR KO: 3.60 ± 0.40) with a further significant increase after FSK application. Although the time course of
of mid-colonic mucosa for all three genotypes (in μmol cm−2 h−1: WT: 4.45 ± 0.19, F508del: 4.35 ± 0.36 and CFTR KO: 3.60 ± 0.40) with a further significant increase after FSK application. Although the time course of 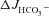 increase after FSK was significantly slower in F508del than WT mucosa, the
increase after FSK was significantly slower in F508del than WT mucosa, the 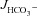 was not different 30 min after FSK addition in the F508del and WT mucosa, but was significantly lower in the CFTR KO colonic mucosa (Fig. 3A, P < 0.05). However, the relative
was not different 30 min after FSK addition in the F508del and WT mucosa, but was significantly lower in the CFTR KO colonic mucosa (Fig. 3A, P < 0.05). However, the relative 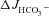 response expressed as a percentage of the basal rate was not significantly different in the three genotypes (Fig. 3B).
response expressed as a percentage of the basal rate was not significantly different in the three genotypes (Fig. 3B).
Figure 3. Basal and FSK-stimulated 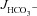 , Isc and resistance (Rt) in mid-colonic mucosa.
, Isc and resistance (Rt) in mid-colonic mucosa.
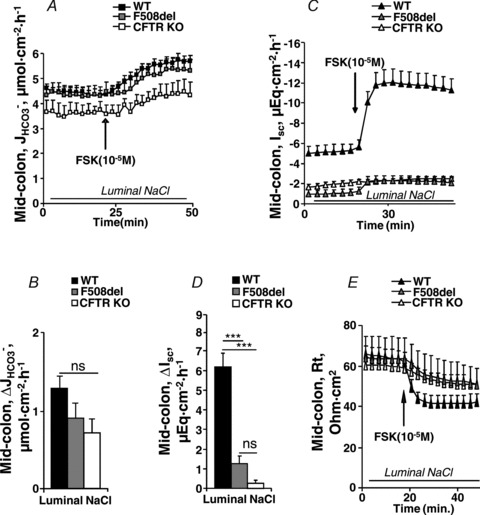
Effects of FSK (10−5
m) on the time course of 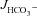 (A), Isc (B) and Rt (E) in isolated mid-colonic mucosa. C and D, individual secretory responses in
(A), Isc (B) and Rt (E) in isolated mid-colonic mucosa. C and D, individual secretory responses in 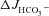 and ΔIsc, for the three different genotypes. In mid-colonic mucosa, the absolute magnitude of FSK-induced
and ΔIsc, for the three different genotypes. In mid-colonic mucosa, the absolute magnitude of FSK-induced 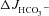 was not significantly different between each genotype. FSK-induced ΔIsc was highly significantly different between WT and the two CF genotypes. See text for further information about basal Isc. ***P < 0.001; ns, no statistical difference; n= 5–11.
was not significantly different between each genotype. FSK-induced ΔIsc was highly significantly different between WT and the two CF genotypes. See text for further information about basal Isc. ***P < 0.001; ns, no statistical difference; n= 5–11.
Basal Isc was markedly more negative in WT compared to F508del and CFTR KO mid-colonic mucosa (in μEq cm−2: WT: −5.20 ± 0.73, F508del: −1.03 ± 0.35 and CFTR KO: −1.93 ± 0.30), suggesting CFTR-mediated electrogenic anion secretion despite the presence of indomethacin and TTX (Fig. 3C, P < 0.001). FSK elicited a strong ΔIsc in WT mucosa, a small ΔIsc in F508del mucosa, and no response in CFTR KO mucosa (Fig. 3B and D). Thus, although we could not directly quantify the F508del CFTR mutant protein in the colonic BBM (less mucosa and fewer enrichment factors for colonic vs. small intestinal BBM prevented unequivocal detection of the mutant protein in colonic preparations), functional evidence for F508del membrane expression was obtained in the colonic mucosa of F508del mice similar to results in the small intestine.
The tissue resistance of colonic mucosa was larger than that of duodenal mucosa, and was not different between the three genotypes (Fig. 3E). It decreased rapidly upon FSK stimulation in the WT mucosa, consistent with the opening of an apical conductance in a relatively ‘tight’ epithelium, and very slowly and to an overall lesser degree in the other genotypes.
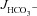 and FSK-induced
and FSK-induced 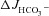 across F508del, CFTR KO and WT duodenum and colon in vivo
across F508del, CFTR KO and WT duodenum and colon in vivo
We next measured 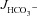 in luminally perfused duodenum and mid-colon before and after FSK perfusion in anaesthetized mice. In the duodenum, basal
in luminally perfused duodenum and mid-colon before and after FSK perfusion in anaesthetized mice. In the duodenum, basal 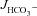 prior to FSK stimulation was significantly lower in CFTR KO compared to WT and F508del mucosa (in μmol cm−1 h−1: WT: 5.26 ± 0.54, F508del: 5.10 ± 0.58 and CFTR KO: 3.97 ± 0.32, Fig. 4A, P < 0.05), whereas WT and F508del mucosa were not statistically different. In contrast, FSK-stimulated
prior to FSK stimulation was significantly lower in CFTR KO compared to WT and F508del mucosa (in μmol cm−1 h−1: WT: 5.26 ± 0.54, F508del: 5.10 ± 0.58 and CFTR KO: 3.97 ± 0.32, Fig. 4A, P < 0.05), whereas WT and F508del mucosa were not statistically different. In contrast, FSK-stimulated 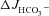 was significantly higher in F508del duodenum than in CFTR KO duodenum, but significantly lower than in WT (Fig. 4A and B).
was significantly higher in F508del duodenum than in CFTR KO duodenum, but significantly lower than in WT (Fig. 4A and B).
Figure 4. Basal and FSK-stimulated duodenal and mid-colonic HCO3− secretion (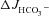 ) in anaesthetized mice.
) in anaesthetized mice.
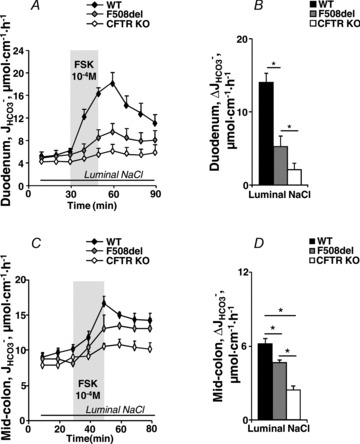
Time course (A and C) and magnitude of HCO3− secretory response (B and D) in luminally perfused duodenum (A and B) and mid-colon (C and D) of anaesthetized WT, F508del and CFTR KO mice. FSK-induced 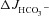 was significantly different in luminally perfused duodenum and mid-colon between each genotype. *P < 0.05; n= 5.
was significantly different in luminally perfused duodenum and mid-colon between each genotype. *P < 0.05; n= 5.
Figure 4C shows the high 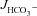 in the mid-colonic mucosa of all three genotypes in vivo (in μmol cm−1 h−1: WT: 9.58 ± 0.48, F508del: 8.56 ± 0.90 and CFTR KO: 7.82 ± 0.64), with a further significant increase after FSK application (Fig. 4C and D). Again, the FSK-induced
in the mid-colonic mucosa of all three genotypes in vivo (in μmol cm−1 h−1: WT: 9.58 ± 0.48, F508del: 8.56 ± 0.90 and CFTR KO: 7.82 ± 0.64), with a further significant increase after FSK application (Fig. 4C and D). Again, the FSK-induced 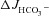 in F508del mid-colon was significantly higher than that in CFTR KO colon, but significantly lower than in WT colon.
in F508del mid-colon was significantly higher than that in CFTR KO colon, but significantly lower than in WT colon.
Effect of luminal Cl− on FSK-induced 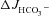 , ΔIsc and Rt in WT, F508del and CFTR KO duodenal mucosa
, ΔIsc and Rt in WT, F508del and CFTR KO duodenal mucosa
We next explored the potential involvement of apical Cl−/HCO3− exchangers in the elevated HCO3− secretion by F508del duodenal mucosa. Replacement of Cl− with gluconate in the luminal bath significantly reduced basal 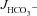 similarly in all three genotypes (in μmol cm−2 h−1: WT: 1.17 ± 0.25 to 0.76 ± 0.20, F508del: 0.87 ± 0.16 to 0.52 ± 0.20 and CFTR KO: 0.94 ± 0.12 to 0.69 ± 0.15, P < 0.05) (Fig. 5A). Subsequent stimulation by FSK in the absence of luminal Cl− did not significantly reduce
similarly in all three genotypes (in μmol cm−2 h−1: WT: 1.17 ± 0.25 to 0.76 ± 0.20, F508del: 0.87 ± 0.16 to 0.52 ± 0.20 and CFTR KO: 0.94 ± 0.12 to 0.69 ± 0.15, P < 0.05) (Fig. 5A). Subsequent stimulation by FSK in the absence of luminal Cl− did not significantly reduce 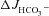 in WT mucosa compared to that in the presence of luminal Cl− (see Figs 2C and Fig. 5C), but abolished the difference in
in WT mucosa compared to that in the presence of luminal Cl− (see Figs 2C and Fig. 5C), but abolished the difference in 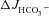 between F508del and CFTR KO mucosa (Fig. 5C). This suggests that the HCO3− secretory response in the F508del mucosa is largely mediated by luminal Cl−/HCO3− exchange, whereas the WT duodenum is capable of Cl−-independent HCO3− secretion.
between F508del and CFTR KO mucosa (Fig. 5C). This suggests that the HCO3− secretory response in the F508del mucosa is largely mediated by luminal Cl−/HCO3− exchange, whereas the WT duodenum is capable of Cl−-independent HCO3− secretion.
Figure 5. Luminal Cl− removal abolished the augmentation of FSK-induced 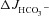 by F508del-CFTR membrane expression in duodenal mucosa.
by F508del-CFTR membrane expression in duodenal mucosa.
Replacement of Cl− in the luminal bath by gluconate caused a slight decrease in the basal 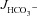 in each genotype (A), and reduced the
in each genotype (A), and reduced the 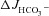 in the F508del mucosa to the
in the F508del mucosa to the 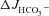 values seen in the CFTR KO mucosa (A and C). These data suggest that FSK (10−5
m) elicits a robust
values seen in the CFTR KO mucosa (A and C). These data suggest that FSK (10−5
m) elicits a robust 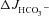 independently of luminal Cl−/HCO3− exchange in WT mucosa, whereas the augmentation of FSK-induced
independently of luminal Cl−/HCO3− exchange in WT mucosa, whereas the augmentation of FSK-induced 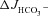 by F508del expression requires luminal Cl−/HCO3− exchange. Basal Isc (B) and Rt (E) increased significantly in all three genotypes after Cl− replacement by gluconate, and the Isc response to FSK (D) was not different from that seen in the presence of luminal Cl− (Fig. 2). ***P < 0.001, *P < 0.05; ns, no statistical difference; n= 5–9.
by F508del expression requires luminal Cl−/HCO3− exchange. Basal Isc (B) and Rt (E) increased significantly in all three genotypes after Cl− replacement by gluconate, and the Isc response to FSK (D) was not different from that seen in the presence of luminal Cl− (Fig. 2). ***P < 0.001, *P < 0.05; ns, no statistical difference; n= 5–9.
The basal Isc of CFTR KO duodenal mucosa in the presence of luminal Cl− was significantly less negative than in F508del and WT mucosa (in μEq cm−2: CFTR KO: 0.19 ± 0.33, F508del: −0.41 ± 0.17 and WT: −0.52 ± 0.23, P < 0.05) (Fig. 5B), but increased to similar values after the removal of luminal Cl− (in μEq cm−2: WT: −1.89 ± 0.38, F508del: −2.06 ± 0.73 and CFTR KO: −1.87 ± 0.38) (Fig. 5C), demonstrating that the increase in Isc after luminal Cl− removal is not primarily due to an increased electrochemical gradient for CFTR-mediated Cl− efflux, but may be a diffusion potential elicited by the charge selectivity of tight junctions. The subsequent FSK-induced ΔIsc did not differ significantly compared to the ΔIsc in the presence of luminal Cl− and was significantly higher in WT than the other two genotypes (Figs 2D and 5D).
Tissue resistance (Rt) increased significantly upon luminal Cl− removal to similar levels in all three genotypes, and the subsequent transient increase in Rt in response to FSK was even slightly enhanced in the WT tissue compared to Cl− containing solutions (Figs 2E and 5E).
Effect of luminal Cl− on FSK-induced 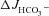 , ΔIsc and Rt in WT, F508del and CFTR KO colonic mucosa
, ΔIsc and Rt in WT, F508del and CFTR KO colonic mucosa
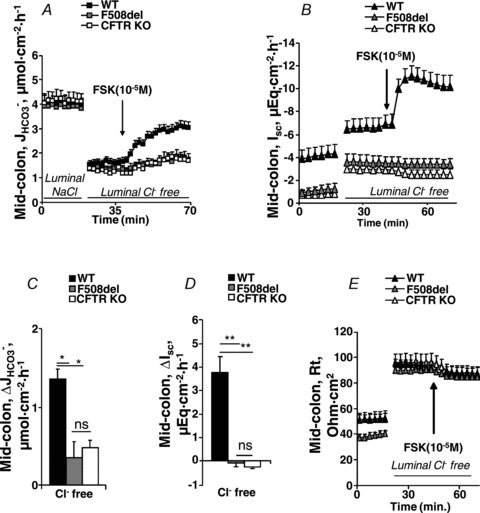
In mid-colonic mucosa, the substitution of luminal Cl− with gluconate resulted in a dramatic decrease in basal 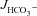 in all three genotypes, consistent with high rates of luminal alkalinization mediated by apical Cl−/HCO3− exchange (in μmol cm−2 h−1: WT: 4.00 ± 0.28 to 1.61 ± 0.16, F508del: 3.90 ± 0.22 to 1.34 ± 0.16 and CFTR KO: 4.13 ± 0.35 to 1.28 ± 0.17) (Fig. 6A). These high luminal alkalinization rates are not dependent on CFTR expression, since both
in all three genotypes, consistent with high rates of luminal alkalinization mediated by apical Cl−/HCO3− exchange (in μmol cm−2 h−1: WT: 4.00 ± 0.28 to 1.61 ± 0.16, F508del: 3.90 ± 0.22 to 1.34 ± 0.16 and CFTR KO: 4.13 ± 0.35 to 1.28 ± 0.17) (Fig. 6A). These high luminal alkalinization rates are not dependent on CFTR expression, since both 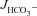 and the decrease in
and the decrease in 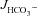 after removal of luminal Cl− were identical in all three genotypes. Cl− removal did not significantly influence subsequent FSK-induced
after removal of luminal Cl− were identical in all three genotypes. Cl− removal did not significantly influence subsequent FSK-induced 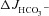 responses in either WT or CFTR KO mucosa, but strongly reduced
responses in either WT or CFTR KO mucosa, but strongly reduced  in the F508del mucosa to that seen in CFTR KO mucosa (Figs 3A and C and 6A and C). This suggests that similar to duodendal mucosa, F508del expression in mid-colonic mucosa elicits an FSK-induced increase in Cl−/HCO3− exchange.
in the F508del mucosa to that seen in CFTR KO mucosa (Figs 3A and C and 6A and C). This suggests that similar to duodendal mucosa, F508del expression in mid-colonic mucosa elicits an FSK-induced increase in Cl−/HCO3− exchange.
Figure 6. Luminal Cl− removal abolished the augmentation of FSK-induced 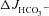 by F508del CFTR membrane expression in mid-colonic mucosa.
by F508del CFTR membrane expression in mid-colonic mucosa.
Replacement of Cl− in the luminal bath by gluconate caused a strong decrease in the basal  in each genotype (A), and reduced the
in each genotype (A), and reduced the 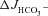 in the F508del mucosa to the
in the F508del mucosa to the 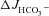 values seen in the CFTR KO mucosa (A and C). Basal Isc (B) and Rt (E) increased significantly in all three genotypes, and the Isc response to FSK (D) was significantly lower in WT and F508del mucosa compared to that in the in the presence of luminal Cl− (Fig. 3). **P < 0.01, *P < 0.05; ns, no statistical difference; n= 5–9.
values seen in the CFTR KO mucosa (A and C). Basal Isc (B) and Rt (E) increased significantly in all three genotypes, and the Isc response to FSK (D) was significantly lower in WT and F508del mucosa compared to that in the in the presence of luminal Cl− (Fig. 3). **P < 0.01, *P < 0.05; ns, no statistical difference; n= 5–9.
Luminal Cl− removal resulted in a strong ΔIsc increase in colonic mucosa from all three genotypes (in μEq cm−2: WT: −3.01 ± 0.57 to −5.27 ± 0.63, F508del: −0.65 ± 0.20 to −2.85 ± 0.46 and CFTR KO: −0.78 ± 0.34 to −2.97 ± 0.34, P < 0.01) (Fig. 6B). Subsequent FSK-elicited ΔIsc was less negative in all genotypes than in the presence of luminal Cl− (Figs 3D and 6D).
Tissue resistance (Rt) increased significantly upon luminal Cl− removal in all three genotypes and FSK application resulted in a small decrease in Rt in all three genotypes (ns) (Fig. 6E).
NHE3 inhibition abolished the FSK-mediated small increase in luminal alkalinisation in CFTR KO mucosa
In CFTR KO duodenal as well as mid-colonic mucosa, FSK application generated a small, but significant, 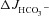 without any change in Isc, suggesting that this
without any change in Isc, suggesting that this 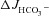 response is unlikely to be mediated by an anion conductance (Figs 2 and 3). Inhibition of the apical Na+/H+ exchanger NHE3 increases duodenal HCO3− secretion in vivo and in vitro (Clarke et al. 2001; Furukawa et al. 2004; Singh et al. 2010). Preincubation of duodenal and mid-colonic mucosa with 20 μm of the NHE3 inhibitor S1611 completely abolished the FSK-induced
response is unlikely to be mediated by an anion conductance (Figs 2 and 3). Inhibition of the apical Na+/H+ exchanger NHE3 increases duodenal HCO3− secretion in vivo and in vitro (Clarke et al. 2001; Furukawa et al. 2004; Singh et al. 2010). Preincubation of duodenal and mid-colonic mucosa with 20 μm of the NHE3 inhibitor S1611 completely abolished the FSK-induced 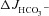 in CFTR KO mucosa, and reduced it in WT and F508del mucosa. The absolute magnitude of decrease was similar in all genotypes (Fig. 7A and B).
in CFTR KO mucosa, and reduced it in WT and F508del mucosa. The absolute magnitude of decrease was similar in all genotypes (Fig. 7A and B).
Figure 7. FSK-induced 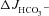 is reduced by NHE3 inhibition and abolished by pharmacological inhibition of CFTR in F508del duodenal and colonic mucosa.
is reduced by NHE3 inhibition and abolished by pharmacological inhibition of CFTR in F508del duodenal and colonic mucosa.
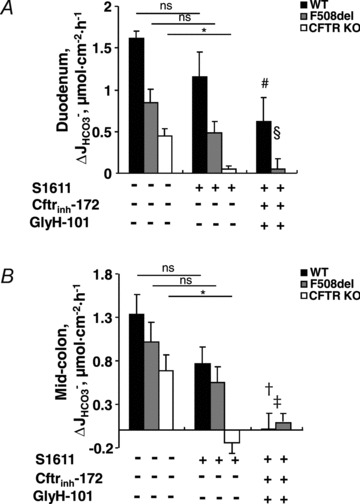
Isolated duodenal (A) and mid-colonic (B) mucosa were preincubated with S1611 (20 μm) (middle column) or with S1611 plus the CFTR inhibitors CFTRinh-172 (20 μm) and GlyH-101 (10 μm) (right column). In the duodenal mucosa, S1611 preincubation alone reduced the effect of FSK on 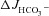 by approximately 25–35% in WT and F508del mucosa, and completely inhibited
by approximately 25–35% in WT and F508del mucosa, and completely inhibited 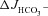 in the CFTR KO mucosa (A, middle column). The CFTR inhibitors decreased the S1611-insensitive FSK-induced
in the CFTR KO mucosa (A, middle column). The CFTR inhibitors decreased the S1611-insensitive FSK-induced 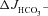 by about 40% in the WT, and abolished
by about 40% in the WT, and abolished 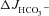 in the F508del mucosa (A, right column). Qualitative similar findings were obtained in the mid-colon (B). This demonstrates that F508del-CFTR channel activity is required for the FSK-induced Cl−/HCO3− exchange in F508del mucosa. ns, no statistical difference; *P < 0.05 vs. CFTR KO (duodenum or mid-colon) without inhibitors, #P < 0.01 vs. WT duodenum without inhibitors, §P < 0.01 vs. F508del duodenum without inhibitors, †P < 0.05 vs. WT mid-colon without inhibitors, ‡P < 0.05 vs. F508del mid-colon without inhibitors; n= 3–5.
in the F508del mucosa (A, right column). Qualitative similar findings were obtained in the mid-colon (B). This demonstrates that F508del-CFTR channel activity is required for the FSK-induced Cl−/HCO3− exchange in F508del mucosa. ns, no statistical difference; *P < 0.05 vs. CFTR KO (duodenum or mid-colon) without inhibitors, #P < 0.01 vs. WT duodenum without inhibitors, §P < 0.01 vs. F508del duodenum without inhibitors, †P < 0.05 vs. WT mid-colon without inhibitors, ‡P < 0.05 vs. F508del mid-colon without inhibitors; n= 3–5.
Can F508del-CFTR-enhanced HCO3− secretion be influenced by CFTR inhibitors?
We next tested whether HCO3− secretion in F508del mutant intestine is sensitive to CFTR inhibitors. Figure 7A and B displays forskolin-induced 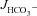 in WT, F508del and CFTR KO duodenal and mid-colonic mucosa without inhibitors (left bars), after NHE3 inhibition with 20 μm S1611 (middle bars), and after addition of the CFTR inhibitors CFTRinh-172 and GlyH-101 to the luminal bath in the continued presence of S1611 (right bars). Any residual FSK-induced
in WT, F508del and CFTR KO duodenal and mid-colonic mucosa without inhibitors (left bars), after NHE3 inhibition with 20 μm S1611 (middle bars), and after addition of the CFTR inhibitors CFTRinh-172 and GlyH-101 to the luminal bath in the continued presence of S1611 (right bars). Any residual FSK-induced 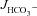 in CFTR KO mucosa was abolished by S1611 (Fig. 7A and B, middle bars). Moreover, any residual S1611-insensitive
in CFTR KO mucosa was abolished by S1611 (Fig. 7A and B, middle bars). Moreover, any residual S1611-insensitive 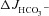 in F508del duodenal, WT mid-colonic and F508del mid-colonic mucosa was abolished by the CFTR inhibitors (Fig. 7A and B, right bars). Since the CFTR inhibitors had little effect on basal
in F508del duodenal, WT mid-colonic and F508del mid-colonic mucosa was abolished by the CFTR inhibitors (Fig. 7A and B, right bars). Since the CFTR inhibitors had little effect on basal 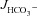 (even in colonic mucosa, where the high
(even in colonic mucosa, where the high 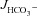 rates are largely Cl−/HCO3− exchange mediated), these results suggest that the CFTR inhibitors act predominantly by preventing F508del-CFTR acting as a Cl− recycling pathway for Cl−/HCO3− exchange. To also test this hypothesis in vivo, we compared the FSK-induced
rates are largely Cl−/HCO3− exchange mediated), these results suggest that the CFTR inhibitors act predominantly by preventing F508del-CFTR acting as a Cl− recycling pathway for Cl−/HCO3− exchange. To also test this hypothesis in vivo, we compared the FSK-induced 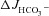 in the colon of anaesthetized F508del mice in the presence of CFTR inhibitors with that seen in the CFTR KO colon (of note, S1611 cannot be used in the in vivo experiments). The CFTR inhibitors reduced the magnitude of FSK-induced
in the colon of anaesthetized F508del mice in the presence of CFTR inhibitors with that seen in the CFTR KO colon (of note, S1611 cannot be used in the in vivo experiments). The CFTR inhibitors reduced the magnitude of FSK-induced 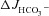 in F508del colon to that seen in CFTR KO colon (Fig. 8). These data suggest that the augmented FSK-induced HCO3− secretory response in F508del compared to CFTR KO intestine may be dependent on the channel activity of the mutant protein. The interpretation of these experiments is complicated by the fact that CFTRinh-172 and GlyH-101 are not completely specific for CFTR (Steward et al. 2011, Caputo et al. 2008; Kelly et al. 2010), nor do they fully prevent CFTR activation in intact WT intestine (see suppl. Fig. 2, and De Boeck et al. 2011).
in F508del colon to that seen in CFTR KO colon (Fig. 8). These data suggest that the augmented FSK-induced HCO3− secretory response in F508del compared to CFTR KO intestine may be dependent on the channel activity of the mutant protein. The interpretation of these experiments is complicated by the fact that CFTRinh-172 and GlyH-101 are not completely specific for CFTR (Steward et al. 2011, Caputo et al. 2008; Kelly et al. 2010), nor do they fully prevent CFTR activation in intact WT intestine (see suppl. Fig. 2, and De Boeck et al. 2011).
Figure 8. Pharmacological inhibition of CFTR abolished the augmentation of FSK-induced 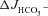 by F508del CFTR membrane expression in the colon of anaesthetized mice in vivo.
by F508del CFTR membrane expression in the colon of anaesthetized mice in vivo.
Luminal application of CFTRinh-172 (20 μm) and GlyH-101 (10 μm) reduced the FSK-induced 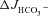 in F508del perfused mid-colon (black and shaded bars) of anaesthetized mice to the level seen in the CFTR KO colon (open bar). The FSK-induced
in F508del perfused mid-colon (black and shaded bars) of anaesthetized mice to the level seen in the CFTR KO colon (open bar). The FSK-induced 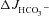 in the CFTR KO colon is due to FSK-induced NHE3 inhibition; however, a sequential application of all inhibitors, as was done in Fig. 7 in isolated mucosa, is not feasible in vivo. *P < 0.05; ns, no statistical difference; n= 3–5.
in the CFTR KO colon is due to FSK-induced NHE3 inhibition; however, a sequential application of all inhibitors, as was done in Fig. 7 in isolated mucosa, is not feasible in vivo. *P < 0.05; ns, no statistical difference; n= 3–5.
Forskolin and luminal Cl− removal induced OSR1 and SPAK phosphorylation in intestinal mucosa in vivo
One puzzling finding of this study is the fact that luminal Cl− removal had no inhibitory effect on FSK-induced 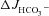 in WT mucosa, but inhibited
in WT mucosa, but inhibited 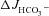 in F508del mucosa. Because this result suggests a high HCO3− permeability of CFTR in WT intestine, we investigated whether luminal Cl− removal or FSK addition activates the WNK downstream kinases OSR1 and SPAK in vivo. Western blot analysis of protein lysate from scraped duodenal mucosa after in vivo perfusion with hypotonic Cl−-free solution, performed with an antibody against the phosphorylated T-loop of OSR1 and SPAK, demonstrated time-dependent phosphorylation of OSR1 and SPAK (Fig. 9A), indicating that this signal pathway is activated in murine duodenum in vivo. We next assessed whether FSK activates SPAK/OSR1. This was indeed the case (Fig. 9B), suggesting that the lack of dependence of FSK-induced
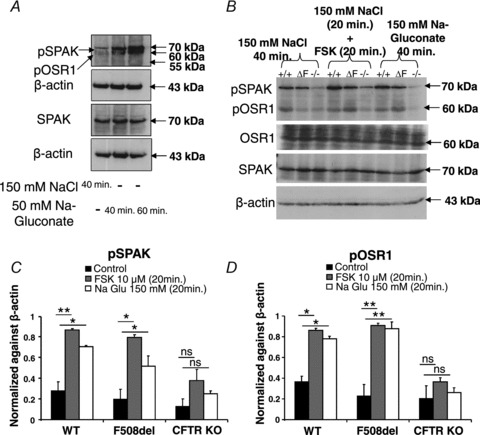
in F508del mucosa. Because this result suggests a high HCO3− permeability of CFTR in WT intestine, we investigated whether luminal Cl− removal or FSK addition activates the WNK downstream kinases OSR1 and SPAK in vivo. Western blot analysis of protein lysate from scraped duodenal mucosa after in vivo perfusion with hypotonic Cl−-free solution, performed with an antibody against the phosphorylated T-loop of OSR1 and SPAK, demonstrated time-dependent phosphorylation of OSR1 and SPAK (Fig. 9A), indicating that this signal pathway is activated in murine duodenum in vivo. We next assessed whether FSK activates SPAK/OSR1. This was indeed the case (Fig. 9B), suggesting that the lack of dependence of FSK-induced 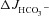 on luminal Cl− in WT intestine may be explained by strong SPAK/OSR1 phosphorylation during FSK stimulation.
on luminal Cl− in WT intestine may be explained by strong SPAK/OSR1 phosphorylation during FSK stimulation.
Figure 9. Rescue of defective WNK signalling in F508del duodenum.
A and B, SPAK/OSR1 phosphorylation was observed in response to luminal perfusion with Cl− free hypotonic solution (A), luminal Cl− removal and FSK stimulation in WT duodenum in vivo (B). Each lane represents total protein lysate from scraped proximal duodenum after in vivo luminal perfusion of one mouse with the indicated perfusate. B, representative blot of three different experiments. C and D, summary of three experiments in individual mice of the different genotypes under the indicated experimental conditions. Values were normalised to β-actin levels. There was significant stimulation of SPAK/OSR1 phosphorylation in WT and F508del, but not CFTR KO mice. The significance was calculated by ANOVA post hoc test. *P≤ 0.05 and **P≤ 0.01; ns, no statistical difference.
Finally, we assessed whether there was a difference in SPAK/OSR1 phosphorylation in WT, F508del and CFTR KO duodenum. While both FSK and luminal Cl− free perfusion quickly increased the amount of phosphorylated OSR1 and SPAK in WT duodenum, CFTR KO mucosa displayed levels of phosphorylated SPAK/OSR1 far below those seen in WT mucosa under control conditions (Fig. 9B–D). Interestingly, a significant increase in phospho-SPAK/OSR1 was also seen in F508del duodenum in response to FSK or luminal Cl− free perfusion (Fig. 9B–D). These data suggest that the low levels of Cl− conductance provided by apical membrane expression of F508del-CFTR are sufficient to restore WNK-signalling to F508del duodenal mucosa.
Discussion
In 1994, Poulsen et al. reported HCO3− permeability of CFTR in patch-clamp experiments (Poulsen et al. 1994), and similar observations were subsequently made by others (Reddy & Quinton, 2003; Tang et al. 2009). What about HCO3− transport in CFTR mutants? Some data suggest a selective loss of the CFTR HCO3− conductance in disease-causing CFTR mutants that are associated with more severe CF phenotypes (Choi et al. 2001, Reddy & Quinton, 2003) or idiopathic pancreatitis (Schneider & Saur, 2011). Other patch-clamp studies of heterogeneously expressed CFTR mutants associated with severe CF phenotypes found both reduced Cl− and reduced HCO3− permeabilities, but no change in their relationship (Tang et al. 2009). The most frequent CF mutation, F508del, has not been studied in this regard. It was therefore unknown whether F508del, when expressed at the apical plasma membrane, can mediate both Cl− and HCO3− secretion.
In order to answer this question, we made use of F508del mice on the FVB/N background, which express a small amount of mature fully glycosylated CFTR in the brush border membrane of the small intestine (French et al. 1996). We bred these mice in parallel with CFTR KO mice on the same congenic FVB/N background, and found approximately 5–10% of WT band C levels in highly enriched small intestine BBM from F508del mice. Interestingly, F508del duodenum and mid-colon, the two segments that are characterized by the highest HCO3− secretory rates in mouse intestine, both displayed significantly higher FSK-induced HCO3− secretory responses compared to CFTR KO mucosa, despite this small amount of mature CFTR protein in the apical membrane, and despite a rather low FSK-induced ΔIsc response. This suggests that even low expression of mature fully glycosylated F508del CFTR mutant protein in the apical membrane enabled the intestinal epithelium to secrete substantial amounts of HCO3− ions.
Our next step was to obtain some insight into the molecular mechanism for this F508del-augmented HCO3− secretory response. Based on the literature, three potential mechanisms are feasible: first, the activated F508del-CFTR conductance is selective for HCO3− over Cl−. Such a phenomenon has recently been reported for WT CFTR in pancreatic duct cells following activation of the WNK signalling pathway by low intracellular Cl− concentrations (Park et al. 2010). We therefore studied whether FSK activated the WNK signalling pathway in murine intestine by measuring the phosphorylation of the WNK1/4 downstream kinases SPAK and OSR1. We observed a strong induction of SPAK/OSR1 phosphorylation by luminal FSK application in murine intestine in vivo (Fig. 9). FSK activated the WNK signalling pathway to a similar degree to long-term removal of luminal Cl−, which may well be the explanation for the lack of inhibition of agonist-activated HCO3− secretion by luminal Cl− removal, observed in this paper as well as other studies, and in different epithelia and species (Ishiguro et al. 1996, Chan et al. 1996, Guba et al. 1996, Spiegel et al. 2003, Ishiguro et al. 2009). However, we found no significant difference between SPAK/OSR1 phosphorylation in WT and F508del intestine, while SPAK phosphorylation was significantly impaired in CFTR KO intestine. This suggests that WNK signalling, with all the resultant effects on transepithelial Cl− transport, is operative in epithelia that express F508del in their apical membranes. However, because the same strong SPAK/OSR1 phosphorylation was seen in WT intestine, the findings were not sufficient to explain the relatively strong enhancement of FSK-induced HCO3− secretion in F508del compared to CFTR KO intestine, despite the small amount of membrane-resident channel protein in F508del compared with WT intestinal mucosa.
A second possible explanation is that the low amount of F508del CFTR mutant protein stimulates luminal Cl−/HCO3− anion exchangers, either by direct molecular interaction or by providing an apical shunt (recycling) pathway for Cl−. We therefore replaced Cl− in the luminal bath with the impermeant anion gluconate. This manoeuvre did not significantly inhibit FSK-induced HCO3− secretion in the WT mucosa, but reduced 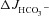 to the level seen in the complete absence of CFTR (which was found to be due to FSK inhibition of NHE3, see Fig. 7). This feature was also observed in colonic mucosa. This suggests that FSK stimulates a robust Cl−-independent HCO3− conductance in WT intestine, whereas the F508del-enhanced HCO3− secretion occurs predominantly via Cl−/HCO3− exchange (despite robust SPAK/OSR1 phosphorylation). The critical finding of this study is, therefore, that the major HCO3− exit pathways appear to differ in WT and F508del murine intestine, at least when this assumption is based on results obtained by the classic manoeuvre of Cl− removal from the luminal bath.
to the level seen in the complete absence of CFTR (which was found to be due to FSK inhibition of NHE3, see Fig. 7). This feature was also observed in colonic mucosa. This suggests that FSK stimulates a robust Cl−-independent HCO3− conductance in WT intestine, whereas the F508del-enhanced HCO3− secretion occurs predominantly via Cl−/HCO3− exchange (despite robust SPAK/OSR1 phosphorylation). The critical finding of this study is, therefore, that the major HCO3− exit pathways appear to differ in WT and F508del murine intestine, at least when this assumption is based on results obtained by the classic manoeuvre of Cl− removal from the luminal bath.
What may be the reason for an enhanced FSK-induced apical Cl−/HCO3− exchange in F508del compared to CFTR KO mucosa? There are three potential reasons for this finding. First, F508del expression, with the residual apical Cl− conductance, may result in better overall tissue integrity, including better Slc26 expression levels. Similar expression levels for Slc26a6 and Slc26a3 have previously been demonstrated in CFTR KO and WT intestine by ourselves and others (Simpson et al. 2005; Tuo et al. 2006), making it unlikely that differences would exist between F508del and CFTR KO intestine. Nevertheless, we carefully studied tissue morphology in the duodenum and colon of all three genotypes. Although fine differences in villous length and thickness, crypt depth and lamina propria cell density were seen, a blinded investigator (U.S.) was unable to classify histological specimens into the groups ‘KO, F508del and WT’. This suggests that under our breeding conditions, chronic intestinal changes appear late in the life of F508del and KO mice; such mice were not studied in the present project.
A second potential explanation is that expression of CFTR, even in mutant form, may enhance the expression and/or membrane trafficking of apical anion exchangers irrespective of CFTR channel function. Such an observation has been made in pancreatic cell lines, where heterologous expression of CFTR influenced the expression and/or activity of endogenous Cl−/HCO3− exchangers independent of CFTR channel function (Ignath et al. 2009). We therefore tested whether the relatively specific CFTR inhibitors CFTRinh-172 (Ma et al. 2002) and GlyH-101 (Muanprasat et al. 2004) inhibited duodenal and colonic HCO3− secretion to a higher degree in F508del compared to CFTR KO mucosa. Indeed, luminal CFTRinh-172 and GlyH-101 application decreased FSK-induced HCO3− secretory rates in F508del mucosa to the rates observed in CFTR KO mice. Since the effects of these agents on basal HCO3− secretory rates (which are strongly dependent on apical Cl−/HCO3− exchange, particularly in the mid-distal colon (Xiao et al. 2012), were not significant, we assume that this inhibitory effect may be more related to CFTR channel inhibition than to (direct) Slc26 anion exchange inhibition. However, interference with other pathways cannot be fully ruled out.
A third concept that best explains our findings is that Cl− recycling at the apical membrane, as a means to activate luminal Cl−/HCO3− exchange, requires CFTR channel function (Greger et al. 2001; Simpson et al. 2005; Singh et al. 2010). One may speculate that Cl− exit via CFTR, coupled to Cl− uptake in exchange for HCO3− exit via an electroneutral Cl−/HCO3− exchanger, results an equivalent rise in Isc to that of the titrated HCO3− in the luminal bath. However, the apical Cl−/HCO3− exchangers in the intestine may, in part, be electrogenic (Chernova et al. 2005; Lamprecht et al. 2005, 2006; Ohana et al. 2009; Yamaguchi et al. 2009; Alper et al. 2011). Other conductances are also activated by FSK (Sørensen et al. 2010), and the time courses of the Isc and HCO3− secretory responses may be different due to technical reasons. Thus, FSK-induced ΔIsc and 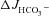 need not be numerically identical for Cl− recycling to explain enhanced intestinal epithelial HCO3− secretion. In addition, the activation of an apical anion conductance (i.e. membrane-resident F508del) may affect membrane potential, cell volume and pHi; these events in turn may affect apical and basolateral HCO3− transporters and result in an overall increase in epithelial HCO3− secretion.
need not be numerically identical for Cl− recycling to explain enhanced intestinal epithelial HCO3− secretion. In addition, the activation of an apical anion conductance (i.e. membrane-resident F508del) may affect membrane potential, cell volume and pHi; these events in turn may affect apical and basolateral HCO3− transporters and result in an overall increase in epithelial HCO3− secretion.
What may explain the observation that the magnitude of FSK-mediated 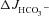 was not significantly influenced by the absence of luminal Cl− in WT mucosa, while it was reduced to levels seen in CFTR KO intestine in F508del mucosa? Our working hypothesis was that in WT mucosa, the FSK-induced activation of apical anion efflux via CFTR may cause cell shrinkage and a decrease in [Cl−]i (Valverde et al. 1993; MacLeod et al. 1994; Bachmann et al. 2003; Gawenis et al. 2004), which in turn may activate the WNK signalling pathway, resulting in the phosphorylation of the downstream kinases OSR1 and SPAK (Kahle et al. 2005; Park et al. 2010), and a change of the anion selectivity of CFTR from Cl− to HCO3− selectivity (Park et al. 2010). This would explain the Cl−-independent stimulation of
was not significantly influenced by the absence of luminal Cl− in WT mucosa, while it was reduced to levels seen in CFTR KO intestine in F508del mucosa? Our working hypothesis was that in WT mucosa, the FSK-induced activation of apical anion efflux via CFTR may cause cell shrinkage and a decrease in [Cl−]i (Valverde et al. 1993; MacLeod et al. 1994; Bachmann et al. 2003; Gawenis et al. 2004), which in turn may activate the WNK signalling pathway, resulting in the phosphorylation of the downstream kinases OSR1 and SPAK (Kahle et al. 2005; Park et al. 2010), and a change of the anion selectivity of CFTR from Cl− to HCO3− selectivity (Park et al. 2010). This would explain the Cl−-independent stimulation of 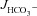 in WT mucosa, which we observed. To explain the dependence of FSK-mediated
in WT mucosa, which we observed. To explain the dependence of FSK-mediated 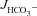 on luminal Cl− in F508del mucosa, we further speculated that differences in WNK signalling may occur between WT and F508del intestine, because we anticipated that the shrinkage and loss of [Cl−]i in F508del (and CFTR KO) epithelial cells would be insufficient to activate the WNK signalling pathway. However, this was not found to be the case for F508del mucosa, in which SPAK/OSR1 phosphorylation upon either FSK addition or luminal Cl− removal was similar to WT, whereas it was significantly attenuated in CFTR KO mucosa.
on luminal Cl− in F508del mucosa, we further speculated that differences in WNK signalling may occur between WT and F508del intestine, because we anticipated that the shrinkage and loss of [Cl−]i in F508del (and CFTR KO) epithelial cells would be insufficient to activate the WNK signalling pathway. However, this was not found to be the case for F508del mucosa, in which SPAK/OSR1 phosphorylation upon either FSK addition or luminal Cl− removal was similar to WT, whereas it was significantly attenuated in CFTR KO mucosa.
Despite substantial SPAK/OSR1 phosphorylation in F508del intestine, FSK-stimulated HCO3− secretion was nevertheless dependent on luminal Cl− and thus likely to a large part be mediated by Cl−/HCO3− exchange. This does not preclude a HCO3− permeability of F508del itself upon WNK activation, which may have simply been too small to detect, and which may become apparent when pharmacological strategies to deliver more F508del to the membrane become available. It also suggests to us that in WT intestine, luminal Cl−/HCO3− exchange is inhibited by FSK, not so much because of SPAK/OSR1 phosphorylation, but by the large CFTR-mediated HCO3− conductance (which is high, both because the channel is strongly expressed in the membrane and because SPAK/OSR1 is phosphorylated). High CFTR-mediated HCO3− conductance would reduce the driving force for HCO3− efflux via Cl−/HCO3− exchange. However, in F508del intestine, this CFTR-mediated HCO3− conductance is low (because of the small amount of F508del-CFTR protein in the membrane, irrespective of SPAK/OSR1 phosphorylation) and may not be sufficient to result in inhibition of HCO3− efflux via Cl−/HCO3− exchange. On the contrary, apical Cl−/HCO3− exchange may become activated by Cl− efflux via membrane resident F508del to provide the increased HCO3− efflux. Evidence in support of this hypothesis, although not firm proof, is a rapid decrease in pHi in microdissected duodenal villi after FSK application (Y. Qin, unpublished observations).
What may be the clinical significance of this finding? Species differences have been reported in the processing and membrane expression of F508del-CFTR, with better stability in murine than human epithelial cells (Ostedgaard et al. 2007). Thus, the use of a murine F508del model provided us with an epithelium with residual F508del-CFTR membrane expression even without the use of pharmacological agents to deliver more F508del to the plasma membrane. The better survival of the F508del mice with less organ pathology and fewer obstructive episodes has already been demonstrated (French et al. 1996, Wilke et al. 2011) and it stands to reason that this might be linked to the markedly higher intestinal alkalinization rates found in this study. The transporter(s) for this F508del-augmented alkalinization are most likely not predominantly the mutated CFTR, but Slc26a3 and possibly other members of this anion exchanger gene family. If a similar situation exists in CF patients, with F508del-CFTR membrane expression in intestinal mucosa either spontaneously (van Barneveld et al. 2010) or after successful treatment with pharmacological agents (Lukacs & Verkman, 2012), knowledge about the transport properties of these anion exchangers may be important for clinicians when they treat intestinal complications of CF patients.
In summary, we have shown that expression of even small amounts of F508del-CFTR in the brush border membrane significantly enhances the forskolin-induced HCO3− secretory response in both murine duodenum and colon compared to CFTR KO mice. This secretion of HCO3− is dependent on luminal Cl− in both intestinal segments and therefore likely due to Cl−/HCO3− exchange. In addition, the expression of F508del-CFTR partially restores WNK signalling, which is defective in CFTR-deficient intestine. Small molecules that prevent proteasome degradation of F508del-CFTR, resulting in more mutant protein reaching the apical membrane, may therefore have the potential to significantly enhance intestinal HCO3− secretion, improve mucus viscosity and prevent intestinal impactions.
Acknowledgments
This work was supported by a financial grant from Mukoviszidose e.V., Bonn, the German Cystic Fibrosis Association, and the German Science Foundations grants SFB621-C9, and Se460/13-4 (all to U.S.) and grant 2011-0016484 from the National Research Foundation of Korea (to M.G.L.). We thank Regina Engelhardt, Brigitte Rausch, Natascha Cirpka, Silke Thiele and Denise Renner for their help with the animal breeding and genotyping, and Dr med. vet. Matthias Ottinger for help with animal management.
Glossary
- BBM
brush border membrane
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- DRA
down regulated in adenoma
- FSK
forskolin
- HRP
horseradish peroxidase
- NHE3
Na+/H+ exchanger isoform 3
- Rt
electrical resistance
- PEG
polyethylene glycol
- SPAK/OSR1
Ste20-related proline–alanine-rich kinases
- WNK
with no lysine (K) protein kinase
Author contributions
Conception and design of the experiments: F.X., H.J. U.S.; collection, analysis and interpretation of data: F.X., J.L., A.K.S., B.R., J.W., A.S., B.J.S.; drafting the article or revising it critically for important intellectual content: F.X., H.P., M.G.L. G.L., H.J, U.S. All authors approved the final version of the manuscript. The authors have no conflict of interest to disclose.
Author's present address
J. Wang: Department of Surgery, Jinling Hospital, Nanjing University, School of Medicine, 305 East Zhongshan Road, Nanjing 210002, P.R. China.
Supplementary material
Suppl. Fig. 1
suppl. Fig. 2
References
- Alper SL, Stewart AK, Vandorpe DH, Clark JS, Horack RZ, Simpson JE, Walker NM, Clarke LL. Native and recombinant Slc26a3 (downregulated in adenoma, Dra) do not exhibit properties of 2Cl−/1HCO3− exchange. Am J Physiol Cell Physiol. 2011;300:C276–C286. doi: 10.1152/ajpcell.00366.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann O, Rossmann H, Berger UV, Colledge WH, Ratcliff R, Evans MJ, Gregor M, Seidler U. cAMP-mediated regulation of murine intestinal/pancreatic Na+/HCO3− cotransporter subtype pNBC1. Am J Physiol Gastrointest Liver Physiol. 2003;284:G37–45. doi: 10.1152/ajpgi.00209.2002. [DOI] [PubMed] [Google Scholar]
- Beuers U, Hohenester S, de Buy Wenniger LJ, Kremer AE, Jansen PL, Elferink RP. The biliary HCO3− umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology. 2010;52:1489–1496. doi: 10.1002/hep.23810. [DOI] [PubMed] [Google Scholar]
- Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- Chan HC, Ko WH, Zhao W, Fu WO, Wong PY. Evidence for independent Cl− and HCO3− secretion and involvement of an apical Na+-HCO3− cotransporter in cultured rat epididymal epithelia. Exp Physiol. 1996;81:515–524. doi: 10.1113/expphysiol.1996.sp003954. [DOI] [PubMed] [Google Scholar]
- Chen EY, Yang N, Quinton PM, Chin WC. A new role for bicarbonate in mucus formation. Am J Physiol Lung Cell Mol Physiol. 2010;299:L542–549. doi: 10.1152/ajplung.00180.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Chernova MN, Jiang L, Friedman DJ, Darman RB, Lohi H, Kere J, Vandorpe DH, Alper SL. Functional comparison of mouse slc26a6 anion exchanger with human SLC26A6 polypeptide variants: differences in anion selectivity, regulation, and electrogenicity. J Biol Chem. 2005;280:8564–8580. doi: 10.1074/jbc.M411703200. [DOI] [PubMed] [Google Scholar]
- Choi JY, Muallem D, Kiselyov K, Lee MG, Thomas PJ, Muallem S. Aberrant CFTR-dependent HCO3− transport in mutations associated with cystic fibrosis. Nature. 2001;410:94–97. doi: 10.1038/35065099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LL, Harline MC. Dual role of CFTR in cAMP-stimulated HCO3− secretion across murine duodenum. Am J Physiol Gastrointest Liver Physiol. 1998;274:G718–G726. doi: 10.1152/ajpgi.1998.274.4.G718. [DOI] [PubMed] [Google Scholar]
- Clarke LL, Stien X, Walker NM. Intestinal bicarbonate secretion in cystic fibrosis mice. JOP. 2001;2:263–267. [PubMed] [Google Scholar]
- Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq JP, Lazdunski M. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature. 1991;354:526–528. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- De Boeck K, Derichs N, Fajac I, de Jonge HR, Bronsveld I, Sermet I, Vermeulen F, Sheppard DN, Cuppens H, Hug M, Melotti P, Middleton PG, Wilschanski M. New clinical diagnostic procedures for cystic fibrosis in Europe. J Cyst Fibros. 2011;10(Suppl 2):S53–66. doi: 10.1016/S1569-1993(11)60009-X. [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Dorwart MR, Shcheynikov N, Yang D, Muallem S. The solute carrier 26 family of proteins in epithelial ion transport. Physiology (Bethesda) 2008;23:104–114. doi: 10.1152/physiol.00037.2007. [DOI] [PubMed] [Google Scholar]
- French PJ, van Doorninck JH, Peters RH, Verbeek E, Ameen NA, Marino CR, de Jonge HR, Bijman J, Scholte BJ. A delta F508 mutation in mouse cystic fibrosis transmembrane conductance regulator results in a temperature-sensitive processing defect in vivo. J Clin Invest. 1996;98:1304–1312. doi: 10.1172/JCI118917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa O, Bi LC, Guth PH, Engel E, Hirokawa M, Kaunitz JD. NHE3 inhibition activates duodenal bicarbonate secretion in the rat. Am J Physiol Gastrointest Liver Physiol. 2004;286:G102–109. doi: 10.1152/ajpgi.00092.2003. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Yang N, Quinton PM. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator-dependent bicarbonate secretion. J Clin Invest. 2009;119:2613–2622. doi: 10.1172/JCI38662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawenis LR, Boyle KT, Palmer BA, Walker NM, Clarke LL. Lateral intercellular space volume as a determinant of CFTR-mediated anion secretion across small intestinal mucosa. Am J Physiol Gastrointest Liver Physiol. 2004;286:G1015–1023. doi: 10.1152/ajpgi.00468.2003. [DOI] [PubMed] [Google Scholar]
- Greger R, Schreiber R, Mall M, Wissner A, Hopf A, Briel M, Bleich M, Warth R, Kunzelmann K. Cystic fibrosis and CFTR. Pflugers Arch. 2001;443(Suppl 1):S3–7. doi: 10.1007/s004240100635. [DOI] [PubMed] [Google Scholar]
- Gregory RJ, Rich DP, Cheng SH, Souza DW, Paul S, Manavalan P, Anderson MP, Welsh MJ, Smith AE. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol Cell Biol. 1991;11:3886–3893. doi: 10.1128/mcb.11.8.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guba M, Kuhn M, Forssmann WG, Classen M, Gregor M, Seidler U. Guanylin strongly stimulates rat duodenal HCO3− secretion: proposed mechanism and comparison with other secretagogues. Gastroenterology. 1996;111:1558–1568. doi: 10.1016/s0016-5085(96)70018-5. [DOI] [PubMed] [Google Scholar]
- Ignath I, Hegyi P, Venglovecz V, Szekely CA, Carr G, Hasegawa M, Inoue M, Takacs T, Argent BE, Gray MA, Rakonczay Z., Jr CFTR expression but not Cl− transport is involved in the stimulatory effect of bile acids on apical Cl−/HCO3− exchange activity in human pancreatic duct cells. Pancreas. 2009;38:921–929. doi: 10.1097/MPA.0b013e3181b65d34. [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Steward MC, Wilson RW, Case RM. Bicarbonate secretion in interlobular ducts from guinea-pig pancreas. J Physiol. 1996;495:179–191. doi: 10.1113/jphysiol.1996.sp021583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Steward MC, Naruse S, Ko SB, Goto H, Case RM, Kondo T, Yamamoto A. CFTR functions as a bicarbonate channel in pancreatic duct cells. J Gen Physiol. 2009;133:315–326. doi: 10.1085/jgp.200810122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Wilson FH, Lifton RP. Regulation of diverse ion transport pathways by WNK4 kinase: a novel molecular switch. Trends Endocrinol Metab. 2005;16:98–103. doi: 10.1016/j.tem.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Kelly M, Trudel S, Brouillard F, Bouillaud F, Colas J, Nguyen-Khoa T, Ollero M, Edelman A, Fritsch J. Cystic fibrosis transmembrane regulator inhibitors CFTRinh-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition. J Pharmacol Exp Ther. 2010;333:60–69. doi: 10.1124/jpet.109.162032. [DOI] [PubMed] [Google Scholar]
- Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S, Soyombo A, Thomas PJ, Muallem S. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol. 2004;6:343–350. doi: 10.1038/ncb1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht G, Baisch S, Schoenleber E, Gregor M. Transport properties of the human intestinal anion exchanger DRA (down-regulated in adenoma) in transfected HEK293 cells. Pflugers Arch. 2005;449:479–490. doi: 10.1007/s00424-004-1342-x. [DOI] [PubMed] [Google Scholar]
- Lamprecht G, Schaefer J, Dietz K, Gregor M. Chloride and bicarbonate have similar affinities to the intestinal anion exchanger DRA (down regulated in adenoma) Pflugers Arch. 2006;452:307–315. doi: 10.1007/s00424-006-0049-6. [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR, Grinstein S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993;268:21592–21598. [PubMed] [Google Scholar]
- Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med. 2012;18:81–91. doi: 10.1016/j.molmed.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod RJ, Lembessis P, Hamilton JR. Isotonic volume reduction associated with cAMP stimulation of 36Cl efflux from jejunal crypt epithelial cells. Am J Physiol Gastrointest Liver Physiol. 1994;267:G387–392. doi: 10.1152/ajpgi.1994.267.3.G387. [DOI] [PubMed] [Google Scholar]
- Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol. 2004;124:125–137. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchekehu RW, Quinton PM. A new role for bicarbonate secretion in cervico-uterine mucus release. J Physiol. 2010;588:2329–2342. doi: 10.1113/jphysiol.2010.187237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohana E, Yang D, Shcheynikov N, Muallem S. Diverse transport modes by the solute carrier 26 family of anion transporters. J Physiol. 2009;587:2179–2185. doi: 10.1113/jphysiol.2008.164863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329:805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostedgaard LS, Rogers CS, Dong Q, Randak CO, Vermeer DW, Rokhlina T, Karp PH, Welsh MJ. Processing and function of CFTR-ΔF508 are species-dependent. Proc Natl Acad Sci U S A. 2007;104:15370–15375. doi: 10.1073/pnas.0706974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Nam JH, Kim JY, Namkung W, Yoon JS, Lee JS, Kim KS, Venglovecz V, Gray MA, Kim KH, Lee MG. Dynamic regulation of CFTR bicarbonate permeability by [Cl−]i and its role in pancreatic bicarbonate secretion. Gastroenterology. 2010;139:620–631. doi: 10.1053/j.gastro.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Poulsen JH, Fischer H, Illek B, Machen TE. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci U S A. 1994;91:5340–5344. doi: 10.1073/pnas.91.12.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinton PM. Role of epithelial HCO3− transport in mucin secretion: lessons from cystic fibrosis. Am J Physiol Cell Physiol. 2010;299:C1222–1233. doi: 10.1152/ajpcell.00362.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliff R, Evans MJ, Cuthbert AW, MacVinish LJ, Foster D, Anderson JR, Colledge WH. Production of a severe cystic fibrosis mutation in mice by gene targeting. Nat Genet. 1993;4:35–41. doi: 10.1038/ng0593-35. [DOI] [PubMed] [Google Scholar]
- Reddy MM, Quinton PM. Control of dynamic CFTR selectivity by glutamate and ATP in epithelial cells. Nature. 2003;423:756–760. doi: 10.1038/nature01694. [DOI] [PubMed] [Google Scholar]
- Schneider G, Saur D. Mesenchymal stem cells: therapeutic potential for acute pancreatitis. Gastroenterology. 2011;140:779–782. doi: 10.1053/j.gastro.2011.01.026. [DOI] [PubMed] [Google Scholar]
- Seidler U, Blumenstein I, Kretz A, Viellard-Baron D, Rossmann H, Colledge WH, Evans M, Ratcliff R, Gregor M. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca2+-dependent HCO3− secretion. J Physiol. 1997;505:411–423. doi: 10.1111/j.1469-7793.1997.411bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler U, Singh A, Chen M, Cinar A, Bachmann O, Zheng W, Wang J, Yeruva S, Riederer B. Knockout mouse models for intestinal electrolyte transporters and regulatory PDZ adaptors: new insights into cystic fibrosis, secretory diarrhoea and fructose-induced hypertension. Exp Physiol. 2009;94:175–179. doi: 10.1113/expphysiol.2008.043018. [DOI] [PubMed] [Google Scholar]
- Shcheynikov N, Kim KH, Kim KM, Dorwart MR, Ko SB, Goto H, Naruse S, Thomas PJ, Muallem S. Dynamic control of cystic fibrosis transmembrane conductance regulator Cl−/HCO3− selectivity by external Cl−. J Biol Chem. 2004;279:21857–21865. doi: 10.1074/jbc.M313323200. [DOI] [PubMed] [Google Scholar]
- Simpson JE, Gawenis LR, Walker NM, Boyle KT, Clarke LL. Chloride conductance of CFTR facilitates basal Cl−/HCO3− exchange in the villous epithelium of intact murine duodenum. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1241–1251. doi: 10.1152/ajpgi.00493.2004. [DOI] [PubMed] [Google Scholar]
- Singh AK, Riederer B, Chen M, Xiao F, Krabbenhoft A, Engelhardt R, Nylander O, Soleimani M, Seidler U. The switch of intestinal Slc26 exchangers from anion absorptive to HCO3− secretory mode is dependent on CFTR anion channel function. Am J Physiol Cell Physiol. 2010;298:C1057–1065. doi: 10.1152/ajpcell.00454.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Riederer B, Krabbenhöft A, Bonhagen J, Rausch B, Engelhardt R, Soleimani M, Seidler U. Slc26 anion transporters involvement in acid-activated duodenal HCO3− secretion. Proc Physiol Soc. 2009;16:C5. [Google Scholar]
- Singh AK, Sjoblom M, Zheng W, Krabbenhoft A, Riederer B, Rausch B, Manns MP, Soleimani M, Seidler U. CFTR and its key role in in vivo resting and luminal acid-induced duodenal HCO3− secretion. Acta Physiol (Oxf) 2008;193:357–365. doi: 10.1111/j.1748-1716.2008.01854.x. [DOI] [PubMed] [Google Scholar]
- Sjoblom M, Singh AK, Zheng W, Wang J, Tuo BG, Krabbenhoft A, Riederer B, Gros G, Seidler U. Duodenal acidity “sensing” but not epithelial HCO3− supply is critically dependent on carbonic anhydrase II expression. Proc Natl Acad Sci U S A. 2009;106:13094–13099. doi: 10.1073/pnas.0901488106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen MV, Sausbier M, Ruth P, Seidler U, Riederer B, Praetorius HA, Leipziger J. Adrenaline-induced colonic K+ secretion is mediated by KCa1.1 (BK) channels. J Physiol. 2010;588:1763–1777. doi: 10.1113/jphysiol.2009.181933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Phillipper M, Rossmann H, Riederer B, Gregor M, Seidler U. Independence of apical Cl−/HCO3− exchange and anion conductance in duodenal HCO3− secretion. Am J Physiol Gastrointest Liver Physiol. 2003;285:G887–897. doi: 10.1152/ajpgi.00083.2003. [DOI] [PubMed] [Google Scholar]
- Stewart AK, Shmukler BE, Vandorpe DH, Reimold F, Heneghan JF, Nakakuki M, Akhavein A, Ko S, Ishiguro H, Alper SL. SLC26 anion exchangers of guinea pig pancreatic duct: molecular cloning and functional characterization. Am J Physiol Cell Physiol. 2011;301:C289–303. doi: 10.1152/ajpcell.00089.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward MC, Ishiguro H, Case RM. Mechanisms of bicarbonate secretion in the pancreatic duct. Annu Rev Physiol. 2005;67:377–409. doi: 10.1146/annurev.physiol.67.031103.153247. [DOI] [PubMed] [Google Scholar]
- Talbot C, Lytle C. Segregation of Na/H exchanger-3 and Cl/HCO3 exchanger SLC26A3 (DRA) in rodent cecum and colon. Am J Physiol Gastrointest Liver Physiol. 2010;299:G358–367. doi: 10.1152/ajpgi.00151.2010. [DOI] [PubMed] [Google Scholar]
- Tang L, Fatehi M, Linsdell P. Mechanism of direct bicarbonate transport by the CFTR anion channel. J Cyst Fibros. 2009;8:115–121. doi: 10.1016/j.jcf.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Tuo B, Riederer B, Wang Z, Colledge WH, Soleimani M, Seidler U. Involvement of the anion exchanger SLC26A6 in prostaglandin E2- but not forskolin-stimulated duodenal HCO3− secretion. Gastroenterology. 2006;130:349–358. doi: 10.1053/j.gastro.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Tuo B, Wen G, Song P, Xu J, Liu X, Seidler U, Dong H. Genistein stimulates duodenal HCO3− secretion through PI3K pathway in mice. Eur J Pharmacol. 2011;651:159–167. doi: 10.1016/j.ejphar.2010.10.070. [DOI] [PubMed] [Google Scholar]
- Tuo B, Wen G, Seidler U. Differential activation of the HCO3− conductance through the cystic fibrosis transmembrane conductance regulator anion channel by genistein and forskolin in murine duodenum. Br J Pharmacol. 2009;158:1313–1321. doi: 10.1111/j.1476-5381.2009.00398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde MA, O’Brien JA, Sepulveda FV, Ratcliff R, Evans MJ, Colledge WH. Inactivation of the murine cftr gene abolishes cAMP-mediated but not Ca2+-mediated secretagogue-induced volume decrease in small-intestinal crypts. Pflugers Arch. 1993;425:434–438. doi: 10.1007/BF00374869. [DOI] [PubMed] [Google Scholar]
- van Barneveld A, Stanke F, Tamm S, Siebert B, Brandes G, Derichs N, Ballmann M, Junge S, Tümmler B. Functional analysis of F508del CFTR in native human colon. Biochim Biophys Acta. 2010;1802:1062–1069. doi: 10.1016/j.bbadis.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–171. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker NM, Simpson JE, Brazill JM, Gill RK, Dudeja PK, Schweinfest CW, Clarke LL. Role of down-regulated in adenoma anion exchanger in HCO3− secretion across murine duodenum. Gastroenterology. 2009;136:893–901. doi: 10.1053/j.gastro.2008.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zeltwanger S, Hu S, Hwang TC. Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. J Physiol. 2000;524:637–648. doi: 10.1111/j.1469-7793.2000.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- Wilke M, Buijs-Offerman RM, Aarbiou J, Colledge WH, Sheppard DN, Touqui L, Bot A, Jorna H, de Jonge HR, Scholte BJ. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros. 2011;10(Suppl 2):S152–S171. doi: 10.1016/S1569-1993(11)60020-9. [DOI] [PubMed] [Google Scholar]
- Xiao F, Juric M, Li J, Riederer B, Yeruva S, Kumar Singh A, Zheng L, Glage S, Kollias G, Dudeja P, Tian DA, Xu G, Zhu J, Bachmann O, Seidler U. Loss of downregulated in adenoma (DRA) impairs mucosal HCO3− secretion in murine ileocolonic inflammation. Inflamm Bowel Dis. 2012;18:101–111. doi: 10.1002/ibd.21744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Ishiguro H, Steward M, Sohma Y, Yamamoto A, Shimouchi A, Kondo T. Apical Cl−/HCO3− exchanger stoichiometry in the modeling of HCO3− transport by pancreatic duct epithelium. J Med Invest. 2009;56(Suppl):325–328. doi: 10.2152/jmi.56.325. [DOI] [PubMed] [Google Scholar]
- Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, Lukacs GL, Taddei A, Folli C, Pedemonte N, Galietta LJ, Verkman AS. Nanomolar affinity small molecule correctors of defective ΔF508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- Yu YC, Miki H, Nakamura Y, Hanyuda A, Matsuzaki Y, Abe Y, Yasui M, Tanaka K, Hwang TC, Bompadre SG, Sohma Y. Curcumin and genistein additively potentiate G551D-CFTR. J Cyst Fibros. 2011;10:243–252. doi: 10.1016/j.jcf.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.