

Abstract
In the present study it was hypothesized that voluntary aerobic exercise favours a pro-fibrotic phenotype and promotes adverse remodelling in hearts from spontaneously hypertensive rats (SHRs) in an angiotensin II-dependent manner. To test this, female SHRs at the age of 1 year were started to perform free running wheel exercise. Captopril was used to inhibit the renin–angiotensin system (RAS). Normotensive rats and SHRs kept in regular cages were used as sedentary controls. Training intensity, expressed as mean running velocity, was positively correlated with the left ventricular mRNA expression of TGF-β1, collagen-III and biglycan but negatively correlated with the ratio of sarcoplasmic reticulum Ca2+-ATPase (SERCA)2a to Na+–Ca2+ exchanger (NCX). A pro-fibrotic phenotype was verified by Picrosirius red staining. Sixty-seven per cent of SHRs performing free running wheel exercise died either spontaneously or had to be killed during a 6 month follow-up. In the presence of captopril, aerobic exercise did not show a similar positive correlation between training intensity and the expression of fibrotic markers. Moreover, in SHRs receiving captopril and performing free running wheel exercise, a training intensity-dependent reverse remodelling of the SERCA2a-to-NCX ratio was observed. None of these rats died spontaneously or had to be killed. In captopril-treated SHRs performing exercise, expression of mRNA for decorin, a natural inhibitor of TGF-β1, was up-regulated. Despite these differences between SHR-training groups with and without captopril, positive training effects (lower resting heart rate and no progression of hypertension) were found in both groups. In conclusion, high aerobic exercise induces an angiotensin II-dependent adverse remodelling in chronic pressure overloaded hearts. However, high physical activity can potentially induce reverse remodelling in the presence of RAS inhibition.
Key points
Physical exercise is recommended as first line therapy for hypertensive patients. However, studies investigating long-term effects of high intensity exercise on the progression of hypertensive heart disease have revealed conflicting results.
We show that high intensity aerobic exercise accelerates hypertensive heart disease and improves fibrosis.
Surprisingly, high intensity aerobic exercise in the presence of an angiotensin converting enzyme inhibitor not only attenuated training induced mal-adaptation but exerts positive repair processes. These effects were independent of blood pressure effects.
The results of this study provide evidence that high physical activity in hypertensives must be considered as an important risk factor rather than a therapeutic intervention.
Introduction
Cardiovascular disease is the leading cause of death in modern societies and is often associated with hypertension-dependent end organ damage. Advancing age is the major additional risk factor in particular in combination with hypertension. Aerobic exercise is a first-line therapeutic strategy to reduce the risk of cardiovascular disease with hypertension and ageing (Hagberg et al. 2000; Chobanian et al. 2003; Williams et al. 2004; Westhoff et al. 2007). However, the effect of aerobic exercise on heart remodelling and its potential mechanisms has not been examined. The limited available data in experimental animals do not support the idea of favourable effects of aerobic exercise on chronic pressure overloaded hearts (Schultz et al. 2007; van Deel et al. 2011). It remained unclear whether this maladaptive hypertrophy observed in such studies is indeed caused by over-stimulation of classical pro-hypertrophic stimuli such as the renin–angiotensin system.
Increased deposition of collagen I and III are believed to be important mechanisms mediating left ventricular stiffening concomitant with hypertension (Diez et al. 2002). Biglycan, a member of the small leucine rich proteoglycan family, is constitutively expressed in the heart; it triggers matrix assembly and activates TGF-β1 (Latif et al. 2005; Ruoslahti & Yamaguchi, 1991; Bereczki et al. 2007). TGF-β1, a cytokine locally produced in the heart, triggers the expression of collagens (Masague, 1990). Its activity can be attenuated by another small proteoglycan, decorin, which neutralizes TGF-β1 by binding (Ruoslahti, 1988; Hildebrand et al. 1994). TGF-β1 has been identified as a key molecule at the transition of adaptive cardiac hypertrophy to mal-adaptive cardiac hypertrophy (Villarreal & Dillmann, 1992; Boluyt et al. 1994). TGF-β1 plays a critical role due to its pro-fibrotic effects and its influence on the expression of the calcium handling protein SERCA2a (Lijnen et al. 2000; Mufti et al. 2008). SERCA2a is responsible for refilling sarcoplasmatic reticulum during the diastolic phase of a heart cyclus (Bers, 2000). A low SERCA2a expression or activity requires an up-regulation of the Na+–Ca2+ exchanger (NCX) to extrude calcium from the cytosol during the diastolic phase of a heart cycle (Schillinger et al. 2003). Therefore, a reduced SERCA/NCX ratio can be found in failing hearts and is a reliable marker of adverse remodelling.
The effect of aerobic exercise on pressure overloaded hearts is difficult to predict. On the one hand aerobic exercise lowers age-dependent arterial stiffening and thereby reduces afterload (Amaral et al. 2000; Fleenor et al. 2010). On the other hand aerobic exercise is associated with exercise-dependent activation of the sympathetic nervous system that then triggers a release of renin from cells of the juxtaglomerular apparatus and thereby activates the renin–angiotensin system (RAS). Angiotensin II, the main molecule of the RAS cascade, induces the cardiac expression of TGF-β1 (Wenzel et al. 2001). As stated above, this scenario will accelerate fibrosis and malfunction of calcium handling in the left ventricle. In the current study the role of the RAS cascade on exercise-dependent effects of cardiac remodelling was investigated by administration of captopril, an inhibitor of angiotensin converting enzyme (ACE).
In the present study we hypothesized that inhibition of the RAS will attenuate possible mal-adaptive effects caused by aerobic exercise in hypertensives and will shift the response to exercise in a more favourable direction. We further hypothesized that a high training intensity will lead to more detrimental effects than moderate exercise. To test these hypotheses we used spontaneously hypertensive rats (SHRs) at the age of 1 year with established hypertension. SHRs represent a suitable model of long-term adaptation to hypertension and are extensively characterized. Voluntary wheel running was used as a model of voluntary aerobic exercise (Bradley et al. 2008; Fleenor et al. 2010). As voluntary exercise results in different exercise intensities between animals, this allows us to correlate exercise-dependent adaptations with exercise intensity.
Methods
Ethical approval
All rats used in this study were house strains obtained from the animal facility of our institute. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (publication 85-23, revised 1996). The treatment protocols were approved by the local authorities.
Eleven normotensive female Wistar rats at the age of 12 months were used as normotensive control rats. Thirty-four female SHRs were used at the age of 12 months and randomly divided into one of the following four groups: SHR controls (SHR-sed; n= 11), SHR captopril (SHR-sed + Capto, n= 11), SHR exercise (SHR-train, n= 6), and SHR exercise with captopril (SHR-train + Capto; n= 6). Female rats were used because they have a higher spontaneous running activity (Eickelboom & Mills, 1988). Animals had free access to food and water. Rats in exercise groups had free access to running wheels that were connected to a computerized registration of running distances and running times. Where required, captopril was given with the drinking water at 30 mg kg−1 day−1. All rats were monitored on a weekly basis and they were removed from the programme if they developed a constitutive loss of body weight and blood pressure for three consecutive weeks. These rats were killed at that time and analysed in the same way as all other rats as well. All experiments were terminated after 26 weeks if rats did not develop signs of decompensated heart failure. At the end of the experimental period all rats were killed by cervical dislocation, hearts were rapidly excised, and the aorta was cannulated for retrograde perfusion with a 16-gauge needle connected to a Langendorff perfusion system to remove any blood. From the blood free hearts, atria and right ventricles were separated and the remaining left ventricle was weighed and rapidly frozen in liquid nitrogen for later analysis. The tibia lengths were measured and the left ventricular plus septum weight was normalized to tibia lengths and taken as the left ventricular weight. To determine the length of tibia, bones were separated from the muscle tissue and measured with a calliper.
Determination of blood pressure in vivo
For weekly determination of blood pressure, rats were set in a separate chamber (TSE Systems GmbH, Bad Homburg, Germany) and blood pressure (peak systolic blood pressure, diastolic blood pressure and heart rate) was determined via the tail-cuff method. Briefly, the mean of 10 consecutive blood pressure readings was obtained for each animal at weekly intervals. Before starting, rats were conditioned for 1 week by daily measurements and handling before any data were recorded. All blood pressure measurements were performed by the same person to minimize any variations. Body weight was recorded weekly and water consumption daily.
qRT-PCR
Total RNA was isolated from left ventricles using peqGold TriFast (peqlab; Biotechnologie GmbH, Germany) according to the manufacturer's protocol. To remove genomic DNA contamination, isolated RNA samples were treated with 1 U DNase per μg RNA (Invitrogen, Karlsruhe, Germany) for 15 min at 37°C. One microgram of total RNA was used in 10 μl reaction to synthesize cDNA using Superscript RNaseH reverse transcriptase (200 U μg−1 RNA; Invitrogen) and oligo dTs (Roche, Mannheim, Germany) as primers. Reverse transcriptase reactions were performed for 60 min at 37°C. Real-time quantitative PCR was performed using the Icycler IQ detection system (Bio-Rad, Munich, Germany) in combination with the IQ SYBR Green real-time PCR supermix (Bio-Rad). The thermal cycling programme consisted of initial denaturation in one cycle of 3 min at 95°C, followed by 45 cycles of 30 s at 95°C (denaturation), 30 s at primer specific annealing temperature, and 30 s at 72°C (elongation). Primer sequences used for determination are given in Table 1. Data are normalized to Hypoxanthine phosphoribosyltransferase (HPRT) expression that was used as a house-keeping gene in this study. Preliminary experiments with actin and β2 microglobulin, which were alternatively considered as house keeping genes, revealed the lowest variability in the HPRT group. Quantification was performed as described (Livak & Schmittgen, 2001).
Table 1.
Description of all primers used in this study
| Biglycan | Forward: | TGA TTG AGA ATG GGA GCC TGA G |
| Reverse: | CCT TGG TGA TGT TGT TGG AGT G | |
| Product length: | 144 bp | |
| NCBI: | NM_017087 | |
| Collagen- | Forward: | TGG AGT CGG AGG AAT G |
| III | ||
| Reverse: | GCC AGA TGG ACC AAT AG | |
| Product length: | 184 bp | |
| NCBI: | NM_032085 | |
| Decorin | Forward: | GGC AGT CTG GCT AAT GTT C |
| Reverse: | CTT CGG AGA TGT TGT TGT TAT G | |
| Product length: | 133 bp | |
| NCBI: | NM_024129 | |
| HPRT | Forward: | CCA GCG TCG TGA TTA GTG AT |
| Reverse: | CAA GTC TTT CAG TCC TGT CC | |
| Product length: | 132 bp | |
| NCBI: | NM_ 012583 | |
| NCX | Forward: | CCG TAA TCA GCA TTT CAG AG |
| Reverse: | GCC AGG TTC GTC TTC TTA AT | |
| Product length: | 187 bp | |
| NCBI: | NM_019268 | |
| SERCA2a | Forward: | CGA GTT GAA CCT TCC CAC AA |
| Reverse: | AGG AGA TGA GGT AGC GGA TGA A | |
| Product length: | 268 bp | |
| NCBI: | NM_001110139 | |
| TGF-β1 | Forward: | ATT CCT GGC GTT ACC TTG |
| Reverse: | CCT GTA TTC CGT CTC CTT GG | |
| Product length: | 117 bp | |
| NCBI: | NM_021578 | |
| PGC-1α | Forward: | AGT GCT CAG CCG AGG ACA CGA |
| Reverse: | TGC CCC TGC CAG TCA CAG GA | |
| Product length: | 180 bp | |
| NCBI: | NM_031347 | |
| Nrf-1 | Forward: | GGC ATC ACT GGC AGA GGC CG |
| Reverse: | GCT GCT GCG GTT TCC CCA GA | |
| Product length: | 168 bp | |
| NCBI: | NM_001100708 |
Picrosirius red staining
Samples were fixed and pre-incubated with Tissue-Tek (Sakura, Alphen, Netherlands) and sliced in 10 μm pieces. Tissue slices were fixed in Bouin solution and subsequently stained in 0.1% (w/v) Sirius red solution (Sigma-Aldrich Chemie, Steinheim, Germany). Slices were washed by 0.01 n HCl, Aqua destillata and counterstained for nuclei by Mayer's hemalaun solution, washed with Aqua destillata for 5 min and dehydrated with ethanol. Finally, slices were visualized under light microscopy. The amount of stained tissue was quantified via Leica Confocal Software Lite Version 2.6.1 (LCS Lite). The mean of n= 5 preparations was used to quantify the extent of collagen labelling.
Immunoblotting
Protein was extracted from frozen left-ventricular heart tissue in extraction buffer, containing (mmol l−1): Mops 5, sucrose 300, EGTA 1, bovine serum albumin 0.015, and 0.01% (v/v) protease inhibitor cocktail (Sigma, Taufkirchen, Germany). The homogenate was centrifuged at 1000 g at 4°C for 10 min and the supernatant was used for SERCA2a and NCX detection by Western blotting. Protein samples were loaded on NuPAGE Bis-Tris Precast gels (10%; Life Technology, Darmstadt, Germany) and subsequently transferred onto nitrocellulose membranes. Primary antibodies directed against SERCA2a, NCX and cardiac α-actin (loading control) were used as described before (Maxeiner et al. 2010).
Heart function analysis
Left ventricular function was assessed by Langendorff technique ex vivo as described before. Briefly, hearts were quickly removed and connected to a Langendorff perfusion system and a balloon was inserted into the left ventricle. Left ventricular diastolic pressure was adjusted to 10 mmHg and kept constant thereafter. After 20 min of stabilization, hearts were paced at 240 bpm for 5 min and left ventricular developed pressure (systolic pressure – diastolic pressure) and +dp/dt were recorded.
Statistics
Data are given as means ± standard deviation. In all cases in which more than two groups were compared, two-side ANOVA was used to decide about differences among groups followed by the Student–Newman–Keuls test as a post hoc analysis to identify between group differences. In cases where only two groups were compared, Student's t test for unpaired samples was used. Correlation analysis was performed as a linear regression analysis. Exact P values are given for all experiments and differences between groups are indicated by appropriate symbols. The statistical analysis was performed using SPSS v. 17.0.
Results
Animal characteristics are shown in Table 2 for normotensive control rats, sedentary SHRs, and running SHRs. Left ventricular weights were greater in all SHRs compared to normotensive rats. Left ventricular weight was lower in SHR-sed + Capto compared to SHR-sed and SHR-train + Capto compared to SHR-train. SHR-train had greater left ventricular weight than SHR-sed. Among these animals one rat died spontaneously after 10 weeks and could not be used for further analysis, two rats had to be killed after 24 weeks and one rat after 25 weeks. All other rats survived for 26 weeks.
Table 2.
Animal characteristics
| LV/TL (mg mm−1) | Psyst (mmHg) | Pdiast (mmHg) | HR (bpm) | |
|---|---|---|---|---|
| ANOVA | P≤ 0.000 | P≤ 0.000 | P≤ 0.000 | P≤ 0.011 |
| Wistar (n= 11) | 177 ± 22 | 136 ± 10 | 89 ± 9 | 398 ± 34 |
| SHR-sed (n= 11) | 259 ± 15* | 183 ± 17* | 119 ± 10 | 438 ± 24‡ |
| SHR-sed + Capto (n= 11) | 203 ± 16*# | 158 ± 16*# | 105 ± 11 | 422 ± 31 |
| SHR-train (n= 5/6) | 366 ± 55*# | 169 ± 28* | 107 ± 25 | 388 ± 49 |
| SHR-train + Capto (n= 6) | 286 ± 26*#§ | 179 ± 22* | 119 ± 14 | 424 ± 20 |
Data are means ± SD. LV/TL, ratio of left ventricular weight to tibia length; Psyst, systolic blood pressure; Pdiast, diastolic blood pressure; HR, heart rate. *P < 0.05 vs. Wistar; #P < 0.05 vs. SHR-sed; §P < 0.05 vs. SHR-train; ‡P < 0.05 vs. SHR-train.
Absolute blood pressure values are also given in Table 2 as they were analysed 8 weeks after the onset of voluntary exercise or sedentary control period. Systolic blood pressures were greater in all SHRs compared to normotensive control rats. Systolic blood pressure in SHR-sed + Capto was lower than in SHR-sed. Systolic blood pressures were not different between SHR-train + Capto and SHR-train. Diastolic blood pressures were also greater in SHRs compared to normotensive rats but not significantly different between SHR groups.
Training performance and effect on blood pressure
Running distance, running time and mean running velocity did not differ between SHR-train and SHR-train + Capto (Table 3). Heart rates significantly dropped in SHR-train compared to SHR-sed and SHR-train + Capto compared to SHR-sed + Capto (Table 3). This was taken as an indicator of positive training effects. On a molecular level this positive training effect was further confirmed by increased Nrf-1 and PGC-1α expression (Fig. 1). Systolic blood pressure was further raised in SHR-sed and this was significantly different compared to all other SHR groups (Table 3). No significant difference occurred in the development of diastolic blood pressures (Table 3).
Table 3.
Training performance and training-induced changes in SHR
| SHR-sed | SHR-sed + Capto | SHR-train | SHR-train + Capto | P value | |
|---|---|---|---|---|---|
| RD (km per week) | — | — | 48.9 ± 19.8 | 47.3 ± 12.4 | 0.858 |
| RT (h w−1) | — | — | 19.6 ± 6.4 | 22.0 ± 4.0 | 0.460 |
| RV (km per hour) | — | — | 2.43 ± 0.56 | 2.47 ± 0.19 | 0.775 |
| HR (bpm) | +1 ± 24 | +12 ± 40 | −60 ± 38* | −32 ± 27# | 0.001 |
| Psyst (mmHg) | +17 ± 16 | −19 ± 13* | −8 ± 33* | −4 ± 19* | 0.002 |
| Pdiast (mmHg) | +6 ± 14 | −12 ± 10 | −7 ± 27 | −1 ± 14 | 0.084 |
Data are means ± SD; RD, running distance; RT, running time; RV, running velocity; HR, heart rate; Psyst, systolic blood pressure; Pdiast, diastolic blood pressure. Two-side unpaired t test was used for RD, RT and RV. ANOVA and Student–Newman–Keuls post hoc analysis was used for HR, Psyst and Pdiast. *P < 0.05 vs. SHR-sed.; #P < 0.05 vs. SHR-sed + Capto.
Figure 1. Induction of PGC-1α and Nrf-1 by exercise in the left ventricle of SHRs.
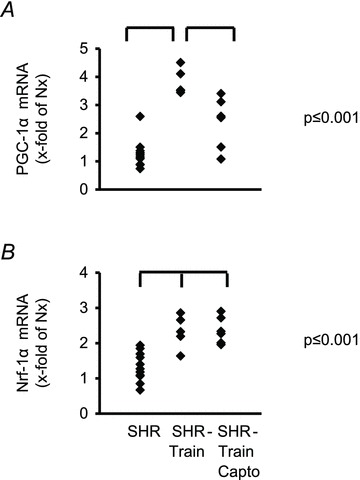
Data points from all rats are normalized to the mean expression of normotensive rats (Nx).
Effect of training intensity on left ventricular expression of fibrotic markers
In general, left ventricular TGF-β1 expression was higher in SHR-sed compared to normotensive rats (2.01 ± 0.73 AU vs. 1.00 ± 0.40 AU; n= 11; P= 0.001) and this was attenuated by captopril (SHR-sed + Capto; 0.82 ± 0.34 AU). This pro-fibrotic effect was significantly increased in SHR-train rats (6.72 ± 3.72 AU; P= 0.037). Moreover, a significant positive correlation between total running distance and running velocity with left ventricular expression of TGF-β1 was observed in SHR-train (Fig. 2). This positive correlation was converted into a negative correlation in SHR-train + Capto although this did not reach the level of significance. The strong correlation between tissue expression of TGF-β1 and training intensity in SHR-train suggests a pro-fibrotic effect of high training intensities. This was confirmed by analysis of collagen III expression. Collagen III is a classical down-stream target of TGF-β1. Again, left ventricular collagen III expression was higher in SHR-sed compared to normotensive rats and this effect was attenuated by captopril (Fig. 3). As assessed by Picrosirius red staining, total collagen was strongly increased in SHR-train and lower in SHR-train + Capto (Fig. 4). The pro-fibrotic influence of running in SHR-train was further stressed by a significant correlation of biglycan expression (Fig. 5). In contrast, decorin expression was not affected by wheel running but was more highly expressed in SHR-train + Capto compared to SHR-train (Fig. 6).
Figure 2. Correlation between training intensity and left ventricular TGF-β1 expression in SHR-train and SHR-train + Capto.
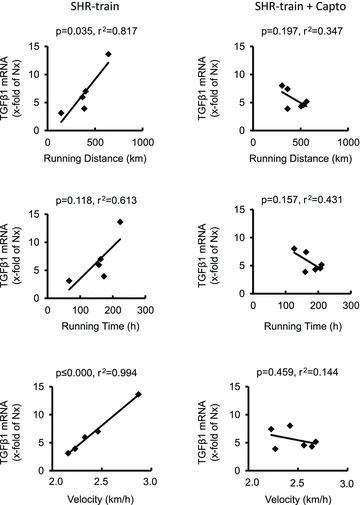
Expression is normalized to the mean expression of normotensive rats (Nx).
Figure 3. Correlation between training intensity and left ventricular collagen-III (Coll III) expression in SHR-train and SHR-train + Capto.
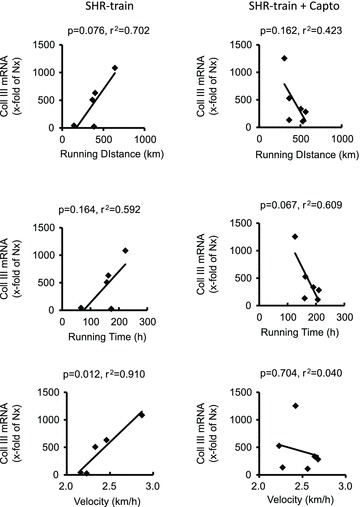
Expression is normalized to the mean expression of normotensive rats (Nx).
Figure 4. Left ventricular collagen expression.
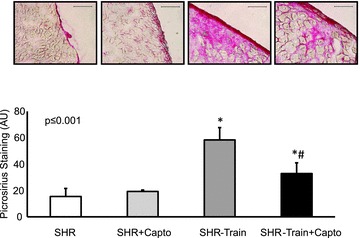
Tissue slices were stained for total collagen (Pircosirius red; top; scale bar 100 μm). Mean staining intensities ± SD are given as bar graphs. *Significant difference from SHRs; #significant difference from SHR-train.
Figure 5. Correlation between training intensity and left ventricular biglycan expression in SHR-train and SHR-train + Capto.
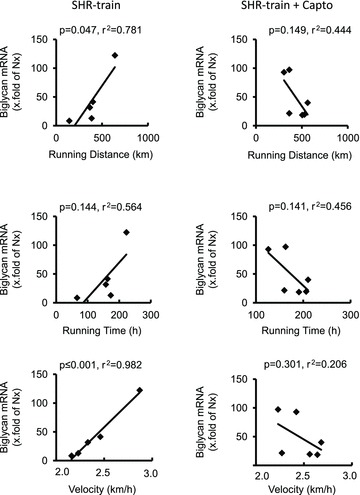
Expression is normalized to the mean expression of normotensive rats (Nx).
Figure 6. Correlation between training intensity and left ventricular decorin expression in SHR-train and SHR-train + Capto.
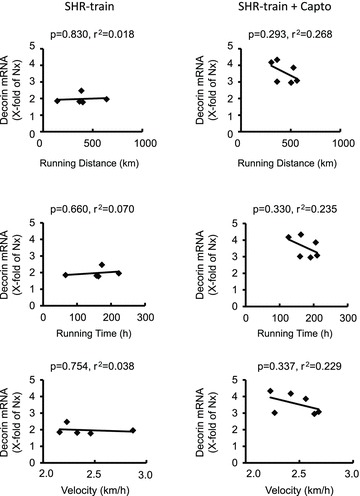
Expression is normalized to the mean expression of normotensive rats (Nx).
Effect of training intensity on left ventricular expression of SERCA2a and NCX
Finally, we evaluated the left ventricular expression of SERCA2a and NCX. Training intensity was negatively correlated with the SERCA2a-to-NCX ratio (Fig. 7). This effect of training intensity was again converted into a significant accentuation of the SERCA2a-to-NCX ratio in SHR-train + Capto (Fig. 7). These data on SERCA2a and NCX mRNA expression were confirmed by Western blot. Protein expression of NCX significantly increased in the SHR-train group (Fig. 8). From the data on fibrosis and calcium handling proteins we expected a significant effect on cardiac function. As indicated in Fig. 9 left ventricular developed pressure (LVDP) and +dp/dtmax were indeed significantly worsened in the SHR-train group and these detrimental effects were normalized by captopril.
Figure 7. Correlation between training intensity and left ventricular SERCA2a-to-NCX ratio in SHR-train and SHR-train + Capto.
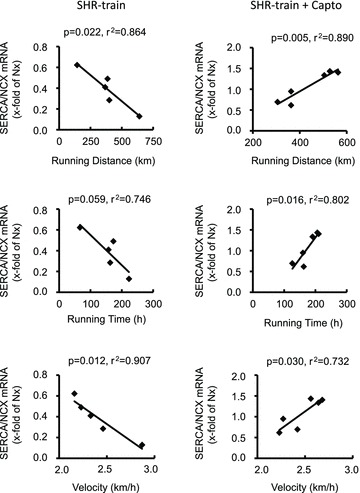
Expression is normalized to the mean expression of normotensive rats (Nx).
Figure 8. Left ventricular protein expression of sodium–calcium exchanger (NCX) and SERCA2a.
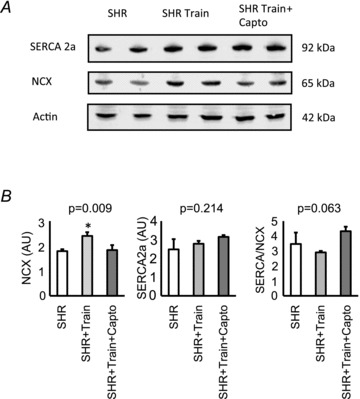
A, original blots; B, mean data ± SD. *Significant difference from SHRs.
Figure 9. Left ventricular function of rat hearts from SHR, SHR-train, and SHR-train + Capto.
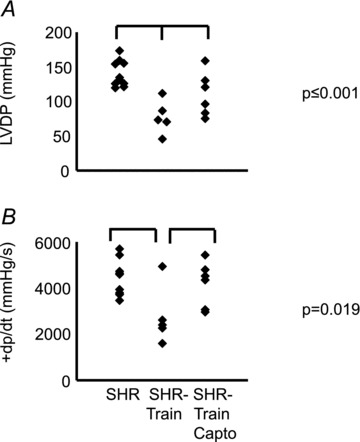
Left ventricular developed pressure (LVDP) and the first derivative (+dp/dt) are given in A and B, respectively.
Discussion
The present study assessed the effect of training intensity on left ventricular fibrosis and on the expression of calcium handling proteins. Both fibrosis and a low SERCA2a-to-NCX ratio favour diastolic heart failure (Diez et al. 2002; Schillinger et al. 2003). The results of the current study show that high training intensity of aerobic exercise in old SHRs with long-term established hypertension produces a pro-fibrotic phenotype with unfavourable expression patterns of calcium handling proteins. Inhibition of the angiotensin converting enzyme (ACE) attenuated any high intensity training-dependent effect on fibrotic markers. A complete new finding of this study is that under the tested conditions, high training intensity induces adverse remodelling as indicated by a correlation between training intensity and expression of pro-fibrotic markers and a highly significant inverse correlation between SERCA2a-to-NCX ratio and high intensity training. Of note, this effect was not accompanied by any significant differences in blood pressure. However, an age-dependent increase in blood pressure in SHRs was attenuated by free running wheel exercise independent of ACE inhibition.
In chronic pressure overload conditions the cytokine TGF-β1 plays a key role at the transition of adaptive cardiac hypertrophy to mal-adaptive hypertrophy because TGF-β1 expression is closely linked to this transition (Villarreal & Dillmann, 1992; Boluyt et al. 1994). Its cardiac expression is controlled by angiotensin II and depends on an activation of the RAS (Wenzel et al. 2001). Physical activity stimulates the sympathetic nervous system and thereby increases the release of renin from the cells of the juxtaglomerular apparatus. Therefore, it can be predicted that high physical activity is associated with strong activation of RAS and this should stress the chronic pressure overloaded heart. In the current study we provide further evidence for this hypothesis. Old SHRs are already at the risk to develop mal-adaptive hypertrophy and heart failure. This process was accelerated in SHRs performing exercise in a training intensity-dependent way. Our study is in line with a previous finding in a two-kidney, one-clip model in which moderate exercise reduced neither blood pressure nor hypertrophy although moderate exercise did not worsen left ventricular remodelling (Boissiere et al. 2008).
It is proposed that myocardial fibrosis is responsible for an increase in myocardial stiffness that may alter left ventricular diastolic properties (Diez et al. 2002). In untreated SHRs performing free running wheel activity, we observed a high mortality during the subsequent 6 months of follow-up. Quantification of heart function indicated reduced cardiac function as monitored by left ventricular developed pressure. After a period of high physical activity with positive training effects, such as reduced resting heart rate and lack of progressive increase in hypertension compared to sedentary SHRs, they spontaneously lost body weight and developed a drop in blood pressure. This led to killing for ethical reasons (3 rats) or sudden death (1 rat). In total, 4 out of 6 rats did not reach the predicted end-point of follow-up. In contrast no such effect occurred in SHRs treated with captopril. Neither the absolute amount of physical activity nor the training effects on heart rate and blood pressure were different between both training groups. The only difference was the subsequent inhibition of the RAS. Among the fibrotic markers we found an attenuation of training intensity-dependent expression of TGF-β1, of collagen III, a down-stream target of TGF-β1 signalling, and biglycan in the captopril running group. TGF-β1 and biglycan are known to be induced in a RAS-dependent manner (Zimmermann et al. 1999). High training intensity in captopril treated SHRs not only reveals a lack of progression of fibrosis but also shows a training intensity-dependent reverse remodelling. The effect of reverse remodelling was more stringent for the effect of SERCA2a-to-NCX ratio. This might be explained by earlier in vitro findings. In isolated and cultured adult rat ventricular cardiomyocytes, angiotensin II reduces the expression of SERCA2a in a TGF-β1-dependent way (Mufti et al. 2008). In contrast, stimulation of adrenergic receptors increases the expression of SERCA2a (Anwar et al. 2005, 2008). High training intensity activates the sympathetic nervous system leading to an activation of cardiac adrenoceptors. In the presence of ACE inhibition, angiotensin II does not repress SERCA2a expression and the remaining net effect of running activity is an improvement of the SERCA2a-to-NCX ratio. The effect of ACE inhibition in SHRs performing free running wheel exercise was less pronounced when fibrotic markers were analysed. However, the strong correlation between training intensity and expression of TGF-β1, collagen III and biglycan was abrogated. TGF-β1 activity is not strictly linked to TGF-β1 expression and modified by small proteoglycans. Decorin normally binds TGF-β1 and by such an immobilization it attenuates its binding to TGF-β1 receptors (Ruoslahti, 1988; Hildebrand et al. 1994). Of note, in training performing SHRs with captopril the expression of decorin was higher than in all other groups. This might contribute to the positive effect of ACE inhibition in this case because decorin is known to ameliorate adverse remodelling by inhibiting TGF-β (Jahanyar et al. 2007). However, for decorin no association between training intensity and expression was found.
In the literature there is a controversy about differences between physiological and pathophysiological hypertrophy and the effect of exercise on these different types of hypertrophy. In general, pathophysiological hypertrophy is considered to be triggered by the calcineurin/NFAT pathway and this seems to be reduced by moderate cardiac hypertrophy (Oliveira et al. 2009; Libonati et al. 2011). In contrast, free running wheel models are known to induce excessive cardiac hypertrophy. Our study points out that activation of the renin–angiotensin system significantly contributed to this effect. We have previously demonstrated that angiotensin II reduces SERCA2a expression leading to reduced myocyte function and the data of this study are in agreement with this finding (Mufti et al. 2008).
It is still a matter of debate whether physical activity is protective against the progression of hypertension. To date modest levels of regular voluntary aerobic exercise are recommended as first-line therapy for preventing cardiovascular disease. Physical activity does not only affect the heart but also age-dependent artery stiffening (Fleenor et al. 2010). Thus overall physical activity may affect more than the heart function only. However, animal experiments do not provide strong evidence that physical activity itself lowers blood pressure and improves the outcome (Schultz et al. 2007; van Deel et al. 2011). Our study is in line with previous animal studies showing detrimental effects of physical activity on chronic pressure overloaded hearts. Our study is also in line with proceeding animal studies that do not reveal a blood pressure lowering effect in SHRs (Schlüter et al. 2010; Coimbra et al. 2008). A main difference between animal studies and patient studies investigating the effect of exercise on blood pressure and heart function may be that the initiation of even moderate physical activity in humans lowers their body weight. The body mass index strongly correlates to blood pressure (Hedeyati et al. 2011). Furthermore, patients participating in life-style programmes have often additional changes in their diet which also influences the body mass index. Another difference between animal studies and clinical studies in this field is that the so-called sedentary rats are not really sedentary. They freely move around in their cages whereas sedentary patients often have very low and probably non-physiological low levels of physical activity. Despite these limitations the current study identifies the RAS as a potential risk in high training intensity and provides some evidence for a training-induced reverse remodelling in the presence of RAS inhibition. It has been reported that RAS inhibition may reverse myocardial fibrosis (Diez et al. 2002). Here we show that additional high aerobic exercise induces reverse remodelling in addition. The study may contribute to optimize treatment regimes of hypertensive patients performing aerobic exercise.
As a limitation of this study, the use of only one specific type of sex, female rats, must be considered. Of course, the data cannot predict the outcome if male rats are used. Female rats were used in this study because female rats have a higher spontaneous running activity (Eickelboom & Mills, 1988). This is a prerequisite of our study. However, in a recent meta-analysis of studies using SHR and exercise we could not find any major differences between male and female rats with respect to the influence of exercise on blood pressure and hypertrophy (Schlüter et al. 2010). Therefore, we do not expect such a difference. However, for the same reason than we used female rats, the majority of comparable studies were also performed with females rats (i.e. Schultz et al. 2007).
Conclusions
The results of this study provide evidence that high physical activity in hypertensives must be considered as an important risk factor rather than a therapeutic intervention. We identified the activation of RAS as a main trigger for adverse remodelling in hearts of SHRs with high training intensity. Moreover, in the presence of ACE inhibition there was a training intensity-dependent reverse remodelling not identified before. Therefore one may predict that in the presence of effective inhibition of RAS voluntary aerobic exercise significantly contributes to reverse remodelling.
Acknowledgments
Grant funding was performed by the Deutsche Forschung-sgemeinschaft as a personal grant to KDS (SCHLU324/7-1) and PROMISE.
Glossary
- ACE
angiotensin converting enzyme
- NCX
Na+–Ca2+ exchanger
- Nrf-1
nuclear respiratory factor-1
- PGC-1α
peroxisome proliferator activated receptor γ coactivator 1α
- RAS
renin–angiotensin system
- SERCA
sarcoplasmatic reticulum Ca2+-ATPase
- SHR
spontaneously hypertensive rat
- TGF-β1
transforming growth factor-β1
Author contributions
The authors contributed to the study as follows: analysis and interpretation of data (R.M.); conception and design (R.S.); drafting the article and final approval of the revision to be published (K.D.S.). All experiments were done at the Physiologisches Institut, JLU Gießen, Germany.
References
- Amaral SL, Zorn TMT, Michelini LC. Exercise training normalizes wall-to-lumen ratio of the gracilis muscle arterioles and reduces pressure in spontaneously hypertensive rats. J Hypertens. 2000;18:1563–1572. doi: 10.1097/00004872-200018110-00006. [DOI] [PubMed] [Google Scholar]
- Anwar A, Taimor G, Korkusuz H, Schreckenberg R, Berndt T, Abdallah Y, Piper HM, Schlüter K-D. PKC-dependent signal transduction pathways increase SERCA2a expression in adult rat cardiomyocytes. J Mol Cell Cardiol. 2005;39:911–919. doi: 10.1016/j.yjmcc.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Anwar A, Schlüter K-D, Heger J, Piper HM, Euler G. Enhanced SERCA2a expression improves contractile performance of ventricular cardiomyocytes of rat under adrenergic stimulation. Pflugers Arch. 2008;457:485–491. doi: 10.1007/s00424-008-0520-7. [DOI] [PubMed] [Google Scholar]
- Bereczki E, Gonda S, Csont T, Korpos E, Zvara A, Ferdinandy P, Santha M. Overexpression of biglycan in the heart of transgenic mice: An antibody microarray study. J Proteom Res. 2007;6:854–861. doi: 10.1021/pr060571b. [DOI] [PubMed] [Google Scholar]
- Bers DM. Calcium flux involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- Boissiere J, Eder V, Machet M-C, Courteix D, Bonnet P. Moderate exercise training does not worsen left ventricle remodelling and function in untreated severe hypertensive rats. J Appl Physiol. 2008;104:321–327. doi: 10.1152/japplphysiol.00442.2007. [DOI] [PubMed] [Google Scholar]
- Boluyt MO, O’Neill L, Meredith AL, Bing OHL, Brooks WW, Conrad CH, Crow MT, Lakatta EG. Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure. Circ Res. 1994;75:23–32. doi: 10.1161/01.res.75.1.23. [DOI] [PubMed] [Google Scholar]
- Bradley RL, Jeon YL, Liu F-F, Maratos-Flier E. Voluntary exercise improves insulin sensitivity and adipose tissue inflammation in diet-induced obese mice. Am J Physiol Endocrinol Metab. 2008;295:E586–E594. doi: 10.1152/ajpendo.00309.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jr, Jones DW, Materson BJ, Oparil S, Wright TJ, Jr, Roccella EJ. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560–2572. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- Coimbra R, Sanchez LS, Potenza JM, Rossoni LV, Amaral SL, Michelini LC. Is gender crucial for cardiovascular adjustment induced by exercise training in female spontaneously hypertensive rats. Hypertension. 2008;52:514–521. doi: 10.1161/HYPERTENSIONAHA.108.114744. [DOI] [PubMed] [Google Scholar]
- Diez J, Querejeta R, Lopez B, Gonzalez A, Larman M, Ubago JLM. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. 2002;105:2512–2517. doi: 10.1161/01.cir.0000017264.66561.3d. [DOI] [PubMed] [Google Scholar]
- Eickelboom R, Mills R. A microanalysis of wheel running in male and female rats. Physiol Behav. 1988;43:625–630. doi: 10.1016/0031-9384(88)90217-x. [DOI] [PubMed] [Google Scholar]
- Fleenor BS, Marshall KD, Durrant JR, Lesniewski LA, Seals DR. Arterial stiffening with ageing is associated with transforming growth factor-β1-related changes in adventitial collagen: reversal by aerobic exercise. J Physiol. 2010;588:3971–3982. doi: 10.1113/jphysiol.2010.194753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg JM, Park J-J, Brown MD. The role of exercise training in the treatment of hypertension. Sports Med. 2000;30:193–206. doi: 10.2165/00007256-200030030-00004. [DOI] [PubMed] [Google Scholar]
- Hedeyati SS, Elsayed EF, Reilly RF. Non-pharmacological aspects of blood pressure management: what are the data. Kidney Int. 2011;79:1061–1070. doi: 10.1038/ki.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand A, Romaris M, Rasmussen LM, Heinegard D, Twardzik DR, Border WA, Ruoslahti E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodullin with transforming growth factor beta. Biochem J. 1994;302:527–534. doi: 10.1042/bj3020527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahanyar J, Joyce DL, Southard RE, Loebe M, Noon GP, Koerner MM, Torre-Amione G, Youker KA. Decorin-mediated transforming growth factor-β inhibition ameliorates adverse cardiac remodelling. J Heart Lung Transplan. 2007;26:34–40. doi: 10.1016/j.healun.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Latif N, Sarathchandra P, Taylor PM, Antoniw J, Yacoub MH. Localization and pattern of expression of extracellular matrix components in human heart valves. J Heart Valve Dis. 2005;14:542–548. [PubMed] [Google Scholar]
- Libonati JR, Sabri A, Xiao C, MacDonnell SM, Renna BF. Exercise training improves systolic function in hypertensive myocardium. J Appl Physiol. 2011;111:1637–1643. doi: 10.1152/japplphysiol.00292.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lijnen PJ, Petrov VV, Fagard FH. Induction of cardiac fibrosis by transforming growth factor-β1. Mol Genet Metab. 2000;71:418–435. doi: 10.1006/mgme.2000.3032. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[–ΔΔCT] Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Massagué J. The transforming growth factor-β family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- Maxeiner H, Krehbiel N, Müller A, Woitasky N, Akintürk H, Müller M, Weigand MA, Abdallah Y, Kasseckert S, Schreckenberg R, Schlüter K-D, Wenzel S. New insights into paracrine mechanisms of human cardiac progenitor cells. Eur J Heart Fail. 2010;12:730–737. doi: 10.1093/eurjhf/hfq063. [DOI] [PubMed] [Google Scholar]
- Mufti S, Wenzel S, Euler G, Piper HM, Schlüter K-D. Angiotensin II-dependent loss of cardiac function: Mechanisms and pharmacological targets attenuating this effect. J Cell Physiol. 2008;217:242–249. doi: 10.1002/jcp.21501. [DOI] [PubMed] [Google Scholar]
- Oliveira RSF, Ferreira JCB, Gomes ERM, Paixao NA, Rolim NPL, Medeiros A, Guatimosim S, Brum PC. Cardiac anti-remodelling effect of aerobic training is associated with a reduction in the calcineurin/NFAT signalling pathway in heart failure mice. J Physiol. 2009;587:3899–3910. doi: 10.1113/jphysiol.2009.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E. Structure and biology of proteoglycans. Annu Rev Cell Biol. 1988;4:229–255. doi: 10.1146/annurev.cb.04.110188.001305. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Yamaguchi Y. Proteoglycans as modulators of growth factor activities. Cell. 1991;64:867–869. doi: 10.1016/0092-8674(91)90308-l. [DOI] [PubMed] [Google Scholar]
- Schillinger W, Fiolet JW, Schlotthauer K, Hasenfuss G. Relevance of Na+-Ca2+ exchange in heart failure. Cardiovasc Res. 2003;57:921–933. doi: 10.1016/s0008-6363(02)00826-x. [DOI] [PubMed] [Google Scholar]
- Schlüter K-D, Schreckenberg R, da Costa Rebelo RM. Interaction between exercise and hypertension in spontaneously hypertensive rats: a meta-analysis of exerpimental studies. Hypertens Res. 2010;33:1155–1161. doi: 10.1038/hr.2010.155. [DOI] [PubMed] [Google Scholar]
- Schultz RL, Swallow JG, Waters RP, Kuzman JA, Redetzke RA, Said S, de Escobar GM, Gerdes AM. Effects of excessive long-term exercise on cardiac function and myocyte remodeling in hypertensive heart failure rats. Hypertension. 2007;50:410–416. doi: 10.1161/HYPERTENSIONAHA.106.086371. [DOI] [PubMed] [Google Scholar]
- Van Deel ED, de Boer M, Kuster DW, Boontje NM, Holemans P, Sipido KR, van der Velden J, Duncker DJ. Exercise training does not improve cardiac function in compensated or decompensated left ventricular hypertrophy induced by aortic stenosis. J Mol Cell Cardiol. 2011;50:1017–1025. doi: 10.1016/j.yjmcc.2011.01.016. [DOI] [PubMed] [Google Scholar]
- Villarreal FK, Dillmann WH. Cardiac hypertrophy-induced changes in mRNA levels of TGF-β1, fibronectin, and collagen. Am J Physiol Heart Circ Physiol. 1992;262:H1861–H1866. doi: 10.1152/ajpheart.1992.262.6.H1861. [DOI] [PubMed] [Google Scholar]
- Wenzel S, Taimor G, Piper HM, Schlüter K-D. Redox-sensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-β expression in adult ventricular cardiomyocytes. FASEB J. 2001;15:2291–2293. doi: 10.1096/fj.00-0827fje. [DOI] [PubMed] [Google Scholar]
- Westhoff TH, Franke N, Schmidt S, Vallbracht-Israng K, Meissner R, Yildirim H, Schlattmann P, Zidek W, Dimeo F, van der Giet M. Too old to benefit from sports? The cardiovascular effects of exercise training in elderly subjects treated for isolated systolic hypertension. Kidney Blood Press Res. 2007;30:240–247. doi: 10.1159/000104093. [DOI] [PubMed] [Google Scholar]
- Williams B, Poulter NR, Brown MJ, Davis M, McInnes GT, Potrter JF, Sever PS, McThom S. Guidelines for management of hypertension: report of the fourth working party of the British Hypertension Society BHS IV. J Hum Hypertens. 2004;18:139–185. doi: 10.1038/sj.jhh.1001683. [DOI] [PubMed] [Google Scholar]
- Zimmermann R, Kastens J, Linz W, Wiemer G, Schölkens BA, Schaper J. Effect of long-term ACE inhibition on myocardial tissue in hypertensive stroke-prone rats. J Mol Cell Cardiol. 1999;31:1447–1156. doi: 10.1006/jmcc.1999.0976. [DOI] [PubMed] [Google Scholar]