

Abstract
The Kir4.1 channel is expressed in the brainstem, retina and kidney where it acts on K+ transportation and pH-dependent membrane potential regulation. Its heteromerization with Kir5.1 leads to K+ currents with distinct properties such as single-channel conductance, rectification, pH sensitivity and phosphorylation modulation. Here we show that Kir5.1 also enables S-glutathionylation to the heteromeric channel. Expressed in HEK cells, an exposure to the oxidant H2O2 or diamide produced concentration-dependent inhibitions of the whole-cell Kir4.1–Kir5.1 currents. In inside-out patches, currents were inhibited strongly by a combination of diamide/GSH or H2O2/GSH but not by either alone. The currents were also suppressed by GSSG and the thiol oxidants pyridine disulfides (PDSs), suggesting S-glutathionylation. In contrast, none of the exposures had significant effects on the homomeric Kir4.1 channel. Cys158 in the TM2 helix of Kir5.1 was critical for the S-glutathionylation, which was accessible to intracellular but not extracellular oxidants. Site-directed mutagenesis of this residue (C158A or C158T) abolished the Kir4.1–Kir5.1 current modulation by oxidants, and eliminated almost completely the biochemical interaction of Kir5.1 with GSH. In tandem Kir4.1–Kir5.1 constructs, the channel with a single Cys158 was inhibited to the same degree as the wild-type channel, suggesting that one glutathione moiety is sufficient to block the channel. Consistent with the location of Cys158, GSSG inhibited the channel only when the channel was open, indicating that the channel inhibition was state dependent. The finding that the heteromeric Kir4.1–Kir5.1 channel but not the homomeric Kir4.1 is subject to the S-glutathionylation thus suggests a novel Kir4.1–Kir5.1 channel modulation mechanism that is likely to occur in oxidative stress.
Key points
K+ channels are the primary regulators of membrane potentials and cellular excitability, dysfunction of which may occur under several pathophysiological conditions affecting cellular function and stress responses, such as oxidative stress known to play an important role in the inflammation state.
In the study, we find evidence for the modulation of a K+ channel by several oxidants. The underlying mechanism for the oxidant-mediated channel modulation appears to be mediated by S-glutathionylation, a newly recognized protein modification process.
A cysteine residue in the second transmembrane domain of the channel protein seems to be the target of S-glutathionylation.
Introduction
Inward rectifier K+ (Kir) channels regulate cellular excitability and neurotransmission (Hibino et al. 2010). The accessibility for K+ to enter the cell through the channels allows some Kir channels including Kir1.1 and Kir4.1 to act as K+ transporters (Butt & Kalsi, 2006). The Kir4.1 channel can be expressed as homomeric and heteromeric channels in the kidney, retina and central nervous system (CNS) (Hibino et al. 2004; Lachheb et al. 2008; Zhang et al. 2011). The channel composed of Kir4.1 and Kir5.1 subunits has distinct functional properties such as single-channel conductance, time-dependent activation and gating (Bond et al. 1994; Yang et al. 2000; Casamassima et al. 2003; Shang et al. 2009). The homomeric Kir4.1 is pH sensitive with pKa∼6.0 (Yang & Jiang, 1999; Xu et al. 2000), whereas the pH sensitivity of the heteromeric Kir4.1–Kir5.1 channel is much higher (pKa∼7.4) (Yang & Jiang, 1999; Casamassima et al. 2003), allowing it to respond to the changes in pH and 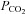 at physiological levels (Tanemoto et al. 2000; Cui et al. 2001; Pessia et al. 2001; Wenker et al. 2010; Yang & Jiang, 1999). The heteromeric Kir4.1–Kir5.1 channel but not the homomeric Kir4.1 is also modulated by several Gq-protein coupled neurotransmitters including serotonin, substance P and thyrotropin releasing hormone (Rojas et al. 2008). The channel modulation is mediated by Kir5.1-dependent phosphorylation by protein kinase C (Rojas et al. 2007).
at physiological levels (Tanemoto et al. 2000; Cui et al. 2001; Pessia et al. 2001; Wenker et al. 2010; Yang & Jiang, 1999). The heteromeric Kir4.1–Kir5.1 channel but not the homomeric Kir4.1 is also modulated by several Gq-protein coupled neurotransmitters including serotonin, substance P and thyrotropin releasing hormone (Rojas et al. 2008). The channel modulation is mediated by Kir5.1-dependent phosphorylation by protein kinase C (Rojas et al. 2007).
While the heteromerization of Kir4.1 with Kir5.1 clearly benefits the renal, retinal and nervous tissues in meeting their functional needs in physiological conditions, the channels may allow cellular responses to several pathophysiological conditions as well, such as oxidative stress known to be a prominent contributor to inflammation and neurodegenerative diseases (Griendling & FitzGerald, 2003; Lin & Beal, 2006; Miller et al. 2011). Since the kidney, retina and CNS are some of the most vulnerable tissues to these disease conditions (Feldman, 2003; Schiffrin et al. 2007; Jager et al. 2008; Giacco & Brownlee, 2010), it is possible that the reactive oxygen species (ROS) and other oxidants act on certain Kir channels that are expressed in the tissues. The abnormalities in the channels may cause disruptions in membrane potentials and K+ ionic homeostasis, contributing to cell injuries in oxidative stress. To test the hypothesis that the Kir4.1–Kir5.1 channel is targeted by ROS and other oxidants, and to understand the mechanism underlying the channel modulation, we performed the studies described here. Our results show that the Kir4.1–Kir5.1 channel, but not the Kir4.1, is susceptible to oxidant challenges, and the molecular basis for the channel modulation appears to be S-glutathionylation at Cys158 of the Kir5.1 subunit.
Methods
Chemicals
Chemicals used in this study were purchased from Sigma unless otherwise stated. Solutions containing H2O2 and glutathione (GSH) were freshly made and used within 4 h. Solutions with other reagents were prepared as concentrated stocks in double-distilled water or dimethyl sulfoxide (DMSO). In cases where DMSO was used, the final concentration of DMSO in the solution was 0.1% (v/v). At this concentration, DMSO did not affect the Kir4.1–Kir5.1 or Kir4.1 channel.
Molecular biology
Rat Kir4.1 cDNA (GenBank no. X83585) and rat Kir5.1 cDNA (GenBank no. X83581) were gifts of Dr John Adelman at the Oregon Health Science University, Portland, OR, USA. These cDNAs were subcloned into the eukaryotic expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA, USA). For co-expression of Kir4.1 with Kir5.1, a tandem dimer of these two cDNAs was constructed using the overlapping extension technique, in which full length Kir4.1 and Kir5.1 sequences were obtained using Pfu DNA polymerase (Stratagene, La Jolla, CA, USA) chain reaction (PCR). The PCR products were joined to each other at the 3′ end of Kir4.1 and 5′ end of Kir5.1. Correct constructions were confirmed with DNA sequencing. Site-specific mutations were made using a site-directed mutagenesis kit (Stratagene). Correct constructions and mutations were confirmed with DNA sequencing.
Tandem-tetrameric channels were constructed as we reported previously (Wang et al. 2007). In brief, cohesive ends of EcoRI site and MfeI site were created at the 5′ end and 3′ end of each subunit, respectively. These sites are complementary, which allows mutual DNA ligation. Both restriction sites were lost after ligation, and the construct still contained only one EcoRI site at the 5′ end and one MfeI site at the 3′ end. This allows construction of the tandem-multimers using the same strategy, in which only one stop codon was retained in the last subunit. The correct orientation of the constructs was confirmed by identification of appropriate restriction fragments with HindIII, and the correct DNA sequencing.
Channel expression
The cDNA constructs were expressed in human embryonic kidney (HEK) cells (CRL-1573, Batch no. 2187595, ATCC, Rockville, MD, USA) as detailed in our previous reports. At 37°C with a 5% CO2 atmosphere, the HEK cells were cultured as a monolayer in Dulbecco's modified Eagle's medium (DMEM)–F12 with 10% fetal bovine serum and penicillin–streptomycin added. Split twice weekly, the HEK cells were transfected using Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA) with 3.5 μg Kir4.1–Kir5.1 dimer cDNA. To facilitate the identification of positively transfected cells, 0.5 μg green fluorescent protein (GFP) cDNA (pEGFP-N2, Clontech, Palo Alto, CA, USA) was added to the cDNA mixture. Cells were disassociated from the monolayer using 0.25% trypsin ∼24 h post-transfection. A few drops of the cell suspension were added on to 5 × 5 mm cover slips in a 35 mm Petri dish. The cells were then incubated at 37°C for an additional 24–48 h before experiments.
Electrophysiology
Single-electrode whole-cell voltage clamp was performed on the HEK cells at room temperature as described previously (Shi et al. 2007; Yang et al. 2010; Zhang et al. 2011). In brief, fire-polished patch pipettes were made of 1.2 mm borosilicate capillary glass (Sutter Instruments, Novato, CA, USA). Tight seals (>1 GΩ before breaking into the whole-cell mode) were obtained with the transfected cells. The patch electrodes had an open tip resistance of 2–4 MΩ. Currents were recorded with the amplifier Axopatch 200B (Molecular Devices, Sunnyvale, CA, USA). The series resistance of ∼10 MΩ was not compensated. Recordings were terminated whenever significant increase (>20%) in series resistance occurred. Current records were lowpass filtered (2 kHz, Bessel, 4-pole filter, −3 dB), digitized (20 kHz, 16-bit resolution), and stored on computer disk for later data analysis using the pCLAMP 9 software (Molecular Devices). Recordings were performed using solutions containing equal concentrations of K+ applied to the bath and recording pipettes. This solution contained (in mm): KCl 40, potassium gluconate 90, potassium fluoride 5, sodium vanadate 0.1, potassium pyrophosphate 10, adenosine diphosphate (ADP) 0.2, Pipes 10, glucose 10 and spermine 0.1, and EGTA 1 (pH 7.4).
Inside-out patch clamp was performed on HEK cells at room temperature (∼24°C). As described previously (Yang et al. 2000), multiple-channel Kir currents were recorded with symmetric high K+ in bath (intracellular) and pipette (extracellular) solutions. The solutions contained (in mm): KCl 40, potassium gluconate 90, potassium fluoride 5, sodium vanadate 0.1, potassium pyrophosphate 10, EGTA 1, ADP 0.2, Pipes 10 and glucose 10 at pH 7.4. To elicit inward rectification, 0.1 mm spermine was added to the bath solution. Giant inside-out patches were obtained using recording pipettes of 1–2 MΩ. Since these Kir channels are pH sensitive, pH was measured and titrated to pH 7.4 in solutions before experiments. Single-channel currents were recorded with a constant single voltage of −60 mV. The single-channel conductance was measured with slope command potentials from −100 mV to 100 mV. The open-state probability (NPopen) was calculated by counting all active channels from a given patch by using Clampfit 10 software (Axon Instruments) having a total duration of 40–60 s as described previously (Yang & Jiang, 1999; Lei et al. 2011).
Western blotting and immunoprecipitation of biotinylated proteins
The methods of Western blot and immunoprecipitation with a Streptavidin Pull-down Assay were described in our previous study (Yang et al. 2010, 2011). In brief, HEK cells expressing wild-type (WT) Kir4.1–Kir5.1 or mutant Kir4.1–Kir5.1 (C158A) channels were incubated with combination of biotinylated glutathione ethyl ester (BioGEE, 250 μm, Invitrogen) and followed by a 750 μm H2O2 treatment. The excessive BioGEE was removed by three washes with phosphate buffered saline (PBS). The RIPA (radio-immunoprecipitation assay) buffer (100 μl, Sigma) was used for cell lysis. Protein concentration was measured by using a bicinchoninic acid protein assay system (Thermo Scientific). All of the protein samples were diluted to comparable concentrations (1 mg ml−1). Whole-cell extracts of both WT and mutant channels (15 μl) were used for Western blot. The samples were run on a 10% SDS-polyacrylamide reducing gel, and then transferred to a polyvinylidene fluoride membrane (Bio-Rad). Rabbit primary antibodies against Kir5.1 (1:1000) (Sigma), mouse primary antibodies against glyceraldehyde 3-phoshate dehydrogenase (GAPDH) (Sigma), and secondary antibodies conjugated with horseradish peroxidase (1:3000) (Bio-Rad) were used. Signal was visualized with the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). Striptavidin-dynabeads (Invitrogen) were used to pull down the biotin–GSH-conjugated proteins. The same amounts of WT and mutant channel protein were incubated with beads for 30 min at room temperature. After washing with PBS 3 times, the sample was boiled for at least 5 min, and then loaded and run in the SDS-PAGE gel parallel with whole cell extract proteins as described above. All the bands densities were quantified by ImageJ (National Institutes of Health, Bethesda, MD, USA).
Structural modelling
The native Kir4.1 and Kir5.1 structure in their closed state were generated by I-TASSER protein structure prediction (Wu et al. 2007; Zhang, 2007, 2008) using prokaryotic Kir3.1 channel as the templates (Kuo et al. 2003; Nishida et al. 2007). The native Kir5.1 structural model was further modified by incorporating the GSH molecule onto the thiol group of Cys158. The sequence alignment was generated by ClustalW2. Structural images were generated with PyMol molecular graphics system (http://www.pymol.org).
Data analysis
Data are presented as the mean ± SEM of each group. Differences in means were tested with Student's t test for paired data and one-way ANOVA with post hoc Tukey's pairwise comparisons for three groups or more. The differences were accepted as significant if P ≤ 0.05.
Results
Oxidants inhibited the heteromeric Kir4.1–Kir5.1 channel but not the homomeric Kir4.1
The Kir4.1–Kir5.1 and Kir4.1 channels were transiently expressed in HEK cells. Whole-cell voltage clamp was performed on GFP-positive cells. Both the bath and pipette solutions contained 145 mm K+. The membrane potential was held at 0 mV. Step depolarizing and hyperpolarizing command pulses were given to the cell in a range from −100 to 100 mV with a 20 mV increment in the duration of 400 ms for each pulse. The cells transfected with Kir4.1–Kir5.1 or Kir4.1 showed typical inward rectifying currents that were easily distinguished from each other as well as the endogenous currents in HEK cells (Fig. 1A, D and F), as reported previously (Bond et al. 1994; Yang & Jiang, 1999; Pessia et al. 2001). A 5 min application of various concentrations of H2O2 to the cells produced marked inhibition of the whole-cell Kir4.1–Kir5.1 currents (Fig. 1A). The channel inhibition showed a clear concentration dependence with 100 μm to 1 mm H2O2 (Fig. 1B). At the maximum effect (measured within the last 100 ms at −100 mV) the Kir4.1–Kir5.1 currents were inhibited by 55 ± 5% (n= 5) with 1 mm H2O2. The channel inhibition reached a plateau in 5 min, and was only partially reversible (Fig. 1C).
Figure 1. Oxidants inhibit the heteromeric Kir4.1–Kir5.1 channel but not the homomeric Kir4.1.
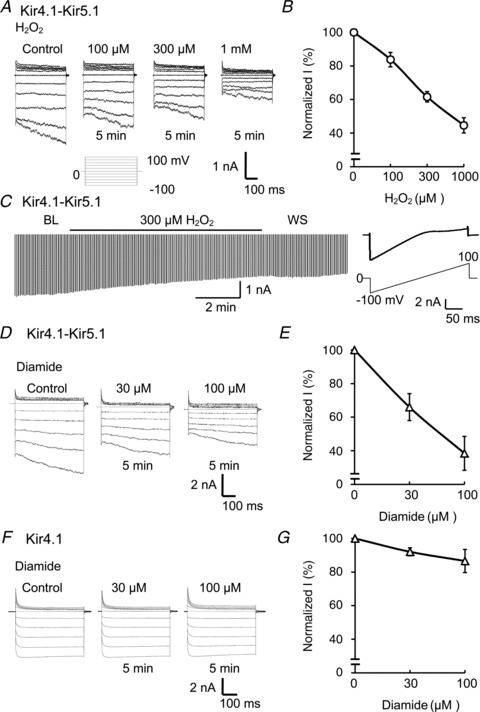
A, whole-cell currents were studied in an HEK cell expressing Kir4.1–Kir5.1 channel. A series of voltage commands (from −100 to 100 mV with a 20 mV increment with a duration of 400 ms each at a holding potential of 0 mV) were applied to the cell in a solution containing 145 mm K+ applied to both the bath and pipette. Treatments with H2O2 (100 μm to 1 mm) inhibited the Kir4.1–Kir5.1 channel dose-dependently. B, summary of the effects of H2O2 on whole-cell Kir4.1–Kir5.1 currents (n= 5). C, whole-cell recording also was performed with ramp potentials form −100 to 0 mm (right); current was strongly inhibited by 300 μm H2O2, and the maximum inhibition was reached in 5 min. D and E, exposure of the Kir4.1–Kir5.1 channel to another oxidant, diamide (30 and 100 μm), also resulted in strong channel inhibition (n= 5). E and F, diamide had very little effect on the homomeric Kir4.1 channel (n= 4). Currents were measured within the last 100 ms at 100 mV. Data are shown as means ± SEM.
The channel inhibition was not limited to H2O2, and another oxidant, diamide, had a similar inhibitory effect. Exposures of the Kir4.1–Kir5.1 channel to diamide (30 and 100 μm) for 5 min also resulted in concentration-dependent inhibition of inward rectifying currents (Fig. 1D and E). The maximum channel inhibition by 100 μm diamide was 67 ± 11% (n= 5, Fig. 1E) similar to the effect of H2O2.
In contrast, the treatment with these oxidants had very little effect on the homomeric Kir4.1 channel. The Kir4.1 channel was inhibited by 7 ± 5% (n= 4) with 100 μm diamide (Fig. 1F and G), and by 8 ± 15% (n= 4) with 1 mm H2O2.
Inhibition of the Kir4.1–Kir5.1 channel by thiol oxidants
The channel inhibitory effects of these oxidants suggest the involvement of thiol oxidation in the channel protein. To test this possibility, we studied the effect of pyridine disulfides (PDSs), reagents that oxidize selectively the sulfhydryl group of cysteine residues. 2,2′-Dithiodipyridine (2-DTP, 50 μm), a membrane permeant PDS (Yoshida et al. 2006; Yang et al. 2010), inhibited the whole-cell Kir4.1–Kir5.1 currents by 56 ± 6% (n= 5) (Fig. 2A and B). A similar effect was observed with another PDS (see below).
Figure 2. 2,2′-Dithiodipyridine inhibits the Kir4.1–Kir5.1 channels.
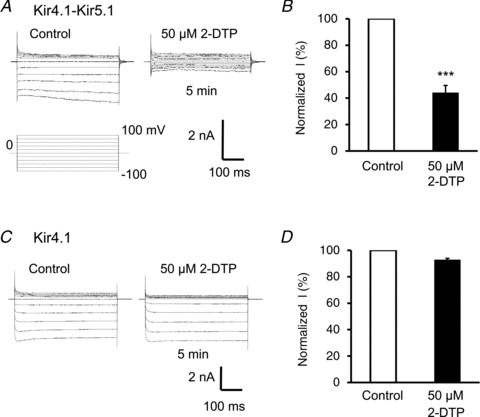
A, in whole-cell recording, the pyridine disulfide 2,2′-dithiodipyridine (2-DTP, 50 μm) markedly inhibited the Kir4.1–Kir5.1 currents. B, summary of the effect of 2-DTP on Kir4.1–Kir5.1 currents in whole-cell recording. Currents were measured within the last 100 ms at −100 mV. The currents were significantly inhibited by 56 ± 6% (n= 5) (***P < 0.001; Student's t test). C and D, application of 50 μm 2-DTP had no effect on the Kir4.1 currents.
The same concentration of 2-DTP inhibited the homomeric Kir4.1 channel by only 7 ± 1% (n= 6) (Fig. 2C and D), which was significantly different from the inhibition of the heteromeric Kir4.1–Kir5.1 channel (P < 0.001, n= 5). These results therefore suggest that the channel sensitivity to oxidants may be mediated by oxidation of thiol groups, and the critical subunit for the oxidant sensitivity is likely to be located in the Kir5.1 subunit.
The critical protein domain was located on the intracellular side
The finding of the PDS effect is encouraging, as the availability of PDSs with different membrane permeability may allow the determination of the intra- vs. extracellular location of potential thiol oxidation sites. Therefore, we examined the Kir4.1–Kir5.1 currents with another two PDSs. Like 2-DTP, the membrane-permeant PDS 2,2′-dithiobis-5-nitropyridine (DTNP, 50 μm) inhibited the whole-cell Kir4.1–Kir5.1 currents by 49 ± 5% (n= 5) (Fig. 3A and C) in a similar manner as 2-DTP. Under the same recording condition, however, a high concentration of 5,5′-dithiobis-2-nitrobenzoic acid (DTNB, 200 μm), a membrane-impermeant PDS (Lin & Beal, 2006; Yang et al. 2010), inhibited the Kir4.1–Kir5.1 currents by only 1 ± 5% (Fig. 3B and C). The distinct effects of the membrane-permeant and membrane-impermeant PDSs were not due to their different potencies, as they both showed the same levels of channel inhibition in inside-out patches (Fig. 3D–F). These results suggest that the critical protein domain for the oxidant sensitivity is located on the cytosolic side of cellular membranes.
Figure 3. Inhibition of Kir4.1–Kir5.1 currents by pyridine disulfides (PDSs) with different membrane permeancies.
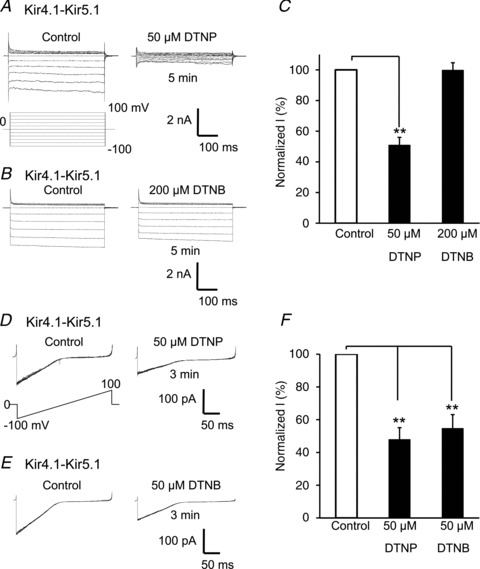
A, whole-cell Kir4.1–Kir5.1 currents were strongly inhibited by the membrane-permeant PDS 2,2′-dithiobis-5-nitropyridine (DTNP, 50 μm). B, under the same recording condition, a high concentration of 200 μm 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), a membrane-impermeant PDS, did not inhibit the channel currents significantly when applied extracellularly. C, summary of effects of DTNP and DTNB on Kir4.1–Kir5.1 currents in whole-cell recording. **P < 0.01 (n= 5; one-way ANOVA). D–F, currents were studied in inside-out patches. Membrane potential was held at 0 mV and commanded with ramp protocol from −100 to 100 mV. Both 50 μm DTNP and 50 μm DTNB inhibited the Kir4.1–Kir5.1 channels, by 52%± 7 and 45%± 8, respectively (n= 4–6). Note that each panel in D and E was made of 8 superimposed recordings.
Glutathione was involved in the oxidant-mediated channel inhibition
The effects of the PDSs also suggest that the channel inhibition may be produced by introducing a thiol adaptor to certain cysteine residue(s) rather than the formation of disulfide bonds. If this is the channel modulation mechanism, the Kir4.1–Kir5.1 channel should be inhibited by cell-endogenous substances. Therefore, we tested potential S-glutathionylation of the heteromeric Kir4.1–Kir5.1 channel in inside-out patches. A combined application of 300 μm H2O2 and 300 μm GSH produced an inhibition of the Kir4.1–Kir5.1 currents by 46 ± 4% (n= 5) (Fig. 4A and G). There was no significant effect on the currents when the same concentration of GSH or H2O2 was applied alone (Fig. 4C, E and G). Similarly, the exposure of internal patch membranes to 30 μm diamide together with 300 μm GSH inhibited the currents by 49 ± 5% (n= 6) (Fig. 4D and G). A treatment with 30 μm diamide alone did not inhibit the currents (Fig. 4D and G).
Figure 4. Glutathione is critical for the oxidant-mediated channel inhibition.
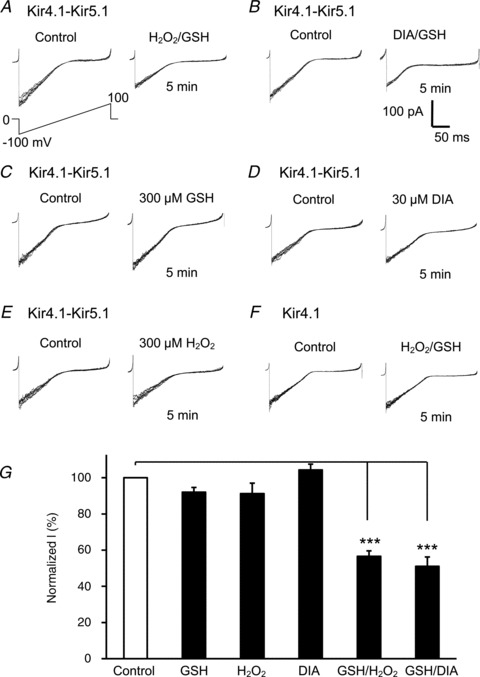
A and B, in giant inside-out patches, a combined application of 300 μm GSH with 300 μm H2O2 (H2O2/GSH) or 300 μm GSH with 30 μm diamide (DIA/GSH) markedly inhibited the Kir4.1–Kir5.1 currents. C–E, none of the 300 μm GSH, 300 μm H2O2 and 30 μm DIA produced evident channel inhibition when applied alone. G, summary of effects of these reagents (***P < 0.001; n= 5–6; one-way ANOVA). Note that each panel in A–E was made of 8 superimposed traces.
We also investigated the effects of oxidized glutathione (GSSG), another S-glutathionylation inducer. In inside-out patches an exposure of internal patch membranes to 2.5 mm and 5.0 mm GSSG suppressed the Kir-4.1–Kir5.1 currents by 16 ± 4% (n= 5) and 58 ± 7% (n= 5), respectively (Fig. 5A and C).
Figure 5. The effect of GSSG.
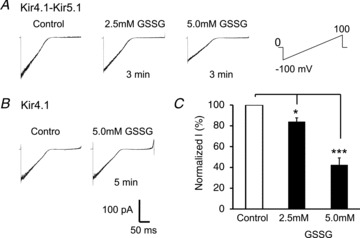
A, in an inside-out patch, exposure of internal patch membranes to 2.5 mm and 5.0 mm oxidized glutathione (GSSG) inhibited the Kir4.1–Kir5.1 currents in a concentration-dependent manner. B, the Kir4.1–Kir5.1 channel was suppressed by 16 ± 4% (n= 5) with 2.5 mm GSSG, and by 58 ± 7% (n= 5) with 5.0 mm GSSG (*P < 0.05; ***P < 0.001; one-way ANOVA).
In contrast, a treatment with 300 μm H2O2 and 300 μm GSH or with 5.0 mm GSSG did not inhibit the Kir4.1 currents significantly in inside-out patches (Figs 4F and 5B). The exposure of internal patch membranes to H2O2/GSH and GSSG inhibited the Kir4.1 currents by 6 ± 5% (n= 4) and 6 ± 3% (n= 4), respectively. The involvement of GSH in the oxidant-mediated channel inhibition strongly suggests S-glutathionylation. The differential sensitivity of Kir4.1 vs. Kir4.1–Kir5.1 suggests that a critical site (or sites) for S-glutathionylation regulation is located on the Kir5.1 subunit.
The effects of GSSG on single channel conductance and open-state probability (NPopen) were also studied. As shown in Fig. 6A, at baseline Kir4.1–Kir5.1 channel showed a high NPopen 1.9 ± 0.56 (n= 5 patches), and the single channel conductance averaged 54.0 ± 0.9 pS (n= 6) (Fig. 6C). Exposure of the internal membranes to 5 mm GSSG decreased the NPopen to 0.79 ± 0.23 (n= 5, P < 0.05) (Fig. 6E), but did not change the single channel conductance significantly (Fig. 6F).
Figure 6. GSSG decreases NPopen but not the unitary conductance.
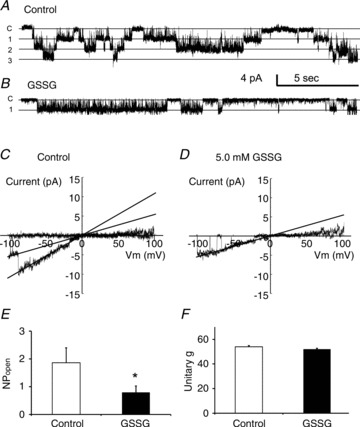
A, single channel currents of Kir4.1–Kir5.1 channel were recorded in an inside-out patch at the membrane potential of −60 mV. At baseline, three active channels were seen (NPopen 1.9 ± 0.56, n= 5 patches). B, in the presence of 5 mm GSSG only one active channel remained (NPopen 0.79 ± 0.23, n= 5, P < 0.05). C and D, single-channel conductance was measured with a ramp voltage from −100 mV to 100 mV. Straight lines represent a unitary conductance 54.0 ± 0.9 pS (n= 7). E and F, on average, 5 mm GSSG inhibited NPopen significantly (*P < 0.05; n= 5, Student's t test), but did not change the unitary conductance in Kir4.1–Kir5.1 channel (54.0 ± 0.9 and 52.0 ± 0.7, respectively; n= 6).
Cys158 in Kir5.1 was critical for S-glutathionylation
To reveal the S-glutathionylation site, we compared the amino acid sequences of Kir4.1 and Kir5.1 subunits, and found eight cysteine residues (4 in the intracellular domains) that exist only in Kir5.1. Among them is Cys158 in the second transmembrane (TM2) helix of Kir5.1, on which further studies were focused as we have recently shown that a similar cysteine residue in Kir6.1 plays a critical role in S-glutathionylation modulation of the vascular KATP channels (Yang et al. 2010, 2011). Thus, we performed site-directed mutagenesis at this site. Mutation of the Cys158 to threonine (the amino acid at the corresponding site in Kir4.1) or alanine (C158T, C158A) eliminated the effects of 5.0 mm GSSG and 50 μm DTNP on the currents in inside-out patches (Fig. 7), although the mutations did not affect the Kir4.1–Kir5.1 channel expression and the channel gating by acidic pH. With an internal solution of pH 6.8, the Kir4.1–Kir5.1 (C158T) channel was inhibited by 76 ± 3% (n= 6), and Kir4.1–Kir5.1 (C158A) was inhibited by 84 ± 4% (n= 6), similar to the WT channel (82 ± 6%, n= 6).
Figure 7. Cys158 of the Kir5.1 in S-glutathionylation.
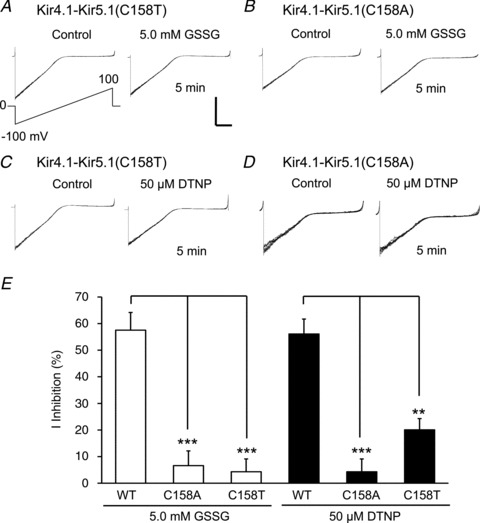
A and B, in inside-out patches, the Kir4.1–Kir5.1 (C158T) (A) and Kir4.1–Kir5.1 (C158A) (B) mutations eliminated the responses of the channels to 5.0 mm GSSG. C and D, the mutant Kir4.1–Kir5.1 channels also lost sensitivity to 50 μm DTNP. Note that each panel was made of 8 superimposed traces. Calibration: 100 pA and 50 ms for D, and 200 pA and 50 ms for the rest. E, statistically, the mutant responses to GSSG and DTNP were significantly different from the WT channel (**P < 0.01; ***P < 0.001, n= 6, one-way ANOVA).
Immune precipitation experiments were performed to show biochemical evidence for the Cys158-dependent S-glutathionylation of the Kir5.1. As shown in Fig. 8A, the anti-Kir5.1 antibodies detected strong Kir5.1 reactive bands of ∼50 kDa in whole-cell extracts obtained from both WT and Kir5.1 (C158A) mutant channels, but not in the extracts from HEK cells transfected with pcDNA3.1 alone or HEK cells without any transfection. The WT and Kir5.1 (C158A) bands showed similar densities, suggesting that the C158A mutation in Kir5.1subunit did not change the protein expression. After pull-down, the WT Kir5.1 pretreated with BioGEE and H2O2 showed a clear Kir5.1-reactive band, but the Kir5.1 (C158A) mutant displayed a much weaker band than the WT. Similar results were obtained in another six experiments. In comparison with WT Kir5.1, the density of Kir5.1 (C158A) mutant was decreased by 74 ± 12% (P < 0.001, n= 7) (Fig. 8B and C).
Figure 8. Western blot and streptavidin pull-down.
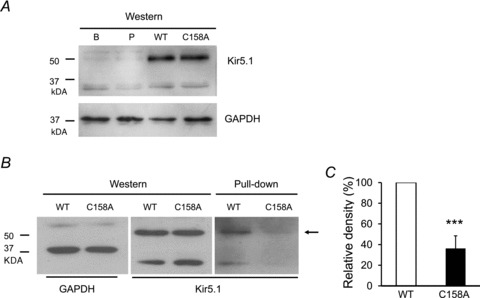
A, Western blot showing Kir4.1–Kir5.1 protein expression in the HEK cells expressing wild-type Kir4.1–Kir5.1 (WT), mutant Kir4.1–Kir5.1 (C158A), pcDNA3.1 vector (P) and non-transfected HEK cells (B) (upper). GAPDH is loading control (down). B, Western blot showed a similar density in reaction with anti-Kir5.1 antibodies (middle) with GAPDH loading control (left). After pull-down under the same experimental condition, a clear ∼50 kDa band indicated by the arrow (right) was seen with WT Kir5.1. In comparison, the density of the Kir5.1 (C158A) mutant was much weaker. C, the band densities of pull down were compared between WT and Kir5.1 (C158A) mutant after normalization of the WT Kir5.1 as 100%. The Kir5.1 (C158A) mutation lowered the band density by 74 ± 12% (n= 7 experiments; ***P < 0.001; Student's t test).
Incorporation of one glutathione moiety was adequate for the channel inhibition
The Kir4.1–Kir5.1 channel is made of two Kir4.1 and two Kir5.1 subunits that are arranged in an alternate pattern (Bond et al. 1994). Therefore, there are two Cys158 in each functional channel. To understand whether S-glutathionylation of one of the cysteines is adequate for channel inhibition, we constructed a tandem tetrameric channel, in which only one of the Kir5.1 subunits carried the Cys158 mutation. The construct expressed functional currents identical to the WT Kir4.1–Kir5.1. The whole-cell currents of the mutant was inhibited by diamide (Fig. 9A), DTNP (Fig. 9C) and GSSG (Fig. 9E) to the same degree as the WT channel (Fig. 9B, D and F), suggesting that only one glutathione appears sufficient to block the channel.
Figure 9. Oxidant responses of the tandem-tetrameric channel with a single C158A mutation.
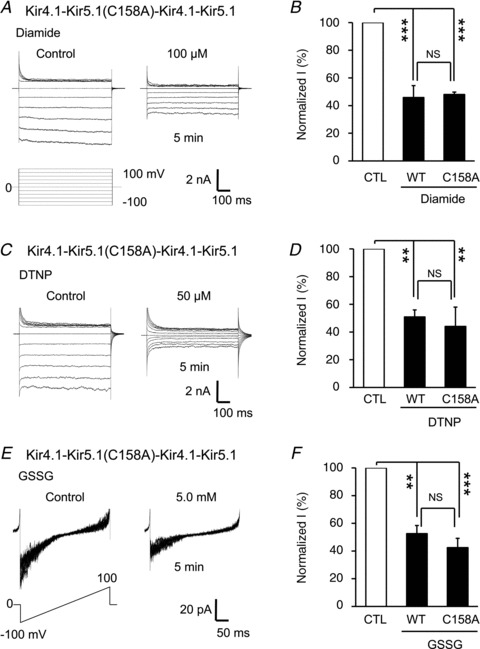
A and C, in whole-cell recording, a single C158 mutation in a tandem-tetramer of Kir4.1–Kir5.1 channel was inhibited by 100 μm diamide and 50 μm DTNP to the same degree as the WT channel. E, similar results were obtained with GSSG in an inside-out patch. B, D and F, statistical analysis indicated that the Kir4.1–Kir5.1 channel with one Cys158 had no significant difference from the WT channel with two Cys158 in their responses to diamide. DNTP and GSSG (**P < 0.01; ***P < 0.001; NS, no significant difference was found; n= 4–6; one-way ANOVA). Note that each panel in E was made of 8 superimposed traces.
State-dependent S-glutathionylation
The recovery time was studied after a channel inhibition with 5.0 mm GSSG and acidic internal solution (pH 6.5). When the channel was inhibited with acid first followed by GSSG, the recovery time was very fast. The currents resumed ∼100% of the control levels within 1 min after washout of both acid and GSSG (Fig. 10A and C), consistent with our previous studies in the absence of GSSG (Xu et al. 2000; Yang et al. 2000). When the patches were exposed to GSSG before acidic pH, two steps of current inhibition were seen. Although the inhibition by acid recovered fast, the GSSG-mediated current inhibition did not show evident recovery in 5 min (Fig. 10B and D). These results strongly suggest that the S-glutathionylation occurs only when the channel is in its open state. The state-dependent S-glutathionylation thus is consistent with the location of the cysteine residue targeted by glutathione.
Figure 10. State-dependent S-glutathionylation.
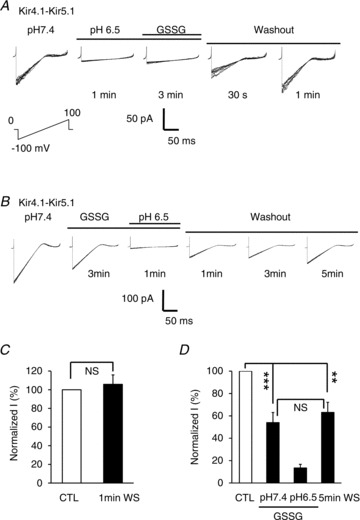
A, in an inside-out patch, the Kir4.1–Kir5.1 currents were strongly suppressed by an acidic internal solution (pH 6.5). The channel was totally inhibited, and not affected by additional 5.0 mm GSSG. The currents resumed ∼50% and 100% of the control (CTL) levels 0.5 and 1 min after washout, respectively. B, similar experiment was done with the reversal of the acid and GSSG application order. The Kir4.1–Kir5.1 currents were suppressed partially with the application of 5.0 mm GSSG first and then strongly with acidic internal solution. After washout, the inhibition by acid recovered fast, but the GSSG-mediated current inhibition did not show any evident recovery in 5 min. C, a complete recovery of the channel inhibition was seen 1 min after washout (WS) when the channel was inhibited with acid first. D, the GSSG-induced channel inhibition did not recover 5 min after washout (NS, no significant difference was found; **P < 0.01; ***P < 0.001; one-way ANOVA). Note that each panel in A and B was made of 8 superimposed traces.
Discussion
In the present study, we have demonstrated a novel modulation mechanism of Kir4.1–Kir5.1 channel. The modulation mechanism is mediated by S-glutathionylation of the Kir5.1 subunit, allowing the heteromeric Kir4.1–Kir5.1 channel to sense and respond to oxidative stress.
Oxidant sensitivity of the heteromeric Kir4.1–Kir5.1 channel
Oxidative stress is a condition when the concentration of oxidants overwhelms the cellular antioxidant systems. Several species of ROS are overly produced during oxidative stress. They can act on proteins, lipids and nucleotides, causing functional and structural damage (Griendling & FitzGerald, 2003; Miller et al. 2011). Oxidative stress is seen in several physiological and pathophysiological conditions such as inflammation, atherosclerosis, ageing, diabetic retinopathy, ischaemia–reperfusion, diabetic renal complications, neurodegenerative diseases, autoimmune diseases, etc. The Kir4.1 and Kir4.1–Kir5.1 channels are known to play an important role in some of the tissues including the retina, CNS and kidney (Tanemoto et al. 2000; Pessia et al. 2001; Wenker et al. 2010). The abnormalities of these channels thus may have an impact on the function and stress responses of the tissues.
Our present studies have shown that the heteromeric Kir4.1–Kir5.1 channel is indeed targeted by several oxidants. Activity of the channel is strongly inhibited by H2O2, diamide, PDS and GSSG. Interestingly, none of these oxidants has significant effect on the homomeric Kir4.1 channel. This finding not only is a surprise, but also has an important implication. Depending on the presence of Kir5.1 subunit, functional channels may be sensitive or insensitive to oxidants. The oxidant sensitivity tends to cause a major loss of activity (by over 50%) of the Kir4.1–Kir5.1 channel. Since the K+ channel regulates membrane potentials and transepithelial K+ transport, the channel inhibitions by endogenous and exogenous oxidants can lead to depolarization, an increase in cellular excitability and a disruption of K+ ionic homeostasis, contributing to the development of retinopathy, neurodegeneration and renal diseases seen in diabetic complications where oxidative stress manifests itself (Feldman, 2003; Lin & Beal, 2006; Schiffrin et al. 2007; Giacco & Brownlee, 2010).
S-Glutathionylation
Several lines of evidences indicate that the oxidant sensitivity of the Kir4.1–Kir5.1 channel is mediated by S-glutathionylation of the Kir5.1 subunit. (1) The channel is inhibited by the oxidants H2O2 and diamide more strongly in the presence of GSH than in the absence of GSH, suggesting the involvement of GSH. (2) Low concentrations of H2O2 (300 μm) and diamide (30 μm) have no effect on Kir4.1–Kir5.1 currents in inside-out patch in the absence of GSH, but inhibit the currents in whole-cell recording where GSH is expected to exist. (3) GSSG is a potent S-glutathionylation inducer, and also strongly inhibits the channel. (4) The Kir4.1–Kir5.1 channel is inhibited by PDS, known to act on cysteine residues similarly to GSH. (5) The oxidant sensitivity of the heteromeric channel relies on a specific cysteine residue where S-glutathionylation takes place.
Critical residue for the S-glutathionylation
The cysteine residue Cys158 is critical for the oxidant sensitivity, mutation of which eliminates almost totally the channel inhibition by the oxidants, and diminished robustly biochemical interaction of the channel protein with BioGEE. Therefore, it is very likely that the incorporation of a glutathione moiety at Cys158 prevents the channel from a full opening, as we have recently shown in the vascular KATP channel (Yang et al. 2010, 2011). The Cys158 is located in the TM2. We have examined its accessibility to extracellular and cytosolic solutions. Using DTNB, a membrane-impermeant PDS, we have found that the channel is not inhibited when DTNB is applied extracellularly, while the channel is strongly inhibited when applied intracellularly.
This residue is inaccessible to intracellular GSSG when the channel is closed. In the open state, GSSG appears to enter the pore and interacts with Cys158 via a covalent bond that is not readily reversible with washout. The residue becomes inaccessible to intracellular GSSG when the channel is brought to the closed state by acidic pH. Under this condition, the Cys158 may not be exposed to GSSG to form S-glutathionylation, so that the acid-induced channel inhibition recovers normally as seen in the absence of GSSG (Xu et al. 2000; Yang et al. 2000). The state-dependent S-glutathionylation therefore is consistent with the location of Cys158 in the ion-conducting pathway (Doyle, 2004).
Using crystallographic structures of the bacterial Kir channel, we modelled the transmembrane helices of Kir5.1 as we did previously (Yang et al. 2011). Our results showed that this residue faced the inner cavity of the ion-conductive pore with its thiol-containing side group projecting into the pore (Fig. 11). The location of S-glutathionylation is supported by our current results of single-channel recording showing that GSSG inhibits NPopen but not single-channel conductance. Besides Cys158, there are another eight cysteine residues in the Kir5.1. Five of them are found in the intracellular protein domains. Although some of them may possibly be S-gluthionylated as suggested in the pulldown assay in Fig. 8, their contribution to channel gating seems modest. The Kir4.1–Kir5.1 channel loses its oxidant sensitivity almost completely after mutation of the Cys158 alone when the rest of the cysteines remain intact. Therefore, it is reasonable to believe that functionally the Cys158 is the determinant residue for S-glutathionylation of the channel.
Figure 11. Structural model of transmembrane helices of Kir5.1.
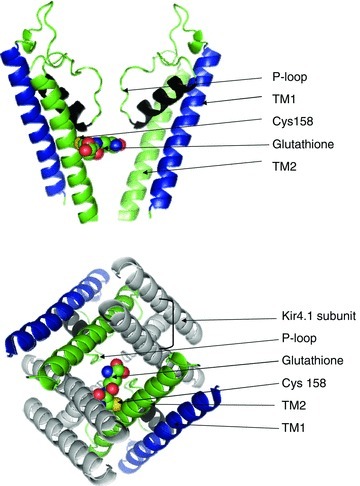
A, structural model of two monomers of Kir5.1 pore-forming domain in the closed state. A glutathione moiety was placed on one Cys158 in arbitrary orientation. B, bottom view of glutathione incorporated into two Cys158 of the TM2 in Kir4.1–Kir5.1 channel. Atom colour: green, carbon; red, oxygen; yellow, sulfur; blue, nitrogen.
Insight into the stoichiometry of the S-glutathionylation
The critical cysteine residue is missing in Kir4.1. Thus, each tetrameric channel consisting of two Kir4.1 and two Kir5.1 subunits has two cysteine residues at the location. We have examined the stoichiometry of these cysteine residues in the oxidant sensitivity of the heteromeric channel. Our results suggest that mutation of a single cysteine does not produce any significant reductions in the channel sensitivity to oxidants. Therefore, S-glutathionylation on one of the cysteines appears sufficient to produce the channel inhibition. Indeed, the space of the inner cavity can accommodate only one glutathione moiety, according to our modelling (Fig. 11). Such a glutathione moiety might not allow TM2 to function appropriately rather than sealing the inner mouth of the ion-conducting pathway. Therefore, with the glutathione moiety, the ion-conducting pathway might not be fully open in the open state, leading to the channel inhibition. The glutathione moiety might also prevent the channel from entering the full closed state, leading to an incomplete channel inhibition due to a steric hindrance effect.
In conclusion, the heteromeric Kir4.1–Kir5.1 channel but not the homomeric Kir4.1 is strongly inhibited by oxidants. The oxidant sensitivity seems to be mediated by S-glutathionylation at Cys158 in the TM2 helix. Thus, the heteromerization of Kir4.1 with Kir5.1 enables the heteromeric channel to sense and respond to oxidants. Such a modulation mechanism may allow the Kir4.1–Kir5.1 channel to be involved in cellular responses to oxidative stress in renal, retinal and nervous tissues where the channel is expressed.
Acknowledgments
This work was supported by the NIH (HD060959, NS073875), and the International Rett Syndrome Research Foundation. L.Y. was partially supported by a scholarship of Harbin Medical University. S.Z. is an MBD fellow of Georgia State University, and Y.Y. was supported by a B&B fellowship of Georgia State University.
Glossary
- DIA
diamide
- 2-DTP
2,2′-dithiodipyridine
- DTNB
5,5′-dithiobis-2-nitrobenzoic acid
- DTNP
2,2′-dithiobis-5-nitropyridine
- GSH
glutathione
- GSSG
oxidized glutathione
- Kir
inward rectifier potassium channels
- PDS
pyridine disulfide
- ROS
reactive oxygen species
Author contributions
Design of study and drafting of the manuscript were by X.J., Y.L. and C.J. X.J. and Y.L. contributed to collection, analysis, interpretation of data, and drafting of the manuscript. All authors approved the final version of the manuscript for publication.
Supplementary material
Supporting Information
References
- Bond CT, Pessia M, Xia XM, Lagrutta A, Kavanaugh MP, Adelman JP. Cloning and expression of a family of inward rectifier potassium channels. Receptors Channels. 1994;2:183–191. [PubMed] [Google Scholar]
- Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casamassima M, D’Adamo MC, Pessia M, Tucker SJ. Identification of a heteromeric interaction that influences the rectification, gating, and pH sensitivity of Kir4.1/Kir5.1 potassium channels. J Biol Chem. 2003;278:43533–43540. doi: 10.1074/jbc.M306596200. [DOI] [PubMed] [Google Scholar]
- Cui N, Giwa LR, Xu H, Rojas A, Abdulkadir L, Jiang C. Modulation of the heteromeric Kir4.1-Kir5.1 channels by PCO2 at physiological levels. J Cell Physiol. 2001;189:229–236. doi: 10.1002/jcp.10021. [DOI] [PubMed] [Google Scholar]
- Doyle DA. Structural changes during ion channel gating. Trends Neurosci. 2004;27:298–302. doi: 10.1016/j.tins.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Feldman EL. Oxidative stress and diabetic neuropathy: a new understanding of an old problem. J Clin Invest. 2003;111:431–433. doi: 10.1172/JCI17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation. 2003;108:2034–2040. doi: 10.1161/01.CIR.0000093661.90582.c4. [DOI] [PubMed] [Google Scholar]
- Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J Biol Chem. 2004;279:44065–44073. doi: 10.1074/jbc.M405985200. [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358:2606–2617. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, et al. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, et al. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am J Physiol Renal Physiol. 2008;294:F1398–1407. doi: 10.1152/ajprenal.00288.2007. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–4015. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia M, Imbrici P, D’Adamo MC, Salvatore L, Tucker SJ. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J Physiol. 2001;532:359–367. doi: 10.1111/j.1469-7793.2001.0359f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Cui N, Su J, Yang L, Muhumuza JP, Jiang C. Protein kinase C dependent inhibition of the heteromeric Kir4.1-Kir5.1 channel. Biochim Biophys Acta. 2007;1768:2030–2042. doi: 10.1016/j.bbamem.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Su J, Yang L, Lee M, Cui N, Zhang X, et al. Modulation of the heteromeric Kir4.1-Kir5.1 channel by multiple neurotransmitters via Gαq-coupled receptors. J Cell Physiol. 2008;214:84–95. doi: 10.1002/jcp.21169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116:85–97. doi: 10.1161/CIRCULATIONAHA.106.678342. [DOI] [PubMed] [Google Scholar]
- Shang L, Ranson SV, Tucker SJ. Kir5.1 underlies long-lived subconductance levels in heteromeric Kir4.1/Kir5.1 channels from Xenopus tropicalis. Biochem Biophys Res Commun. 2009;388:501–505. doi: 10.1016/j.bbrc.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Wu Z, Cui N, Shi W, Yang Y, Zhang X, et al. PKA phosphorylation of SUR2B subunit underscores vascular KATP channel activation by β-adrenergic receptors. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1205–1214. doi: 10.1152/ajpregu.00337.2007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanemoto M, Kittaka N, Inanobe A, Kurachi Y. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J Physiol. 2000;525:587–592. doi: 10.1111/j.1469-7793.2000.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zhang X, Cui N, Wu J, Piao H, Wang X, et al. Subunit-stoichiometric evidence for kir6.2 channel gating, ATP binding, and binding-gating coupling. Mol Pharmacol. 2007;71:1646–1656. doi: 10.1124/mol.106.030528. [DOI] [PubMed] [Google Scholar]
- Wenker IC, Kreneisz O, Nishiyama A, Mulkey DK. Astrocytes in the retrotrapezoid nucleus sense H+ by inhibition of a Kir4.1-Kir5.1-like current and may contribute to chemoreception by a purinergic mechanism. J Neurophysiol. 2010;104:3042–3052. doi: 10.1152/jn.00544.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Skolnick J, Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Cui N, Yang Z, Qu Z, Jiang C. Modulation of Kir4.1 and Kir5.1 by hypercapnia and intracellular acidosis. J Physiol. 2000;524:725–735. doi: 10.1111/j.1469-7793.2000.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shi W, Chen X, Cui N, Konduru AS, Shi Y, et al. Molecular basis and structural insight of vascular KATP channel gating by S-glutathionylation. J Biol Chem. 2011;286:9298–9307. doi: 10.1074/jbc.M110.195123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shi W, Cui N, Wu Z, Jiang C. Oxidative stress inhibits vascular KATP channels by S-glutathionylation. J Biol Chem. 2010;285:38641–38648. doi: 10.1074/jbc.M110.162578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Jiang C. Opposite effects of pH on open-state probability and single channel conductance of Kir4.1 channels. J Physiol. 1999;520:921–927. doi: 10.1111/j.1469-7793.1999.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Xu H, Cui N, Qu Z, Chanchevalap S, Shen W, et al. Biophysical and molecular mechanisms underlying the modulation of heteromeric Kir4.1-Kir5.1 channels by CO2 and pH. J Gen Physiol. 2000;116:33–45. doi: 10.1085/jgp.116.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- Yu L, Jin X, Yang Y, Cui N, Jiang –. Rosiglitazone inhibits vascular KATP channels and coronary vasodilation produced by isoprenaline. Br J Pharmacol. 2011;164:2064–2072. doi: 10.1111/j.1476-5381.2011.01539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Su J, Cui N, Gai H, Wu Z, Jiang C. The disruption of central CO2 chemosensitivity in a mouse model of Rett syndrome. Am J Physiol Cell Physiol. 2011;301:C729–738. doi: 10.1152/ajpcell.00334.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69(Suppl 8):108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.