

Abstract
Background & objectives:
Prostate cancer (CaP) is the fifth most common cancer among Indian men. Tumour protein p53 (TP53) gene increases the fidelity of DNA replication and homologous recombination by transcriptional transactivation of mismatch repair (MMR) genes. DNA repair thus has a potential role in molecular carcinogenesis of CaP. The aim of the present study was to identify mutations, and polymorphisms in TP53 gene and MMR protein expression in CaP in Indian male population.
Methods:
TP53 codon 72 polymorphism was analysed in 105 CaP, 120 benign prostatic hyperplasia (BPH) cases and 106 normal controls. Mutational analysis of TP53 was done in DNA extracted from formalin fixed paraffin embedded tissue of 80 CaP and 24 BPH cases. Expression of MMR proteins viz. hMLH1, hMSH2, hPMS1 and hPMS2 was studied in 80 CaP, 15 prostatic intraepithelial neoplasia (PIN) and 15 BPH cases.
Results:
A somatic C/A variation at the intronic boundary of exon 7 in TP53 gene was observed in one each biopsy samples from CaP and BPH. A significant association of codon 72 TP53 Pro/Pro genotype was observed with the risk of CaP (OR, 2.59, P=0.02) and BPH (OR, 6.27, P<0.001). Immunohistochemical analysis of MMR proteins showed maximum loss of hPMS1 expression in cases of CaP and PIN while no loss in expression of MMR proteins was observed in BPH cases. The study also identified a significant loss of hPMS2 protein in poorly differentiated tumours (Gleason score >7) than in well differentiated tumours (Gleason score 3-6) (P<0.05).
Interpretation & conclusions:
The results of the present study demonstrate that TP53 codon 72 polymorphism plays significant role in the pathogenesis and susceptibility to CaP and BPH. Also, an aberrant MMR protein expression could be involved in progression of prostate cancer through PIN, early CaP to aggressive CaP. The loss of hPMS2 protein expression may serve as a marker for progression of CaP.
Keywords: hMLH1, hMSH2, hPMS1, hPMS2, prostate cancer, TP53
Prostate cancer (CaP) is the second most frequently diagnosed cancer of men and the fifth most common cancer worldwide. Incidence rates of CaP vary by more than 25-fold worldwide, the highest rates are in Australia/New Zealand, Europe, Northern America, and the lowest age-standardised incidence rate is estimated in South-Central Asia1. In India, it is the fifth most common cancer among men2. Although, the incidence of CaP in India is lower as compared to the western population, a substantial increase in its incidence has been witnessed during the last two decades3.
Relatively little is known about the specific aetiology of CaP, though the burden of CaP on public health has been increasing. Multiple aetiologic risk factors have been identified including age, ethnicity, family history, genetic profile, diet, inflammation, steroid hormones and environmental factors. Variations in exposure to these risk factors may explain inter-individual differences in CaP risk. A robust DNA repair capacity may however, diminish any risks conferred by mutations from these risk factors as different DNA repair pathways exist to reverse the different types of DNA damage4.
The integrity of genetic information depends on the fidelity of DNA replication and on the efficiency of several different DNA repair processes. Among many types of DNA repair, the highly conserved genomic restoration mechanism, DNA mismatch repair (MMR) mechanism is responsible for correcting base substitution mismatches and insertion-deletion mismatches (IDLs) generated during DNA replication5. MMR is also involved in apoptosis and checkpoint activation in response to various forms of DNA damage. For efficient functioning of MMR, MutL heterodimer binding to MutS heterodimers is required. The MutS heterodimer, hMSH2/hMSH6, recognizes and binds most base/base mismatches and small insertion deletions loops (IDLs), whereas the hMSH2/hMSH3 preferentially binds to small and large IDLs. For MutL heterodimer, hMLH1 protein product forms a heterodimer with hPMS2, or hPMS1 and it has been shown that hMLH1/hPMS2 heterodimer provides the majority of repair activity6. Tumour protein (TP53) increases the fidelity of DNA replication and homologous recombination (HR) by transcriptional transactivation of MMR genes. Additionally, the TP53 target genes hMSH2, hMLH1, and hPMS2 are able to signal cell cycle arrest and apoptosis after certain types of DNA damage via TP53 or its homologue p737. Consequences of defects in this DNA repair pathway are evidenced by microsatellite instability (MSI), elevated mutation frequency throughout the genome and enhanced recombination events8. The resulting mutator phenotype caused by MMR deficiency has been associated with increased mutation rates in genes that suppress cancer initiation or progression.
In majority of the cancers, TP53 dysfunction is caused through a direct mutation within the DNA-binding domain of the gene. Four mutation “hot spots” have been identified in exons 5 to 8, which coincide with the four most highly conserved regions of TP53 gene. Moreover, mutations in exons 5 to 8 of TP53 comprise 94.2 per cent of all somatic mutations in the International Agency for Research on Cancer (IARC) database, version R119. In addition, exon 4 of TP53 harbours a common G/C nucleotide variation encoding the amino acids arginine (CGC) or proline (CCC) at codon 72 (Arg/Pro). This most frequent TP53 polymorphism at codon 72 lies in a proline-rich region, which is important for its ability to induce apoptosis10.
An accumulation of genetic abnormalities and a decline in DNA repair during ageing may lead to CaP11. Therefore, investigating the abnormalities of MMR system and mutations/polymorphisms in TP53 is important in understanding the pathogenesis of CaP. The present study was thus, aimed to identify mutations in “hotspot” exons 5 to 8 of TP53 gene and TP53 codon 72 polymorphisms and their possible association with the risk of CaP. For evaluation of prognostic relevance of MMR protein expression in CaP, the immunohistochemical expression was investigated in CaP, PIN and BPH patients for hMLH1, hMSH2, hPMS1 and hPMS2 proteins. The association of TP53 polymorphism and MMR protein expression with clinicopathological parameters was also analyzed.
Material & Methods
Two hundred and seventy nine randomly selected cases of prostate enlargement referred from Department of Urology, Safdarjung hospital, New Delhi, between November 2005 and July 2009 were enrolled in Tumour Biology Laboratory, National Institute of Pathology (ICMR), New Delhi for the present preliminary study. After a thorough clinical examination, all cases of prostate enlargement underwent uroflowmetry, digital rectal examination (DRE) and total serum prostate-specific antigen (PSA). Thirty four patients with serum PSA levels greater than 1 ng/ml and less than 4 ng/ml were excluded from the study and were advised to seek diagnostic follow up. Two hundred and forty five patients with serum PSA levels greater than 4ng/ml, were subjected to sextant or sextant plus site specific or 12 core transrectal ultrasound (TRUS) guided prostate biopsy. Of the 245 patients, 105 cases of CaP and 120 BPH cases were finally selected following confirmation by histopathology, wherein absence of basal cell layer was the defining feature for CaP12. Eight previously treated cases and 12 cases with history of malignancy in other organs were excluded from the study. Factors such as age of the patient at the time of diagnosis and serum PSA levels at first diagnosis were recorded and PSA was measured every 6 months thereafter, for a follow up period of six months to three years. One hundred and sixteen normal healthy volunteers without any clinical features of urinary incontinence were investigated for serum PSA levels. Of these, 106 age-matched individuals with serum PSA < 1 ng/ml were recruited as ‘controls’. Controls were ruled out for previous history of any malignancy or urological disorders. Informed consent was obtained from all the participating patients and healthy controls and the study was carried out with the approval of Ethical Review Committee of Safdarjung Hospital, New Delhi.
PCR-RFLP assay for TP53 codon 72 polymorphism analysis: Peripheral blood sample (5 ml) was collected for TP53 polymorphism from 105 CaP patients, 120 BPH patients and 106 normal age-matched healthy controls. DNA extraction from blood samples was done using standard phenol-chloroform method13. TP53 codon 72 polymorphism was determined using PCR-RFLP assay. Briefly, 100 ng DNA was amplified using primers: 5’ -TTG CCG TCC CAA GCA ATG GAT GA-3’ (forward) and 5’ TCT GGG AAG GGA CAG AAG ATG AC-3’ (reverse) (Biolinkk, India). Amplification was performed by initial denaturation at 94oC for 5 min, followed by 35 cycles at 94°C for 45 sec, 58°C for 45 sec, 72°C for 45 sec, and a final extension at 72°C for 10 min. The PCR product was digested using five units of BstUI (MBI, Fermentas). When BstUI restriction site (Arg allele) was present, the 199-bp fragment was digested into two 113 bp and 86 bp fragments. The Pro allele was not cleaved by BstUI, and had a single band of 199 bp. The heterozygous genotype (Arg/Pro) had three bands (199, 113, and 86 bp). The genotypes were confirmed by direct sequencing using Big Dye Terminator Cycle sequencing kit (Applied Biosystems, Foster City, CA).
Mutational analysis of TP53 hot spot exons: Mutational analysis of TP53 gene was studied in 80 patients with CaP. Formalin-fixed, paraffin-embedded surgical tissue from CaP (80 specimens) and BPH (24 specimens) patients were derived from transurethral resection of prostate (TURP) and transrectal ultrasound (TRUS) guided biopsy. The tissue after being fixed in 10 per cent buffered-formalin was embedded in paraffin under standard conditions. Blocks and sections with tumour cells were selected for sectioning for immunohistochemistry and DNA extraction. All evaluations were done in the absence of any identifying information. Clinical follow up data of each patient was obtained from the medical records.
One block of each case of CaP and BPH that contained mostly tumour tissue was selected. Depending on the size of the tumour, 10 μm sections were cut from each sample. For deparaffinization, sections were dewaxed in a series of xylene, washed twice with 100 per cent ethanol to remove the solvent, and dried at room temperature. DNA extraction was performed using standard phenol-chloroform method with slight modification in incubation temperature and time, and two different DNA extraction kits; DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA), and RBC tissue DNA extraction kit (Real Biotech Corporation, Taiwan), to assess the best method to be employed for further extraction procedure. Efforts were made to minimize the contamination of normal tissue in tumour specimens.
PCR amplification of “hotspot” exons 5-8 of TP53 was performed using the following primers (Microsynth, Balgach, Switzerland): exon 5-6 (467bp) was amplified with (5-6_FP) 5’-TGT TCA CTT GTG CCC TGA CT-3’ and (5-6_RP) 5’-TTA ACC CCT CCT CCC AGA GA-3’, for exon 7 (196bp) (7_FP) 5’-CTT GCC ACA GGT CTC CCC AA-3’ and (7_RP) 5’-TGT GCA GGG TGG CAA GTG GC-3’ oligos were used and exon 8 (231bp) was amplified with (8_FP) 5’-TTC CTT ACT GCC TCT TGC TT-3’ and (8_RP) 5’-AGG CAT AAC TGC ACC CTT GG-3’. Each amplification was carried out separately in a 25 μl reaction volume containing 100 ng of template DNA, 0.2 μM of each primer, 0.2 mM of each deoxyribonucleotide (dNTP), 1.5 mM of MgCl2, 1 × PCR buffer and 1 unit of Platinum Taq DNA polymerase (Invitrogen, Groningen, The Netherlands). The reaction mixture was heated up at 94°C for 5 min, followed by 35 cycles of amplification at 94°C for 30 sec, 52-58°C for 30 sec and 72°C for 30 sec with a final extension step at 72°C for 10 min. No template control was included in each PCR to exclude contamination. After PCR amplification, 10 μl PCR products were purified using Exonuclease I (0.067 units) and Shrimp alkaline phosphatase (0.67 units) for 1 h at 37°C followed by 15 min at 85°C to denature the enzymes, and then sequenced directly with big dye dideoxy terminator cycle sequencing kit on an ABI 3130 xl automatic DNA sequencer (Applied Biosystems, Foster City, CA).
Immunohistochemical analysis of MMR: Adequate tissue for immunohistochemical study could be obtained from 80 patients with CaP and 15 with BPH. Additionally fifteen cases of prostatic intraepithelial neoplasia (PIN) were also included in the study to analyse protein expression of four MMR genes (hMLH1, hMSH2, hPMS1, and hPMS2) by immunohistochemical staining. Cases with inadequate tumour tissue were eliminated. Immunohistochemical staining was performed using primary antibodies against hMLH1 (clone G168-728, 1/40; BD Biosciences Pharmingen, San Diego, USA); hMSH2 (clone G219-1129, 1/100; BD Biosciences Pharmingen); hPMS2, (clone H-300, 1/100; Santa Cruz Biotech); and hPMS1 (clone C-20, 1/40; Santa Cruz Biotech). For each case, six sections (3–4 μm thick) were cut and mounted on to poly-l-lysine coated slides and dried overnight at 37°C. Antigenic site retrieval was accomplished by microwaving each slide for 15 min in 0.01 M citric acid buffer (pH 6.0). Sections were incubated with antibody for 2 h at room temperature. Antibody binding was detected using the Elite Vectastain ABC kit (Vector Laboratories Ltd, Peterborough, UK), which is based on the biotin–avidin system, using the manufacturer's protocol. The reaction was visualised using a VIP substrate kit for peroxidase (Vector Laboratories Ltd., UK). Negative control sections were treated in an identical manner with the exception of omission of primary antibody. Normal prostate tissue and stromal cells adjacent to the respective tumour were used as internal positive controls. Loss of MMR proteins expression was defined as complete absence of nuclear staining in tumour cells (but maintained in normal epithelial and stromal cells). Immunohistochemical staining was analysed by two pathologists.
Statistical analysis: χ2 test of significance was applied to assess fulfillment of Hardy-Weinberg equilibrium (HWE) of the genotypic frequencies of TP53 codon 72 polymorphism among patients with CaP, BPH and controls. Logistic regression analysis was performed for the risk assessment of TP53 codon 72 polymorphism. Bonferroni corrections for the multiple comparison was applied to TP53 codon 72 polymorphism analysis which gave an adjusted P value as k (k-1)/2, here the factor k=3 accounted for three groups (CaP, BPH and Controls) being tested in the study hence the adjusted P value for the multiple comparison came out as 0.02 to be significant at the 0.05 level. Additionally, the literature on TP53 codon 72 polymorphism genotype frequencies and genotype based odds ratio (OR) in cases of CaP, BPH and controls was available from the study by Henner et al10. The estimated OR for the comparative groups CaP vs. controls and BPH vs. controls with the genotype frequencies of Arg/Pro in both of these groups were 46 and 44.5 per cent, respectively. Taking the estimate as 1.2, a minimum of 65 samples was needed to estimate this reported OR with 95% confidence interval (CI) and relative precision of 50%14. Associations between TP53 codon 72 genotype and clinical variables were assessed using χ2 tests. Analysis of variance (ANOVA) was performed to evaluate the variation among the markers under study. Kendall's tau-b statistics was applied to obtain the product moment correlation matrix between the five biomarkers (TP53 codon 72 polymorphism and four MMR markers viz., hMLH1, hMLH2, hPMS1 and hPMS2). The association of TP53 codon 72 polymorphism was assessed individually, along with the coexpression of all four MMR markers. Statistical analysis was done using SPSS 17.0 version software (SPSS Inc. Chicago, USA).
Results
TP53 codon 72 polymorphism analysis: The study subjects for TP53 codon 72 polymorphism included 105 CaP cases, 120 BPH cases and 106 normal healthy controls. The mean age ± SD [median (IQR)] was similar for all groups viz. CaP 68.63 ± 9.83 [70 (63-75)], BPH 67.12 ± 9.10 [66.5 (60-73)] and controls 62 ± 10.62 [62 (55-69)] yr. The mean ± SD pre-operative serum PSA levels were 55.47 ± 47.51 ng/ml in CaP, and 25.18 ± 22.49 ng/ml in BPH cases. Control group had serum PSA levels less than 1 ng/ml.
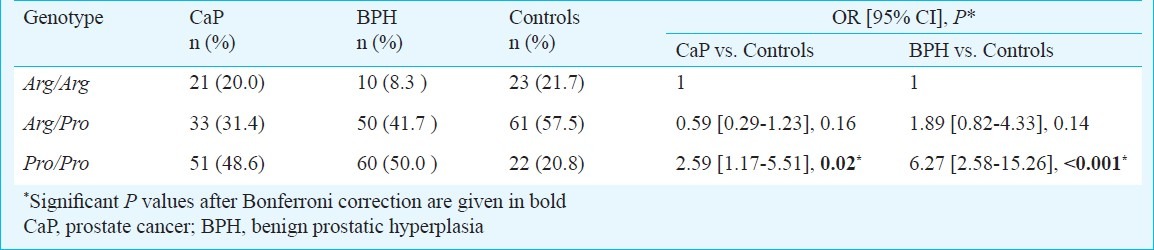
The frequencies of TP53 codon 72 polymorphism of Arg/Arg, Arg/Pro and Pro/Pro in the CaP cases were 21 (20.0%), 33 (31.4%), and 51 (48.6%) and in BPH cases, frequencies were 10 (8.3%), 50 (41.7%), and 60 (50.0%), respectively. The genotype frequencies among controls were found to be in agreement with the expected Hardy-Weinberg equilibrium frequencies of 21.7 per cent Arg/Arg, 57.5 per cent Arg/Pro, and 20.8 per cent Pro/Pro for the codon 72 polymorphism in TP53. The frequencies of the Pro allele were 64.3 per cent in CaP cases, 70.8 per cent in BPH cases and 49.5 per cent in controls whereas the frequencies for Arg allele were 35.7, 29.2, and 50.5 per cent in CaP, BPH and controls, respectively.
The frequencies of TP53 codon 72 polymorphism showed significant difference between CaP and controls (P=0.02). The subjects carrying Pro/Pro genotype had significantly high risk of developing CaP (OR, 2.59; 95% CI, 1.17-5.51, p= 0.02) and BPH (OR, 6.27; 95% CI, 2.58-15.26, P<0.001) as compared to the control group (Table I).
Table I.
Genotypic distribution of codon 72 polymorphism in TP53 gene
Mutational analysis of TP53 hot spot exons: Of the 80 CaP specimens, PCR amplification for all the hot spot exons (5-8) of TP53 could be obtained in 48 CaP and in 24 BPH specimens which were then subjected to direct DNA sequencing. In the current study, no sequence variation or alteration was observed in exon 5, 6 and 8 of TP53 gene. However, a somatic C/A variation in chromosomal position 7577644bp, at the intronic boundary of exon 7 in TP53 gene was observed in genomic DNA obtained from FFPE tissue sections of one each of CaP (n=48) and BPH (n=24) biopsy samples (Fig. 1). This variation was also evaluated in genomic DNA obtained from peripheral blood samples of normal healthy individuals (n=80) but the variant was not observed in samples screened. The C/A variation found in this study has been uploaded in NCBI SNP database (dbSNP) according to Human Genome Variation Society (HGVS) standards under the handle name TB_SS with batch submission ID, Variation_TP53_Ex7 and has been assigned sub-snp number, ss 262860127.
Fig. 1.
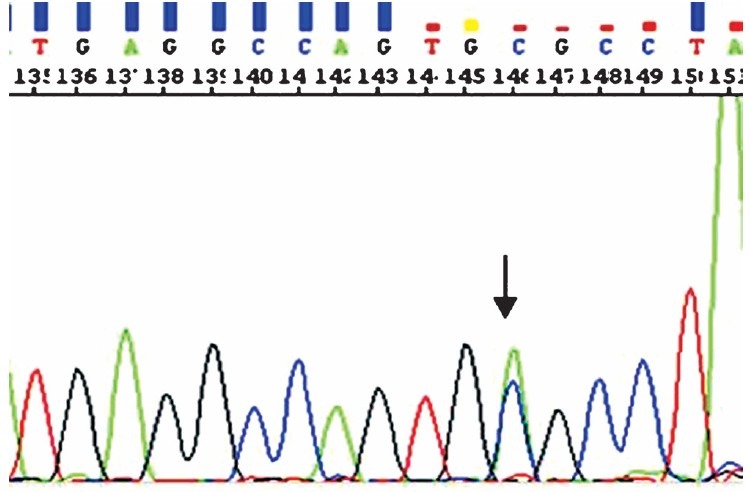
Electrogram showing the C>A variant (arrow) at intronic boundary of exon 7 in TP53 in biopsy sample of a CaP case.
Immunohistochemical analysis of MMR: Immunohistochemical analysis was performed for four MMR proteins (hMLH1, hMSH2, hPMS1, and hPMS2) in 80 cases of CaP, 15 cases of PIN and 15 cases of BPH. Among CaP, 28 cases (35%) had Gleason score > 7, while 52 cases (65%) had Gleason score 3-6. In benign prostate tissue, hMLH1, hMSH2, and hPMS2 were predominantly detected in the nuclei of glandular luminal epithelium, basal cells, and some stromal cells of the prostate gland, while, hPMS1 protein expression was present in the nuclei of basal cells and some stromal cells but absent in the luminal cell nuclei. No loss of MMR proteins was observed in any of the BPH tissues (Table II).
Table II.
Mismatch repair (MMR) protein expression in cases of benign prostatic hyperplasia (BPH), prostatic intraepithelial neoplasia (PIN) and prostate cancer carcinoma prostate (CaP)
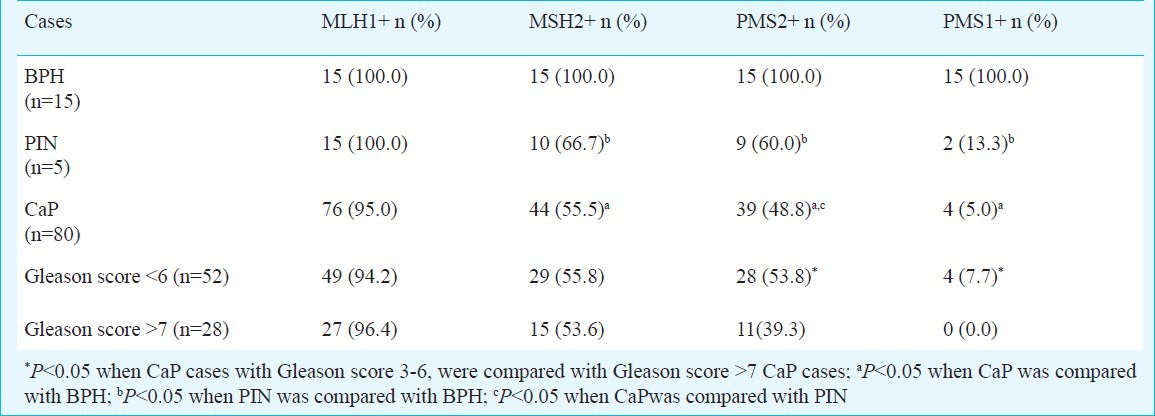
None of the 80 CaP cases showed a complete loss of MMR expression, (hMSH2-/hMLH1-/hPMS2-/hPMS1-). All cases of CaP showed loss of one or the other MMR protein, as profile of hMSH2+/hMLH1+/hPMS2+/hPMS1+ was not observed in a single case. Further, analysis of immunohistochemical patterns revealed that 33 (41.3%) cases of CaP had hMSH2+/hMLH1+/hPMS2-/hPMS1- profile, which was found as the most frequent immunohistochemical pattern. In CaP cases, hMLH1 expression was found in 76 (95%), hMSH2 in 44 (55%), hPMS2 in 39 (48.8%) and hPMS1 in 4 (5%) cases. There was a hierarchy of MMR loss in CaP, hMLH1<hMSH2<hPMS2<hPMS1 i.e. negligible loss of hMLH1 expression and maximum loss of hPMS1 expression (P<0.001) was observed (Table II; Fig. 2).
Fig. 2.
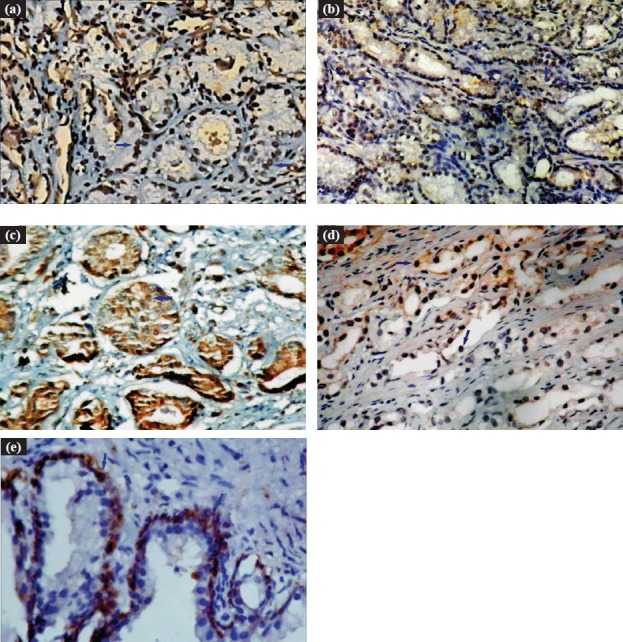
a. Immunohistochemical expression of hMLH1 protein (Blue arrow showing nuclear immunoreactivity) in CaP (Gleason score 5) 200X. b. Immunohistochemical expression of hMSH2 protein (Blue arrow showing nuclear immunoreactivity) in CaP (Gleason score 6) 100X. c. Immunohistochemical expression of hPMS2 protein (Blue arrow showing nuclear immunoreactivity) in CaP (Gleason score 6) 200X. d. Immunohistochemical expression of hPMS1 protein (Blue arrow showing nuclear immunoreactivity) in CaP (Gleason score 6) 200X. e. Immunohistochemical expression of hPMS1 protein in BPH (only in basal cells) (Blue arrow showing nuclear immunoreactivity) 400X.
Comparison of immunohistochemical expression of hMSH2, hMLH1, hPMS2 and hPMS1 in histological grades of CaP showed significantly higher hPMS2 expression in well differentiated (Gleason score 3-6) tumours than in poorly differentiated (Gleason score > 7) tumours (P=0.05) (Fig. 3). A significant loss of hPMS1 was observed in majority of CaP cases (P=0.007), and the expression was observed in only small percentage (7.7%) of well differentiated CaP. In cases of prostatic intraepithelial neoplasia, there was no loss of expression of hMLH1, while loss of hMSH2 was observed in 5 (33.3%), hPMS2 in 6 (40%), and hPMS1 in 13 (86.7%) cases. Amongst MMR proteins studied, maximum loss of hPMS1 expression was observed in CaP and PIN cases. However, significant loss of hPMS2 was observed in CaP compared to PIN (Table II).
Fig. 3.
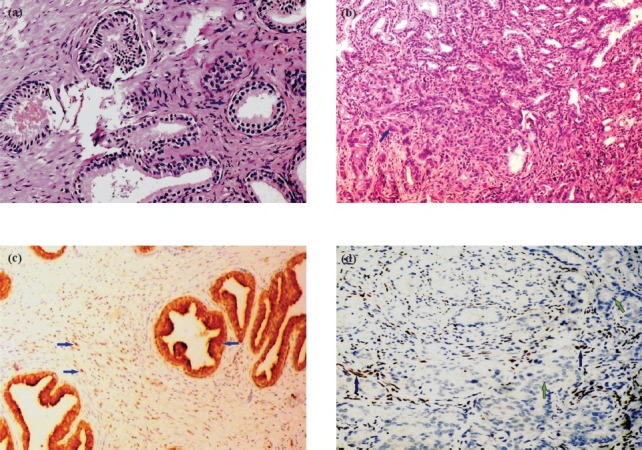
a and b. Normal prostate tissue (200X) and adjacent prostate cancer tissue (Gleason score 7) (100X) paired from the same section, stained with Haematoxylin and eosin. c and d. Normal prostate tissue (100X) and adjacent CaP (Gleason score 7) (200X) paired from the same section, depicting immunohistochemical expression of hPMS2 protein (Blue arrow) in normal prostate epithelium as well as stromal cells (Blue arrow), and absent in prostate cancer epithelial cells.
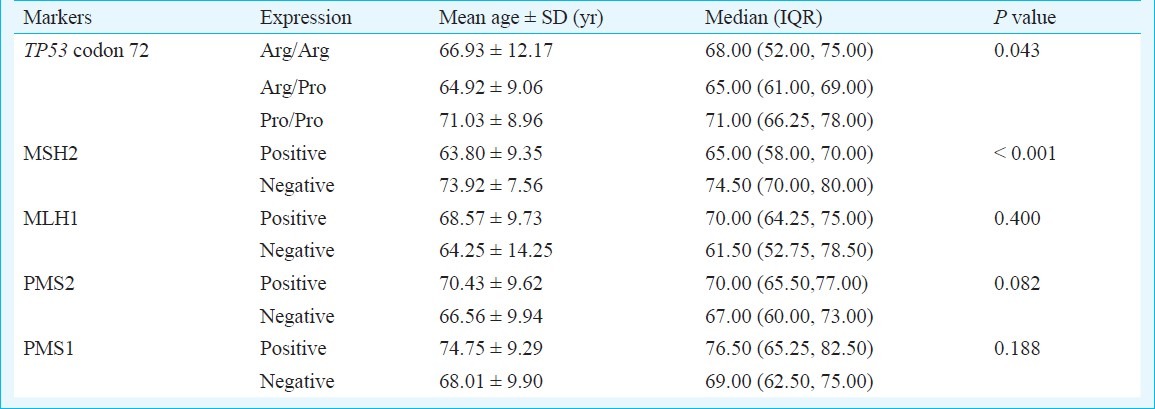
Association of age with TP53 polymorphism and MMR expression: A significant association with mean age at the time of diagnosis was observed with different genotypes of TP53 codon 72 polymorphism and MSH2 expression in CaP. While Pro/Pro genotype was significantly associated with higher mean age (71.03 ± 8.96 yr) at diagnosis (P=0.04), the MSH2 positive expression was associated significantly with lower age (63.80 ± 9.35) (P < 0.001) at diagnosis (Table III).
Table III.
Association of age with TP53 codon 72 genotypes and MMR expression
Association of TP53 and MMR proteins: Correlation between the TP53 codon 72 genotype, and the expression of MMR proteins did not show any statistical significance when TP53 codon 72 genotypes were cross classified with MMR markers (hMSH2, hMLH1, hPMS2 or hPMS1) (data not shown).
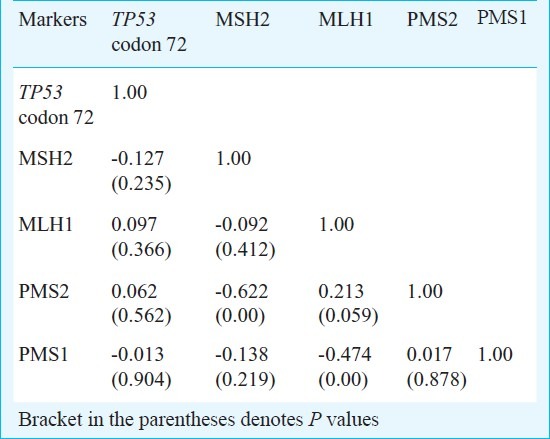
Product moment correlation was done to measure the strength of association of cross tabulation of all the markers (TP53 codon 72 polymorphism, hMSH2, hMLH1, hPMS2 or hPMS1) and the analysis showed a significant inverse interaction of hMLH1 with hPMS1 (P<0.001). Also, hMSH2 showed significant inverse interaction with hPMS2 (P<0.001) (Table IV).
Table IV.
Product moment correlation of markers under study
Discussion
The current study was undertaken to determine the role of mismatch repair genes hMSH2, hMLH1, hPMS2 and hPMS1 in prostate tumorigenesis and to determine if any of these markers may have any potential clinical application. The study also investigated the role of TP53 mutations and TP53 codon 72 polymorphism as potential risk marker for developing CaP.
Several studies15–18 have investigated the role of TP53 codon 72 polymorphism in various cancers, however, there are only a few studies on the impact of TP53 codon 72 polymorphism on CaP10,19. Dumont et al20 have shown that Arg variant functions more efficiently than Pro variant in preventing tumorigenesis, suggesting that individuals with Pro allele have a higher cancer susceptibility compared to those with Arg allele21. The present study also suggests that men carrying Pro variant are more susceptible to CaP and BPH than those carrying Arg variant. Further, an increase in risk of CaP was found associated significantly with TP53 codon 72 variant genotype (Pro/Pro) than from those with the Arg/Pro or Arg/Arg genotypes. The results of the present study are parallel with some case control studies addressing the question of the clinical relevance of TP53 codon 72 polymorphism in various cancers22. Wu et al23 found no association between TP53 codon 72 polymorphism and CaP, Henner et al10 found that men with Pro/Pro genotype have a significantly lower risk of CaP than those with Arg/Arg genotype, with further reduction in risk when the analysis was limited to Caucasians.
Coding single nucleotide polymorphisms (SNP) alter amino acid sequences and can modify structural and biological properties of encoded proteins. Alternatively, regulatory SNPs affect non-coding gene regions such as introns, enhancers, silencers and promoters. These non-coding sequence variants can modify transcription factor binding sites or generate novel binding sites, which may modulate gene expression in an allele-specific manner24. The mutational analysis of hot spot exon of TP53 led to identification of a C>A variation in TP53 at chromosomal position 7577644 in genomic DNA obtained from FFPE sections of one of CaP and one BPH biopsy sample, which was not observed in genomic DNA obtained from peripheral blood samples of normal healthy individuals. Though this variation is located at the intronic boundary (non-coding region) of exon 7 of TP53, the present study did not confirm variation by structural analysis of the TP53 gene.
The present study showed loss of one or the other MMR protein in all the cases studied. Majority (41.3%) of patients with CaP expressed hMSH2+/hMLH1+/hPMS2-/hPMS1-, making this the most frequent immunohistochemical pattern. Loss or absence of hPMS2 and hPMS1 was seen in 51.2 and 95 per cent cases of CaP, respectively. These results are similar to earlier studies reporting loss of hPMS1 expression in 86 per cent patients6,25 suggesting that MMR pathway of carcinogenesis may play a significant role in CaP. The significance of MMR pathway of carcinogeneis is well established in hereditary nonpolyposis colorectal cancer (HNPCC). However, mutational spectrum of CaP is different from that of HNPCC where stronger abnormalities are observed in hMSH2/hMLH1 gene while hPMS2 and hPMS1 mutations are rare6. This weak mutator phenotype may probably also explain the indolent nature of majority of prostate cancers that come to clinical presentation.
Loss of hPMS1 expression was seen in 95 per cent of CaP cases. Among the 15 PIN cases, loss of hPMS1 expression was seen in 86.7 per cent, while all the 15 cases of BPH showed consistent expression for hPMS1. Thus, a significant loss of hPMS1 expression was found with the progression of prostate cancer from BPH to PIN to CaP, as reported by Yian Chen et al6 also. Since hPMS1 is a basal cell specific protein, loss of hPMS1 expression in neoplastic prostate tissue may reflect its potential role in invasion. Neoplastic prostate epithelial cells are postulated to arise from a proliferating subpopulation of stem cells present in the basal compartment in normal glands as well as during malignant transformation26. However, there are conflicting reports on this issue, as it has also been hypothesized that during malignant transformation, some proliferative capacity shifts from basal cells to luminal cells27.
hPMS2 appears to play a critical role in normal MMR activity in some tissues. The hMLH1/hPMS2 heterodimer provides the majority of repair activity while hMLH1/hPMS1 dimer shows milder actions. In the current study, loss of PMS2 expression was found in 51.2 per cent of CaP, 40 per cent of PIN, while no loss was seen in benign prostate tissues. Although significant loss of hPMS2 protein was found between BPH and PIN, no significant difference was found in loss of expression of hPMS2 in CaP and PIN. A comparison of hPMS2 expression in well differentiated (Gleason score 3-6) and poorly differentiated (Gleason score >7) CaP showed a significant loss of hPMS2 expression in poorly differentiated tumors as reported earlier6. hPMS2 loss thus has a potential to serve as a prognostic marker in CaP. Conversely, significantly elevated levels of hPMS2 have also been reported in 52 per cent CaP tissues compared to normal and benign prostate tissues28.
hMSH2 expression was seen in 55 per cent of CaPs, 66.7 per cent of PIN and in all the fifteen BPH. Thus apart from loss of hPMS2 and hPMS1, there was a considerable loss of hMSH2 protein as well. Similarly, Burger et al29 found hMSH2 to be downregulated in 39.6 per cent of CaP cases. In another study, absent to low staining for the hMSH2 protein was documented in 30 per cent of well to moderately differentiated CaP (Gleason scores 5-6) and 29 per cent of poorly differentiated prostate carcinoma (Gleason scores 7-10) specimens25. In view of these observations hMSH2 may also be involved in prostatic carcinogenesis in a subset of prostate cancers. Similar to hPMS2 and hPMS1, there was an early loss of expression of hMSH2 in preneoplastic lesions.
In our study, MLH1 expression was seen in majority of the CaP cases and in all PIN and benign prostate hyperplasia cases. There was no significant loss of hMLH1 in CaP as compared to BPH or PIN suggesting a minor role of hMLH1 in prostate carcinogenesis. Thus of the four MMR proteins, hMLH1 is the least likely to be involved in CaP. This study showed a consistent loss of hPMS2 protein expression in human CaP as well as in PIN, suggesting that the loss of its function occurs early in human prostate neoplasia.
A significant association was observed between TP53 codon 72 variants and age at diagnosis of CaP. As with our study, a significantly higher age at diagnosis was found in individuals who were having (Pro/Pro) than in those who were homozygous for the wild type (Arg/Arg) TP53 genotype. The knowledge of the age at diagnosis of disease in individual carriers of (Pro/Pro) may have potential role in preventive strategies.
A few reports have shown the interaction of MMR system and TP53 in the tumorigenesis. A co-operation between MMR system and TP53 in tumourigenesis has been reported in knockout mice30. However, in the present study, no significant correlation was found between TP53 mutation/polymorphism and MMR expression of the two systems. In addition, the present study showed a significant inverse interaction of hMLH1 with hPMS1, and of hMSH2 with hPMS2 in CaP which may be due to significant loss of hPMS1 and hPMS2 expression.
The major limitation of the present study was the small sample size. The statistical power calculation for the risk analysis could not be reliably determined and this limited our ability to estimate the risk precisely. The results of this analysis should, therefore, be considered empirical observation for studies on larger number of samples. Another limitation to the study is that as loss of MMR protein expression may be a consequence of MMR gene mutation, thus, further studies on MMR mutational spectrum are envisaged to explore the role in complex pathogenesis of CaP.
In conclusion, our results demonstrate a significant loss of hPMS2 expression in poorly differentiated (Gleason score ≥7) tumours than well differentiated (Gleason score 3-6) CaP tumours suggesting its potential as a prognostic maker. As observed in the present study, aberrant MMR expression could be involved in the pathogenesis of prostate cancer through PIN, early CaP to aggressive CaP. Thus, the incorporation of MMR expression to the present biomarker panel may assist in better prognostic evaluation and management. Our findings on codon 72 of TP53 suggest that men carrying Pro/Pro genotype have significantly higher risk of developing CaP and BPH. As TP53 gene influences apoptosis, TP53 gene alteration by causing imbalance between prostate cell proliferation and apoptosis, may have a role to play in the pathogenesis of CaP and BPH. Further studies are required to determine the biologic role of TP53 gene variations in the development and progression of CaP and to determine whether TP53 mutations can be useful as prognostic markers.
Acknowledgment
Authors thank Department of Science and Technology (DST), New Delhi, for providing necessary funds to support this study, also acknowledge the Indian Council of Medical Research (ICMR), New Delhi, for providing financial assistance to second author (AS) in the form a research fellowship.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Sinha R, Anderson DE, McDonald SS, Greenwald P. Cancer risk and diet in India. J Postgrad Med. 2003;49:222–8. [PubMed] [Google Scholar]
- 3.Satyanarayana L, Asthana S. Life time risk for development of ten major cancers in India and its trends over the years 1982 to 2000. Indian J Med Sci. 2008;62:35–44. [PubMed] [Google Scholar]
- 4.Park JY, Huang Y, Sellers TA. Single nucleotide polymorphisms in DNA repair genes and prostate cancer risk. Methods Mol Biol. 2009;471:361–85. doi: 10.1007/978-1-59745-416-2_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Wang J, Fraig MM, Metcalf J, Turner WR, Bissada NK, et al. Defects of DNA mismatch repair in human prostate cancer. Cancer Res. 2001;61:4112–21. [PubMed] [Google Scholar]
- 7.Gatz SA, Wiesmuller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–16. doi: 10.1038/sj.cdd.4401903. [DOI] [PubMed] [Google Scholar]
- 8.Edelbrock M, He H, Schroering A, Fernstrom M, Bathala S, Williams KJ. Recognition and binding of mismatch repair proteins at an oncogenic hot spot. BMC Mol Biol. 2005;6:6. doi: 10.1186/1471-2199-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krypuy M, Ahmed AA, Etemadmoghadam D, Hyland SJ, DeFazio A, Fox SB, et al. High resolution melting for mutation scanning of TP53 exons 5-8. BMC Cancer. 2007;7:168. doi: 10.1186/1471-2407-7-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henner WD, Evans AJ, Hough KM, Harris EL, Lowe BA, Beer TM. Association of codon 72 polymorphism of p53 with lower prostate cancer risk. Prostate. 2001;49:263–6. doi: 10.1002/pros.10021. [DOI] [PubMed] [Google Scholar]
- 11.Malins DC, Johnson PM, Wheeler TM, Barker EA, Polissar NL, Vinson MA. Age-related radical-induced DNA damage is linked to prostate cancer. Cancer Res. 2001;61:6025–8. [PubMed] [Google Scholar]
- 12.Lipski BA, Garcia RL, Brawer MK. Prostatic intraepithelial neoplasia: significance and management. Semin Urol Oncol. 1996;14:149–55. [PubMed] [Google Scholar]
- 13.Blin N, Stafford DW. A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res. 1976;3:2303–8. doi: 10.1093/nar/3.9.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lwanga SK, Lemeshow S. Sample size determination in health studies: A practical manual. Geneva: World Health Organization; 1991. [Google Scholar]
- 15.Storey A, Thomas M, Kalita A, Harwood C, Gardiol D, Mantovani F, et al. Role of a p53 polymorphism in the development of human papillomavirus-associated cancer. Nature. 1998;393:229–34. doi: 10.1038/30400. [DOI] [PubMed] [Google Scholar]
- 16.Zehbe I, Voglino G, Wilander E, Delius H, Marongiu A, Elder L, et al. p53 codon 72 polymorphism and various human papillomavirus 16 E6 genotypes are risk factors for cervical cancer development. Cancer Res. 2001;61:608–11. [PubMed] [Google Scholar]
- 17.Zhang ZW, Laurence NJ, Hollowood A, Newcomb P, Moorghen M, Gupta J, et al. Prognostic value of TP53 codon 72 polymorphism in advanced gastric adenocarcinoma. Clin Cancer Res. 2004;10:131–5. doi: 10.1158/1078-0432.ccr-0853-3. [DOI] [PubMed] [Google Scholar]
- 18.Lattuada D, Vigano P, Somigliana E, Abbiati A, Candiani M, Di Blasio AM. Analysis of the codon 72 polymorphism of the TP53 gene in patients with endometriosis. Mol Hum Reprod. 2004;10:651–4. doi: 10.1093/molehr/gah093. [DOI] [PubMed] [Google Scholar]
- 19.Huang SP, Wu WJ, Chang WS, Wu MT, Chen YY, Chen YJ, et al. p53 Codon 72 and p21 codon 31 polymorphisms in prostate cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:2217–24. [PubMed] [Google Scholar]
- 20.Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33:357–65. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- 21.Manfredi JJ. p53 and apoptosis: it's not just in the nucleus anymore. Mol Cell. 2003;11:552–4. doi: 10.1016/s1097-2765(03)00106-0. [DOI] [PubMed] [Google Scholar]
- 22.Bendesky A, Rosales A, Salazar AM, Sordo M, Peniche J, Ostrosky-Wegman P. p53 codon 72 polymorphism, DNA damage and repair, and risk of non-melanoma skin cancer. Mutat Res. 2007;619:38–44. doi: 10.1016/j.mrfmmm.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Wu WJ, Kakehi Y, Habuchi T, Kinoshita H, Ogawa O, Terachi T, et al. Allelic frequency of p53 gene codon 72 polymorphism in urologic cancers. Jpn J Cancer Res. 1995;86:730–6. doi: 10.1111/j.1349-7006.1995.tb02461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandele OJ, Wang X, Campbell MR, Pittman GS, Bell DA. Human single-nucleotide polymorphisms alter p53 sequence-specific binding at gene regulatory elements. Nucleic Acids Res year? 39:178–89. doi: 10.1093/nar/gkq764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Velasco A, Hewitt SM, Albert PS, Hossein M, Rosenberg H, Martinez C, et al. Differential expression of the mismatch repair gene hMSH2 in malignant prostate tissue is associated with cancer recurrence. Cancer. 2002;94:690–9. doi: 10.1002/cncr.10247. [DOI] [PubMed] [Google Scholar]
- 26.Bonkhoff H. Role of the basal cells in premalignant changes of the human prostate: a stem cell concept for the development of prostate cancer. Eur Urol. 1996;30:201–5. doi: 10.1159/000474170. [DOI] [PubMed] [Google Scholar]
- 27.Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci U S A. 1998;95:1735–40. doi: 10.1073/pnas.95.4.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norris AM, Gentry M, Peehl DM, D’Agostino R, Jr, Scarpinato KD. The elevated expression of a mismatch repair protein is a predictor for biochemical recurrence after radical prostatectomy. Cancer Epidemiol Biomarkers Prev. 2009;18:57–64. doi: 10.1158/1055-9965.EPI-08-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burger M, Denzinger S, Hammerschmied CG, Tannapfel A, Obermann EC, Wieland WF, et al. Elevated microsatellite alterations at selected tetranucleotides (EMAST) and mismatch repair gene expression in prostate cancer. J Mol Med. 2006;84:833–41. doi: 10.1007/s00109-006-0074-0. [DOI] [PubMed] [Google Scholar]
- 30.Kruger S, Bier A, Engel C, Mangold E, Pagenstecher C, von Knebel Doeberitz M, et al. The p53 codon 72 variation is associated with the age of onset of hereditary non-polyposis colorectal cancer (HNPCC) J Med Genet. 2005;42:769–73. doi: 10.1136/jmg.2004.028506. [DOI] [PMC free article] [PubMed] [Google Scholar]