

Abstract
Animal studies have reported dietary salt-induced reductions in vascular function independent of increases in blood pressure (BP). The purpose of this study was to determine if short-term dietary sodium loading impairs cutaneous microvascular function in normotensive adults with salt resistance. Following a control run-in diet, 12 normotensive adults (31 ± 2 years) were randomized to a 7 day low-sodium (LS; 20 mmol day−1) and 7 day high-sodium (HS; 350 mmol day−1) diet (controlled feeding study). Salt resistance, defined as a ≤5 mmHg change in 24 h mean BP determined while on the LS and HS diets, was confirmed in all subjects undergoing study (LS: 84 ± 1 mmHg vs. HS: 85 ± 2 mmHg; P > 0.05). On the last day of each diet, subjects were instrumented with two microdialysis fibres for the local delivery of Ringer solution and 20 mm ascorbic acid (AA). Laser Doppler flowmetry was used to measure red blood cell flux during local heating-induced vasodilatation (42°C). After the established plateau, 10 mm l-NAME was perfused to quantify NO-dependent vasodilatation. All data were expressed as a percentage of maximal cutaneous vascular conductance (CVC) at each site (28 mm sodium nitroprusside; 43°C). Sodium excretion increased during the HS diet (P < 0.05). The plateau % CVCmax was reduced during HS (LS: 93 ± 1 % CVCmax vs. HS: 80 ± 2 % CVCmax; P < 0.05). During the HS diet, AA improved the plateau % CVCmax (Ringer: 80 ± 2 % CVCmax vs. AA: 89 ± 3 % CVCmax; P < 0.05) and restored the NO contribution (Ringer: 44 ± 3 % CVCmax vs. AA: 59 ± 6 % CVCmax; P < 0.05). These data demonstrate that dietary sodium loading impairs cutaneous microvascular function independent of BP in normotensive adults and suggest a role for oxidative stress.
Key points
Pre-clinical studies suggest that acute dietary sodium loading impairs vascular function without alterations in blood pressure; however, human data are lacking.
In this study, normotensive salt-resistant adults participated in a controlled feeding study, in which they consumed a low-sodium diet for 1 week and a high-sodium diet for 1 week, in random order. During each diet, microvascular function was assessed.
Here we report the novel finding of sodium-induced impairments in microvascular function independent of blood pressure in healthy adults.
We additionally show that function was improved by the administration of the anti-oxidant ascorbic acid.
Therefore, in addition to its well-known importance for blood pressure control, lowering sodium intake may have beneficial effects on microvascular function in healthy normotensive adults.
Introduction
Excess dietary sodium consumption has been implicated in the pathogenesis of cardiovascular disease, including hypertension (Meneton et al. 2005). Endothelial dysfunction is a precursor of atherosclerotic vascular disease, an early event in the development and progression of cardiovascular disease (Levy et al. 2001; Cohuet & Struijker-Boudier, 2006; Holowatz et al. 2008). A decrease in endothelial-derived nitric oxide (NO) is detectable in the microvasculature and precedes atherosclerotic plaque development in the conduit vessels (Rossi & Carpi, 2004; Rossi et al. 2006). Impaired microvascular function is a systemic process (IJzeman et al. 2003) and may be the vascular bed in which endothelial dysfunction first develops (Joannides et al. 2006). Recently, the human cutaneous microcirculation has emerged as an accessible and representative vascular bed for the assessment of human microvascular function (Abularrage et al. 2005; Rossi et al. 2006; Holowatz et al. 2008). Given the high level of dietary sodium habitually consumed in modern society (Frassetto et al. 2001) and its potential detrimental influence on cardiovascular function, understanding the effect of dietary sodium on the microvasculature may provide mechanistic insight into early vascular consequences of excess salt intake.
In humans, the detrimental effects of dietary sodium have typically been attributed to increases in blood pressure (BP) (Elliott et al. 1996; Midgley et al. 1996). However, recent studies in rodent models have demonstrated impaired vascular endothelial function during salt loading, without alterations in BP (Lenda et al. 2000; Lenda & Boegehold, 2002; Matavelli et al. 2007; Nurkiewicz & Boegehold, 2007; Zhu et al. 2007). These findings in rodents suggest that salt-induced impairments in vascular function may be independent of BP.
Impairments in endothelial function during acute salt loading, assessed via acetylcholine-induced increases in forearm blood flow, were observed in healthy, normotensive young men (Tzemos et al. 2008). Additionally, this study reported less pronounced vasoconstrictor responses to incremental doses of l-NMMA (competitive inhibitor of endothelial NO synthase) after salt loading, suggestive of a reduction in basal levels of endothelial-derived NO (Tzemos et al. 2008). However, increases in systolic BP during acute salt loading were observed (Tzemos et al. 2008) such that the detrimental effects of salt intake on endothelial function cannot be separated from the effects on BP. Recently, it was demonstrated that the NO component of acetylcholine-induced vasodilatation was reduced following a 5 day high-salt diet in men but not in women (Eisenach et al. 2011). Although the aforementioned studies (Tzemos et al. 2008; Eisenach et al. 2011) indicate impairments in vascular function during salt loading, salt sensitivity status of participants was not assessed. Our laboratory previously reported that local infusion of hypertonic saline (3% NaCl) attenuated cutaneous vasodilatation in response to local heating but did not alter BP (DuPont et al. 2011a), indicating that increases in interstitial sodium concentration may impair microvascular function independent of BP. Given the limited information on the BP-independent effects of dietary salt on microvascular function, further investigation is warranted.
Studies in animal models have demonstrated that dietary salt-induced declines in NO production and endothelium-dependent dilatation are probably the result of increases in reactive oxygen species (ROS) (Lenda et al. 2000), specifically superoxide (Nurkiewicz & Boegehold, 2007; Zhu et al. 2007). ROS reduce NO production and bioavailability by oxidizing NO synthase or critical cofactors essential for NO synthesis and/or by reacting directly with NO itself, forming potent oxidants (Harrison et al. 2007). The deleterious effects of salt on the vasculature in these rodent studies may be a result of increased levels of oxidative stress, and the subsequent decline in NO bioavailability, in the microcirculation.
Therefore, the goal of the current study was to assess cutaneous microvascular function during short-term dietary sodium loading in normotensive adults determined to have salt-resistant BP. Salt sensitivity of BP was individually determined during a controlled dietary sodium perturbation, and microvascular function was assessed. In those determined to have salt-resistant BP, we tested the hypothesis that NO-mediated cutaneous vasodilatation in response to local heating would be attenuated during a high-sodium diet, suggesting sodium-induced impairments in microvascular function independent of changes in BP. We further hypothesized that dietary sodium-induced declines in cutaneous vasodilatation would be ameliorated by the local infusion of ascorbic acid, providing functional evidence for a role of oxidative stress.
Methods
Subjects
Experimental protocols were approved by the Institutional Review Board at the University of Delaware and conformed to the guidelines set forth by the Declaration of Helsinki. Verbal and written consent was obtained from all subjects prior to the study. Twelve young healthy adults determined to have salt-resistant BP participated (see salt sensitivity classification parameters below).
During an initial screening visit, all subjects completed a medical history form, resting brachial BP (Dinamap Dash 2000; GE Medical Systems, Milwaukee, WI) and a resting 12-lead electrocardiogram were recorded, height and weight were measured, and a venous blood sample was collected. Subjects were 22–46 years of age. Older adults were not included because of age-related alterations in the cutaneous vascular responses to local heating (Minson et al. 2002; Black et al. 2008). All subjects were normotensive with a resting seated systolic BP of <120 mmHg and diastolic BP of <80 mmHg, as defined by JNC7 (Chobanian et al. 2003). Subjects were free of any known cardiovascular disease and any evidence of renal, metabolic, pulmonary or neurological disease. Subjects were non-obese (body mass index (BMI) < 30 kg m−2) and did not use tobacco products. Menopausal women were excluded from study participation because salt sensitivity of BP increases with menopause (Schulman et al. 2006).
Dietary salt perturbation
This experiment was a controlled feeding study. All food was prepared by a registered dietitian. There is wide inter-individual variation in typical dietary salt intake (CDC, 2010). Consequently, in order to normalize basal dietary sodium intake, subjects completed a 3- to 7-day run-in diet (100 mmol sodium day−1). After completing the run-in diet, subjects were assigned to 7 days of a low-sodium diet (LS; 20 mmol day−1) and 7 days of a high-sodium diet (HS; 300–350 mmol day−1), in a randomized order. These target sodium intakes were selected to allow us to appropriately classify adults with salt-resistant BP and are consistent with previously published studies (He et al. 2001; Eisenach et al. 2011; Wenner et al. 2011). Food items provided constant daily amounts of carbohydrates (50%), fat (30%) and protein (20%). Potassium intake averaged 75.0 ± 2.6 mmol day−1. The caloric content of the diet was adjusted using the Mifflin–St Jeor equation to maintain constant body weight (Frankenfield et al. 2005). Daily fluid intake was monitored and recorded. Subjects were instructed to maintain their normal activity levels during the dietary sodium perturbation.
On the last day of the LS and HS conditions, a 24 h urine collection was obtained and analysed for total volume, urinary electrolytes (EasyElectrolyte Analyzer; Medica, Bedford, MA, USA) and urine osmolality (Advanced 3D3 Osmometer; Advanced Instruments, Norwood, MA, USA). Urinary sodium and potassium excretion were calculated and normalized to the entire 24 h period. Free water clearance, creatinine clearance and fractional excretion of sodium and chloride were calculated using standard equations. During the concurrent 24 h period, an ambulatory BP monitor (Model 90207; Spacelabs Medical, Issaquah, WA, USA) was worn on the non-dominant arm and measured BP every 20 min during waking hours and every 30 min during sleep. Laboratory BPs were additionally measured using an automated oscillometric sphygmomanometer (Dinamap Dash 2000; GE Medical Systems, Milwaukee, WI, USA) during the experimental visits for the LS and HS diets.
Serum and plasma samples were obtained at enrollment and during the experimental testing day for the LS and HS diet. Haemoglobin (Hb 201+ model, HemoCue, Lake Forest, CA, USA) and haematocrit (Clay Adams Brand, Readacrit Centrifuge, Becton Dickinson, Sparks, MD, USA) were analysed from whole blood. Serum electrolytes (EasyElectrolyte Analyzer) and plasma osmolality (Advanced 3D3 Osmometer, Advanced Instruments, Norwood, MA, USA) were also assessed.
Salt sensitive/resistant status classification
Salt resistance was defined as a ≤5 mmHg change in 24 h mean BP determined while on the LS and HS diets (Schmidlin et al. 2007), an assessment that is reproducible within subjects (Weinberger, 1996). The classification of salt resistance was determined on an individual basis after completion of the full dietary sodium perturbation. Subjects classified as salt sensitive (i.e. a >5 mmHg change in 24 h mean BP determined while on the LS and HS diets) were excluded from analysis, because they could not be used to test the a priori study hypothesis regarding the BP-independent effects of dietary sodium loading.
Instrumentation
On the last day of LS and HS conditions, subjects reported to the laboratory for assessment of cutaneous microvascular function. All protocols were performed with subjects in a semi-recumbent position. Using sterile technique, subjects were instrumented with two microdialysis fibres (10 mm, 30 kDa cut-off membrane, MD 2000; Bioanalytical Systems, West Lafayette, IN, USA) in the ventral side of the left forearm for localized delivery of pharmacological agents, as previously described (DuPont et al. 2011a,b). Briefly, the forearm was temporarily anaesthetized with ice for 10 min prior to fibre insertion (Hodges et al. 2009). Thereafter, a 25-gauge needle was inserted into the skin, with entry and exit points ∼2.5 cm apart, and microdialysis fibres were threaded through the lumen of the needle, which was removed upon placement of the semi-permeable membrane of the fibre. All fibres were then perfused with lactated Ringer solution for 60–90 min, allowing for local hyperaemia due to needle insertion to subside.
Microdialysis sites were perfused with lactated Ringer solution to serve as the control site and 20 mm ascorbic acid (Bioniche Pharma USA; Lake Forest, IL, USA) to locally scavenge ROS. A protocol schematic is illustrated in Fig. 1. All pharmacological solutions were mixed immediately prior to usage, dissolved in lactated Ringer solution, and filtered using syringe microfilters (Whatman Puradisc 13 mm Syringe Filters, Florham Park, NJ, USA). The solutions were also wrapped in foil to inhibit photodegradation of the agents.
Figure 1. Schematic representation of local heating protocol.
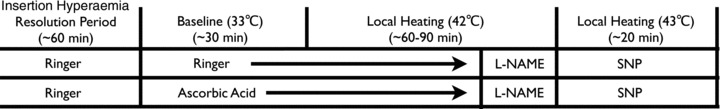
After resolution of the initial insertion hyperaemia, a standard non-painful local heating protocol was employed. Microdialysis sites were perfused with either lactated Ringer solution or AA for baseline measurements at 33°C. Thereafter, the local heater temperature was increased to 42°C. Upon attaining a stable plateau in red blood cell flux, l-NAME was perfused in both microdialysis sites to quantify NO-dependent vasodilatation. Following a new post-heating l-NAME stabilization, local heaters were set to 43°C and SNP was perfused to induce maximal cutaneous vasodilatation. l-NAME, NG-nitro-l-arginine methyl ester; SNP, sodium nitroprusside.
Red blood cell flux, an index of skin blood flow, was measured from ∼1.5 mm2 of skin using multifiber laser Doppler flowmeter probes. The probes were placed in a local heater (MoorLAB, Temperature Monitor SH02; Moor Instruments, Axminster, UK), which was affixed to the skin directly above each microdialysis membrane. The ambulatory BP monitor was placed on the contralateral arm for measurement of BP every 20 min during the protocol.
Local heating protocol
Local skin temperature was set to 33°C upon resolution of the initial insertion trauma hyperaemia and a standard, non-painful heating protocol was used to induce NO-dependent vasodilatation (Kellogg et al. 1999; Minson et al. 2001). Microdialysis sites were perfused at 2 μl min−1 for at least a 30 min baseline period. After baseline measurements, the local heater temperature was increased from 33°C to 42°C at a rate of 0.1°C s−1 and remained at 42°C for the duration of the heating protocol. After red blood cell flux reached a stable plateau (∼40–45 min), 10 mm NG-nitro-l-arginine methyl ester (l-NAME; Sigma-Aldrich, St Louis, MO, USA) was perfused in both microdialysis fibres to quantify NO-dependent vasodilatation at all sites. The phases of the local skin heating response include an initial peak and nadir within the first 10 min, largely due to an axon reflex with a small NO contribution (Minson et al. 2001), followed by a secondary plateau after approximately 30 min, which is predominately mediated by NO (Kellogg et al. 1999; Minson et al. 2001). Following a new post-heating l-NAME stabilization in red blood cell flux, local heaters were set to 43°C and 28 mm sodium nitroprusside (SNP; Nitropress; Hospira, Inc., Lake Forest, IL, USA) was perfused at a rate of 2 μl min−1 in both sites to induce maximal cutaneous vasodilatation (Minson et al. 2001). A dose of 10 mm l-NAME was chosen because this has been shown to sufficiently inhibit NO synthase in the skin (Minson et al. 2001). A dose of 28 mm SNP was chosen because this dose has been shown to elicit maximal vasodilatation in the skin (Minson et al. 2001). A dose of 20 mm ascorbic acid was chosen to be consistent with previous studies conducted in our laboratory (Dupont et al. 2011b).
Data and statistical analysis
Data were collected at 40 Hz using a National Instruments PCI-6221 DAQ board and LabView software (National Instruments Corp., Austin, TX, USA). Cutaneous vascular conductance (CVC) was calculated as red blood cell flux divided by mean BP. CVC data were normalized to a percentage of maximal CVC (%CVCmax), obtained during perfusion of SNP. Baseline, NO plateau and post-heating l-NAME plateau values were obtained over a stable 10 min period. The initial peak CVC was obtained as a 60 s average. The NO contribution to the plateau phase was calculated as the difference between the plateau and the post-heating l-NAME plateau.
Haemodynamic, biochemical and renal function parameters between the LS and HS diets were compared using Student's paired t tests (SPSS 19.0). A 2 × 2 ANOVA was performed for each phase of the local heating response: baseline, initial peak, plateau, and post-heating l-NAME plateau to determine the effect of diet and pharamacological treatment site. Tukey's post hoc comparisons were used when appropriate to determine specific differences. The NO contribution to the plateau during LS and HS diets was compared using Student's paired t tests. The level of significance was set at P < 0.05 and values are reported as means ± SEM. An a priori sample size estimate was conducted based on our primary outcome variable of interest, plateau%CVCmax. Based on our preliminary data, 10 subjects would provide 80% power to detect a 10% reduction in the plateau%CVCmax between the LS and HS diets (GPower 3.1).
Results
Subject characteristics
Baseline subject characteristics are presented in Table 1. All subjects were normotensive and had liver and kidney function within normal limits. Estimated creatinine clearance (Cockcoft-Gault equation) was normal in all subjects (>90 ml min−1). All subjects completed the standardized run-in diet and the subsequent 2 week dietary sodium perturbation.
Table 1.
Baseline subject characteristics
| Baseline characteristic | Value |
|---|---|
| Demographic data | |
| N (M/F) | 12 (5/7) |
| Age (years) | 31 ± 2 |
| Height (cm) | 172 ± 3 |
| Mass (kg) | 71 ± 2 |
| BMI (kg m−2) | 23.3 ± 1 |
| Systolic BP (mmHg) | 110 ± 3 |
| Diastolic BP (mmHg) | 66 ± 3 |
| Heart rate (beats min−1) | 58 ± 3 |
| Biochemical parameters | |
|---|---|
| Haemoglobin (g dl−1) | 13.9 ± 0.5 |
| Haematocrit (%) | 40.8 ± 1.1 |
| Serum sodium (mmol l−1) | 134.1 ± 3.3 |
| Serum potassium (mmol l−1) | 4.1 ± 0.1 |
| Serum chloride (mmol l−1) | 105.6 ± 1.2 |
| Plasma osmolality (mosmol (kg H2O)−1) | 285 ± 1 |
| Serum creatinine (mg dl−1) | 0.84 ± 0.04 |
| Blood urea nitrogen (mg dl−1) | 13.5 ± 0.8 |
| Fasting glucose (mg dl−1) | 80 ± 2.2 |
| Fasting total cholesterol (mg dl−1) | 167.5 ± 8.2 |
| Fasting HDL (mg dl−1) | 60.4 ± 6.8 |
| Fasting LDL (mg dl−1) | 93.1 ± 6.3 |
| Fasting triglycerides (mg dl−1) | 71.1 ± 10.1 |
BMI, body mass index; BP, blood pressure. Values are mean ± SEM.
Dietary sodium perturbation
Haemodynamic, biochemical and renal function parameters during the dietary sodium perturbation are presented in Tables 2 and 3. The HS diet increased urinary sodium excretion (Table 2; P < 0.05). By study design, 24 h mean BP of subjects on the LS to HS diet did not differ (Table 3; P > 0.05). In addition, there were no differences in 24 h systolic BP, diastolic BP or pulse pressure between the LS and HS diets (Table 2; P > 0.05). Therefore, all subjects were characterized as having salt-resistant BP (see Methods section), indicated by a large increase in urinary sodium excretion during the HS diet without a change in mean BP (Tables 2 and 3).
Table 2.
Biochemical and renal responses to dietary sodium perturbation
| Low sodium | High sodium | |
|---|---|---|
| Mass (kg) | 71 ± 2 | 72 ± 2 |
| Haemoglobin (g dl−1) | 14.5 ± 0.5 | 14.0 ± 0.5 |
| Haematocrit (%) | 40.7 ± 1.3 | 38.7 ± 1.0 |
| Serum Na+ (mmol l−1) | 136.7 ± 0.6 | 138.7 ± 0.8 |
| Serum K+ (mmol l−1) | 4.1 ± 0.2 | 3.8 ± 0.1 |
| Serum Cl− (mmol l−1) | 102.1 ± 0.6 | 104.6 ± 1.0* |
| Plasma osmolality (mosmol (kg H2O)−1) | 283 ± 1 | 285 ± 1 |
| Urine osmolality (mosmol (kg H2O)−1) | 411 ± 57 | 437 ± 37 |
| Urine flow rate (ml min−1) | 1.1 ± 0.1 | 1.4 ± 0.1* |
| Urinary Na+ excretion (mmol (24 h)−1) | 32.2 ± 10.0 | 233.0 ± 19.3* |
| Urinary K+ excretion (mmol (24 h)−1) | 41.2 ± 5.2 | 44.0 ± 5.6 |
| FENa (%) | 0.15 ± 0.07 | 0.9 ± 0.08* |
| FECl (%) | 0.4 ± 0.1 | 1.2 ± 0.09* |
| Creatinine clearance (ml−1 min−1 (1.73 cm2)−1) | 131.6 ± 11.1 | 142.5 ± 9.5 |
| Free water clearance (ml min−1) | −0.20 ± 0.1 | 0.65 ± 0.04* |
FENa, fractional excretion of sodium; FECl, fractional excretion of chloride. *P < 0.05 vs. low sodium. Values are mean ± SEM.
Table 3.
Haemodynamic responses to dietary sodium perturbation
| Low sodium | High sodium | |
|---|---|---|
| 24 h systolic BP (mmHg) | 113 ± 2 | 116 ± 2 |
| 24 h diastolic BP (mmHg) | 70 ± 1 | 71 ± 2 |
| 24 h mean BP (mmHg) | 84 ± 1 | 85 ± 2 |
| 24 h pulse pressure (mmHg) | 43 ± 2 | 46 ± 2 |
| 24 h heart rate (beats min−1) | 68 ± 3 | 66 ± 3 |
| Laboratory systolic BP (mmHg) | 117 ± 3 | 120 ± 3 |
| Laboratory diastolic BP (mmHg) | 73 ± 2 | 74 ± 2 |
| Laboratory mean BP (mmHg) | 88 ± 3 | 89 ± 2 |
| Laboratory pulse pressure (mmHg) | 44 ± 2 | 45 ± 2 |
| Laboratory heart rate (beats min−1) | 68 ± 3 | 66 ± 3 |
BP, blood pressure. Values are mean ± SEM.
Microvascular function
Absolute maximal CVC did not differ between LS and HS diets across the sites (Table 4; P > 0.05 for all), indicating that the maximal vasodilatory capacity of the skin microvasculature was not different between diets. There were no differences in baseline%CVCmax at the Ringer site between LS and HS diets (Fig. 2A; P > 0.05). The initial peak%CVCmax was lower during the HS diet but did not reach statistical significance (Fig. 2A; P > 0.05). The HS diet attenuated the plateau%CVCmax at the Ringer site (Fig. 2A; P < 0.05). Post-heating l-NAME attenuated the plateau to the same extent during both LS and HS diets (Fig. 2A). A reduction in the NO contribution to the plateau during the HS diet did not reach statistical significance (LS: 55 ± 5%CVCmax vs. HS: 44 ± 3%CVCmax; P= 0.075).
Table 4.
Absolute maximal cutaneous vascular conductance values
| Low sodium | High sodium | |
|---|---|---|
| Ringer solution | 2.57 ± 0.4 | 2.36 ± 0.4 |
| AA | 2.20 ± 0.4 | 2.52 ± 0.3 |
AA, ascorbic acid. Values are mean ± SEM.
Figure 2. Maximal cutaneous vascular conductance.
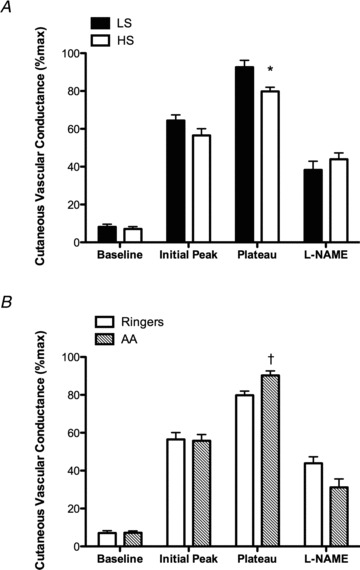
Percentage of maximal cutaneous vascular conductance (%CVCmax) in the microdialysis site perfused with Ringer solution during the low-sodium (LS; filled bars) and high-sodium (HS; open bars) diets during baseline, the initial peak, NO-mediated plateau and post-heating l-NAME plateau (A). The HS diet attenuated the plateau%CVCmax compared with LS. Percentage of maximal cutaneous vascular conductance (%CVCmax) in the Ringer (open bars) and ascorbic acid (AA; hatched bars) sites during the high-sodium (HS) diet during baseline, the initial peak, NO-mediated plateau and post-heating l-NAME plateau (B). During the HS diet, AA augmented the plateau%CVCmax compared with Ringer solution. Values are mean ± SEM. *P < 0.05 vs. LS; †P<0.05 v. Ringer.
During the LS diet, the initial peak and plateau%CVCmax at the AA site were not different from the Ringer site (initial peak: 64 ± 3%CVCmax for Ringer vs. 64 ± 2%CVCmax for AA, P= 0.77; plateau: 93 ± 1%CVCmax for Ringer vs. 91 ± 1%CVCmax for AA, P= 0.33). During the HS diet, there were no differences in baseline or initial peak%CVCmax at the Ringer and AA sites (Fig. 2B; P > 0.05). However, during the HS diet, AA improved the plateau%CVCmax compared with the Ringer site (Fig. 2B; P < 0.05), such that it was not different from the LS diet (P > 0.05). No difference between Ringer and AA sites was observed in the l-NAME attenuation of the plateau (Fig. 2B; P > 0.05). Therefore, on the HS diet, AA restored the NO contribution to the plateau compared with the Ringer site (Fig. 3; P < 0.05).
Figure 3. The NO contribution to the plateau during the HS diet in the Ringer (open bar) and ascorbic acid (AA; hatched bar) sites.
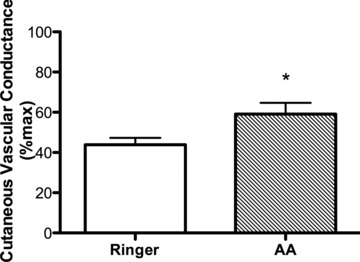
AA restored the NO contribution to the plateau during the HS diet. Values are mean ± SEM. *P < 0.05 vs. Ringer solution.
Discussion
The novel findings of the current study are twofold. First, in salt-resistant adults, the plateau phase of cutaneous vasodilatation in response to local heating was attenuated during a high-sodium diet. Second, dietary sodium-induced impairments in cutaneous vasodilatation in response to local heating were improved by the local infusion of ascorbic acid. Collectively, these findings indicate that dietary sodium-induced declines in microvascular function are independent from BP and suggest a role for oxidative stress in contributing to this impairment.
Endothelial dysfunction is thought to be a precursor to clinical manifestations of cardiovascular disease (Joannides et al. 2006). Excess dietary salt consumption, even in the absence of its effect on BP, may be one contributing factor to vascular endothelial dysfunction. In rodents, dietary salt loading caused impaired endothelium-dependent vasodilatation, independently of BP (Boegehold, 1993; Nurkiewicz & Boegehold, 2007; Zhu et al. 2007). Further, mice fed a high-salt diet demonstrated reduced arteriolar responsiveness to acetylcholine, an endothelium-dependent vasodilator, compared with mice fed a low-salt diet (Nurkiewicz & Boegehold, 2007). Our findings translate these previous reports in experimental animal models to healthy humans. In order to experimentally control for salt-induced alterations in BP, a controlled 2 week dietary sodium perturbation was employed using a within-subjects design, and only those normotensive adults determined to have salt-resistant BP were included in the present study. This study demonstrates that dietary sodium loading generates ascorbate-sensitive oxidative stress in the microvasculature, impairing function independently of BP in normotensive adults. Our results confirm the sodium-dependent reduction in vascular function in humans but importantly extend these findings by indicating that the resultant functional impairments occur independently of BP.
In humans, although there is no standardized methodology for the assessment of the microvasculature, the minimally invasive technique of intradermal microdialysis coupled with laser Doppler flowmetry of the skin provides a viable technique to study mechanisms underlying microvascular function (Cracowski et al. 2006). It is well-established that local skin heating results in an initial peak in cutaneous blood flow, largely mediated by an axon reflex (Minson et al. 2001) via activation of TRPV-1 channels on sensory fibres (Wong & Fieger, 2010), followed by a sustained plateau, primarily mediated by NO (Kellogg et al. 1999; Minson et al. 2001). In the forearm, endothelial NO synthase (eNOS) is thought to be the primary isoform mediating cutaneous NO-dependent vasodilatation in response to local heating (Kellogg et al. 1999, 2008; Bruning et al. 2012), though this does not appear to be the case in the calf (Stewart et al. 2007). We observed an attenuation in the NO-mediated plateau phase during the high dietary sodium condition. Importantly, absolute maximal CVC did not differ between diets, indicating that dietary sodium does not influence the maximal vasodilatory capacity of the skin microvasculature. A dietary sodium-induced reduction in the initial peak of the cutaneous blood flow response to local heating did not reach statistical significance. We previously demonstrated an attenuation of the initial peak response during the local infusion of hypertonic saline (DuPont et al. 2011a). However, local infusion of hypertonic saline probably increases local sodium concentration to a greater degree than does increased dietary sodium consumption.
A secondary aim of the present investigation was to examine a role for oxidative stress in contributing to dietary sodium-induced declines in microvascular function. Disruption of the balance between ROS production and scavenging results in oxidative stress, a pathophysiological condition presumably contributing to the development of cardiovascular disease. Several animal studies have demonstrated that resting superoxide levels are higher in resistance arteries (Lenda et al. 2000; Lenda & Boegehold, 2002; Zhu et al. 2007) and venules (Lenda et al. 2000; Lenda & Boegehold, 2002) of rodents fed a high-salt diet, potentially indicating that higher levels of superoxide during dietary salt loading interfere with NO bioavailability. Studies in rodent models further demonstrated that declines in NO release and endothelium-dependent dilatation during dietary salt loading result from an increase in ROS (Boegehold, 1995; Lenda et al. 2000; Zhu et al. 2007). In normotensive rats fed a high-salt diet, acetylycholine-induced endothelium-dependent dilatation was reduced compared with rats fed a low-salt diet, but exposure of the vascular bed to ROS scavengers reversed the suppressed arteriolar responsiveness (Lenda et al. 2000). Superoxide appears to be the primary source of ROS during dietary salt loading (Nurkiewicz & Boegehold, 2007; Zhu et al. 2007), and its production is probably via NAD(P)H oxidase (Lenda et al. 2000; Zhu et al. 2007), xanthine oxidase (Lenda et al. 2000; Zhu et al. 2007) and eNOS (Nurkiewicz & Boegehold, 2007). In rats fed a short-term high-salt diet, increased vascular superoxide production and the accompanying impairment in endothelium-dependent dilatation are both ameliorated by NAD(P)H and xanthine oxidase inhibition (Zhu et al. 2007). In the present investigation, during the high-sodium diet, the local infusion of ascorbic acid improved the NO-mediated plateau phase of the cutaneous blood flow response to local heating to levels observed during the low-salt diet. These findings suggest that oxidant mechanisms sensitive to ascorbic acid infusion contribute to the sodium-induced impairments in microvascular function in humans with salt-resistant BP.
Interestingly, local infusion of ascorbic acid did not improve the initial peak phase of the skin blood flow response to local heating. This finding could potentially be due to several reasons, including salt-induced alterations in sympathetic vasoconstrictor nerve function (Houghton et al. 2006; Hodges et al. 2008), reductions in cofactor tetrahydrobiopterin (BH4) availability or other non-specific effects of salt on the axon reflex that do not involve NO.
Ascorbic acid is a non-specific scavenger of superoxide radicals and other ROS (Holowatz, 2011); therefore, further investigations are necessary to determine the source of salt-induced increases in ROS. In addition to direct scavenging of ROS, ascorbic acid may have improved salt-induced impairments in cutaneous vasodilatation by promoting the degradation of the endogenous NO synthase inhibitor asymmetrical dimethyl l-arginine (ADMA) (Sydow & Munzel, 2003), protecting the NO synthase cofactor BH4 from oxidation (Heller et al. 2001; Toth et al. 2002) or by inhibiting the arginase pathway (Santhanam et al. 2007).
High dietary salt consumption can potentially increase plasma sodium levels from 2 to 4 mmol l−1 (de Wardener et al. 2004). In the current investigation, serum sodium increased ∼2 mmol l−1 during dietary sodium loading (P= 0.07). Physiological increases in sodium concentration directly increase vascular endothelial cell stiffness and decrease endothelial NO release (Oberleithner et al. 2007). The deleterious effects of sodium on endothelial cell function are reversed by increases in extracellular potassium concentration (Oberleithner et al. 2007). A recent clinical trial reported an increased cardiovascular disease risk in individuals with a high sodium-to-potassium ratio (Cook et al. 2009), suggesting an important interaction between dietary intake of sodium and potassium. It is important to note that in the current study, we did not manipulate dietary potassium intake during the 2 week dietary sodium perturbation, and therefore 24 h urinary potassium excretion was not different between LS and HS diets. Additionally, serum potassium concentration was not different between LS and HS diets and remained within the normal range. Future studies are needed to determine the potential protective effects of increasing dietary potassium intake on the impairments in microvascular function that occur during dietary sodium loading.
Habitual salt intake in modern society remains high (CDC, 2010), despite American Heart Association recommended guidelines to consume less than 2300 mg sodium per day (CDC, 2010). Experimental studies in both animals and humans indicate that a high-salt diet leads to functional changes in the microcirculation that may compromise the ability of the vasculature to respond to a variety of physiological stimuli (Boegehold, 1995; Frisbee & Lombard, 1999; Liu et al. 1999; Tzemos et al. 2008; Eisenach et al. 2011). The cumulative data suggest that along with improved BP control in adults with salt-sensitive hypertension (Chobanian et al. 2003), lowering dietary salt intake may have beneficial BP-independent effects on microvascular function in healthy, normotensive adults.
Acknowledgments
The time and effort expended by all the volunteer subjects are greatly appreciated. We also thank Dr Allen Prettyman and Dr Michael Stillabower for assistance with medical screening of participants, Gabrielle Synder-Marlow, RD for all food preparation, and Carol Catanese, MS for assistance in the conduction of the study. This research was supported by NIH Grants R01 HL104106, 5P20RR016472-12 and 8P20 GM103446-12. Dr Sanders is supported by NIH Grants R01 DK046199, P30 DK079337, and by the Medical Research Service of the Department of Veterans Affairs.
Glossary
- AA
ascorbic acid
- BP
blood pressure
- CVC
cutaneous vascular conductance
- HS
high sodium
- LS
low sodium
- l-NAME
NG-nitro-l-arginine methyl ester
- ROS
reactive oxygen species
- SNP
sodium nitroprusside
Author contributions
The experiments of this study were conducted in the Department of Kinesiology and Applied Physiology at the University of Delaware, Newark, DE, USA. J.L.G. contributed to data acquisition, data analysis, data interpretation, and wrote the first draft of the manuscript. J.J.D. contributed to data acquisition, data analysis, data interpretation, and critical review of the manuscript. S.L.L.-E. contributed to study design, data interpretation, and critical review of the manuscript. P.W.S. contributed to study design, data interpretation, and critical review of the manuscript. D.G.E. contributed to study design, data acquisition, data analysis, data interpretation, and critical review of the manuscript. W.B.F. contributed to study design, data acquisition, data analysis, data interpretation, and critical review of the manuscript. All authors approved the final version of the manuscript.
References
- Abularrage CJ, Sidawy AN, Aidinian G, Singh N, Weiswasser JM, Arora S. Evaluation of the microcirculation in vascular disease. J Vasc Surg. 2005;42:574–581. doi: 10.1016/j.jvs.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Black MA, Green DJ, Cable NT. Exercise prevents age-related decline in nitric-oxide-mediated vasodilator function in cutaneous microvessels. J Physiol. 2008;586:3511–3524. doi: 10.1113/jphysiol.2008.153742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boegehold MA. Effect of dietary salt on arteriolar nitric oxide in striated muscle of normotensive rats. Am J Physiol Heart Circ Physiol. 1993;264:H1810–H1816. doi: 10.1152/ajpheart.1993.264.6.H1810. [DOI] [PubMed] [Google Scholar]
- Boegehold MA. Flow-dependent arteriolar dilation in normotensive rats fed low- or high-salt diets. Am J Physiol Heart Circ Physiol. 1995;269:H1407–H1414. doi: 10.1152/ajpheart.1995.269.4.H1407. [DOI] [PubMed] [Google Scholar]
- Bruning RS, Santhanam L, Stanhewicz AE, Smith CJ, Berkowitz DE, Kenney WL, Holowatz LA. Endothelial nitric oxide synthase mediates cutaneous vasodilation during local heating and is attenuated in middle-aged human skin. J Appl Physiol. 2012;112:2019–2026. doi: 10.1152/japplphysiol.01354.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) Sodium intake among adults – United States, 2005–2006. MMWR Morb Mortal Wkly Rep. 2010;59:746–749. [PubMed] [Google Scholar]
- Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jr, Jones DW, Materson BJ, Oparil S, Wright JT, Jr, Roccella EJ. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206–1252. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- Cohuet G, Struijker-Boudier H. Mechanisms of target organ damage caused by hypertension: therapeutic potential. Pharmacol Ther. 2006;111:81–98. doi: 10.1016/j.pharmthera.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Cook NR, Obarzanek E, Cutler JA, Buring JE, Rexrode KM, Kumanyika SK, Appel LJ, Whelton PK. Joint effects of sodium and potassium intake on subsequent cardiovascular disease: the Trials of Hypertension Prevention follow-up study. Arch Intern Med. 2009;169:32–40. doi: 10.1001/archinternmed.2008.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracowski JL, Minson CT, Salvat-Melis M, Halliwill JR. Methodological issues in the assessment of skin microvascular endothelial function in humans. Trends Pharmacol Sci. 2006;27:503–508. doi: 10.1016/j.tips.2006.07.008. [DOI] [PubMed] [Google Scholar]
- de Wardener HE, He FJ, MacGregor GA. Plasma sodium and hypertension. Kidney Int. 2004;66:2454–2466. doi: 10.1111/j.1523-1755.2004.66018.x. [DOI] [PubMed] [Google Scholar]
- DuPont JJ, Farquhar WB, Edwards DG. Intradermal microdialysis of hypertonic saline attenuates cutaneous vasodilatation in response to local heating. Exp Physiol. 2011a;96:674–680. doi: 10.1113/expphysiol.2011.058404. [DOI] [PubMed] [Google Scholar]
- Dupont JJ, Farquhar WB, Townsend RR, Edwards DG. Ascorbic acid or L-arginine improves cutaneous microvascular function in chronic kidney disease. J Appl Physiol. 2011b;111:1561–1567. doi: 10.1152/japplphysiol.00419.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenach JH, Gullixson LR, Kost SL, Joyner MJ, Turner ST, Nicholson WT. Sex differences in salt sensitivity to nitric oxide dependent vasodilation in healthy young adults. J Appl Physiol. 2011;112:1049–1053. doi: 10.1152/japplphysiol.01197.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott P, Stamler J, Nichols R, Dyer AR, Stamler R, Kesteloot H, Marmot M. Intersalt revisited: further analyses of 24 h sodium excretion and blood pressure within and across populations. Intersalt Cooperative Research Group. BMJ. 1996;312:1249–1253. doi: 10.1136/bmj.312.7041.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenfield D, Roth-Yousey L, Compher C. Comparison of predictive equations for resting metabolic rate in healthy nonobese and obese adults: a systematic review. J Am Diet Assoc. 2005;105:775–789. doi: 10.1016/j.jada.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Frassetto L, Morris RC, Jr, Sellmeyer DE, Todd K, Sebastian A. Diet, evolution and aging – the pathophysiologic effects of the post-agricultural inversion of the potassium-to-sodium and base-to-chloride ratios in the human diet. Eur J Nutr. 2001;40:200–213. doi: 10.1007/s394-001-8347-4. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Lombard JH. Acute elevations in salt intake and reduced renal mass hypertension compromise arteriolar dilation in rat cremaster muscle. Microvasc Res. 1999;57:273–283. doi: 10.1006/mvre.1998.2138. [DOI] [PubMed] [Google Scholar]
- Harrison DG, Gongora MC, Guzik TJ, Widder J. Oxidative stress and hypertension. J Am Soc Hypertens. 2007;1:30–44. doi: 10.1016/j.jash.2006.11.006. [DOI] [PubMed] [Google Scholar]
- He FJ, Markandu ND, MacGregor GA. Importance of the renin system for determining blood pressure fall with acute salt restriction in hypertensive and normotensive whites. Hypertension. 2001;38:321–325. doi: 10.1161/01.hyp.38.3.321. [DOI] [PubMed] [Google Scholar]
- Heller R, Unbehaun A, Schellenberg B, Mayer B, Werner-Felmayer G, Werner ER. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J Biol Chem. 2001;276:40–47. doi: 10.1074/jbc.M004392200. [DOI] [PubMed] [Google Scholar]
- Hodges GJ, Chiu C, Kosiba WA, Zhao K, Johnson JM. The effect of microdialysis needle trauma on cutaneous vascular responses in humans. J Appl Physiol. 2009;106:1112–1118. doi: 10.1152/japplphysiol.91508.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges GJ, Kosiba WA, Zhao K, Johnson JM. The involvement of norepinephrine, neuropeptide Y, and nitric oxide in the cutaneous vasodilator response to local heating in humans. J Appl Physiol. 2008;105:233–240. doi: 10.1152/japplphysiol.90412.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowatz LA. Ascorbic acid: what do we really NO. J Appl Physiol. 2011;111:1542–1543. doi: 10.1152/japplphysiol.01187.2011. [DOI] [PubMed] [Google Scholar]
- Holowatz LA, Thompson-Torgerson CS, Kenney WL. The human cutaneous circulation as a model of generalized microvascular function. J Appl Physiol. 2008;105:370–372. doi: 10.1152/japplphysiol.00858.2007. [DOI] [PubMed] [Google Scholar]
- Houghton BL, Meendering JR, Wong BJ, Minson CT. Nitric oxide and noradrenaline contribute to the temperature threshold of the axon reflex response to gradual local heating in human skin. J Physiol. 2006;572:811–820. doi: 10.1113/jphysiol.2005.104067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IJzeman RG, de Jongh RT, Beijk MA, van Weissenbruch MM, Delemarre-van de Waal HA, Serne EH, Stehouwer CD. Individuals at increased coronary heart disease risk are characterized by an impaired microvascular function in skin. Eur J Clin Invest. 2003;33:536–542. doi: 10.1046/j.1365-2362.2003.01179.x. [DOI] [PubMed] [Google Scholar]
- Joannides R, Bellien J, Thuillez C. Clinical methods for the evaluation of endothelial function – a focus on resistance arteries. Fundam Clin Pharmacol. 2006;20:311–320. doi: 10.1111/j.1472-8206.2006.00406.x. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Liu Y, Kosiba IF, O’Donnell D. Role of nitric oxide in the vascular effects of local warming of the skin in humans. J Appl Physiol. 1999;86:1185–1190. doi: 10.1152/jappl.1999.86.4.1185. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Zhao JL, Wu Y. Endothelial nitric oxide synthase control mechanisms in the cutaneous vasculature of humans in vivo. Am J Physiol Heart Circ Physiol. 2008;295:H123–H129. doi: 10.1152/ajpheart.00082.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenda DM, Boegehold MA. Effect of a high-salt diet on oxidant enzyme activity in skeletal muscle microcirculation. Am J Physiol Heart Circ Physiol. 2002;282:H395–H402. doi: 10.1152/ajpheart.0354.2001. [DOI] [PubMed] [Google Scholar]
- Lenda DM, Sauls BA, Boegehold MA. Reactive oxygen species may contribute to reduced endothelium-dependent dilation in rats fed high salt. Am J Physiol Heart Circ Physiol. 2000;279:H7–H14. doi: 10.1152/ajpheart.2000.279.1.H7. [DOI] [PubMed] [Google Scholar]
- Levy BI, Ambrosio G, Pries AR, Struijker-Boudier HA. Microcirculation in hypertension: a new target for treatment. Circulation. 2001;104:735–740. doi: 10.1161/hc3101.091158. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rusch NJ, Lombard JH. Loss of endothelium and receptor-mediated dilation in pial arterioles of rats fed a short-term high salt diet. Hypertension. 1999;33:686–688. doi: 10.1161/01.hyp.33.2.686. [DOI] [PubMed] [Google Scholar]
- Matavelli LC, Zhou X, Varagic J, Susic D, Frohlich ED. Salt loading produces severe renal hemodynamic dysfunction independent of arterial pressure in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;292:H814–H819. doi: 10.1152/ajpheart.00671.2006. [DOI] [PubMed] [Google Scholar]
- Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005;85:679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- Midgley JP, Matthew AG, Greenwood CM, Logan AG. Effect of reduced dietary sodium on blood pressure: a meta-analysis of randomized controlled trials. JAMA. 1996;275:1590–1597. doi: 10.1001/jama.1996.03530440070039. [DOI] [PubMed] [Google Scholar]
- Minson CT, Berry LT, Joyner MJ. Nitric oxide and neurally mediated regulation of skin blood flow during local heating. J Appl Physiol. 2001;91:1619–1626. doi: 10.1152/jappl.2001.91.4.1619. [DOI] [PubMed] [Google Scholar]
- Minson CT, Holowatz LA, Wong BJ, Kenney WL, Wilkins BW. Decreased nitric oxide- and axon reflex-mediated cutaneous vasodilation with age during local heating. J Appl Physiol. 2002;93:1644–1649. doi: 10.1152/japplphysiol.00229.2002. [DOI] [PubMed] [Google Scholar]
- Nurkiewicz TR, Boegehold MA. High salt intake reduces endothelium-dependent dilation of mouse arterioles via superoxide anion generated from nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1550–R1556. doi: 10.1152/ajpregu.00703.2006. [DOI] [PubMed] [Google Scholar]
- Oberleithner H, Riethmuller C, Schillers H, MacGregor GA, de Wardener HE, Hausberg M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci U S A. 2007;104:16281–16286. doi: 10.1073/pnas.0707791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Carpi A. Skin microcirculation in peripheral arterial obliterative disease. Biomed Pharmacother. 2004;58:427–431. doi: 10.1016/j.biopha.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Rossi M, Carpi A, Galetta F, Franzoni F, Santoro G. The investigation of skin blood flowmotion: a new approach to study the microcirculatory impairment in vascular diseases. Biomed Pharmacother. 2006;60:437–442. doi: 10.1016/j.biopha.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Santhanam L, Lim HK, Miriel V, Brown T, Patel M, Balanson S, Ryoo S, Anderson M, Irani K, Khanday F, Di Costanzo L, Nyhan D, Hare JM, Christianson DW, Rivers R, Shoukas A, Berkowitz DE. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res. 2007;101:692–702. doi: 10.1161/CIRCRESAHA.107.157727. [DOI] [PubMed] [Google Scholar]
- Schmidlin O, Sebastian AF, Morris RC., Jr What initiates the pressor effect of salt in salt-sensitive humans? Observations in normotensive blacks. Hypertension. 2007;49:1032–1039. doi: 10.1161/HYPERTENSIONAHA.106.084640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman IH, Aranda P, Raij L, Veronesi M, Aranda FJ, Martin R. Surgical menopause increases salt sensitivity of blood pressure. Hypertension. 2006;47:1168–1174. doi: 10.1161/01.HYP.0000218857.67880.75. [DOI] [PubMed] [Google Scholar]
- Stewart JM, Medow MS, Minson CT, Taneja I. Cutaneous neuronal nitric oxide is specifically decreased in postural tachycardia syndrome. Am J Physiol Heart Circ Physiol. 2007;293:H2161–H2167. doi: 10.1152/ajpheart.00600.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydow K, Munzel T. ADMA and oxidative stress. Atheroscler Suppl. 2003;4:41–51. doi: 10.1016/s1567-5688(03)00033-3. [DOI] [PubMed] [Google Scholar]
- Toth M, Kukor Z, Valent S. Chemical stabilization of tetrahydrobiopterin by L-ascorbic acid: contribution to placental endothelial nitric oxide synthase activity. Mol Hum Reprod. 2002;8:271–280. doi: 10.1093/molehr/8.3.271. [DOI] [PubMed] [Google Scholar]
- Tzemos N, Lim PO, Wong S, Struthers AD, MacDonald TM. Adverse cardiovascular effects of acute salt loading in young normotensive individuals. Hypertension. 2008;51:1525–1530. doi: 10.1161/HYPERTENSIONAHA.108.109868. [DOI] [PubMed] [Google Scholar]
- Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27:481–490. doi: 10.1161/01.hyp.27.3.481. [DOI] [PubMed] [Google Scholar]
- Wenner MM, Edwards DG, Ray CA, Rose WC, Gardner TJ, Stillabower M, Farquhar WB. Celecoxib does not alter cardiovascular and renal function during dietary salt loading. Clin Exp Pharmacol Physiol. 2011;38:543–549. doi: 10.1111/j.1440-1681.2011.05546.x. [DOI] [PubMed] [Google Scholar]
- Wong BJ, Fieger SM. Transient receptor potential vanilloid type-1 (TRPV-1) channels contribute to cutaneous thermal hyperaemia in humans. J Physiol. 2010;588:4317–4326. doi: 10.1113/jphysiol.2010.195511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Huang T, Lombard JH. Effect of high-salt diet on vascular relaxation and oxidative stress in mesenteric resistance arteries. J Vasc Res. 2007;44:382–390. doi: 10.1159/000102955. [DOI] [PubMed] [Google Scholar]