

Abstract
Background & objectives:
Replication of influenza A virus in the respiratory tract leads to cell damage and liberation of cytokines and chemokines. The in vivo cytokine induction and modulation by recombinant transforming growth factor- β1 (rTGF-β1) has not been studied. Therefore, in the present study the effect of rTGF-β1, a potent immunomodulatory cytokine which has anti-inflammatory properties and downregulates the release of inflammatory molecules, against influenza-virus infection in the airway of mice was investigated.
Methods:
rTGF-β1 was administered intravenously to mice with concomitant intranasal infection of influenza A/Udorn/317/72 (H3N2) virus, and the survival rate, virus titre, histopathological changes and levels of factors regulating inflammation in the airway fluid were analysed.
Result:
The immune response to influenza A virus was characterized by an influx of both macrophages and lymphocytes into the lungs of the infected host. rTGF-β1 significantly suppressed virus multiplication and improved the survival rate of mice. rTGF-β1 downregulated infiltration of neutrophils and the release of inflammatory molecules, such as interferon-gamma (IFN-γ), interleukin-1 β (IL-1β) and stimulated release of IL-10 that potentiates anti-inflammatory response into airway.
Interpretation & conclusions:
A generalized pulmonary inflammation does not contribute to viral clearance but represents an immunological background within which antiviral immunity operates. Treatment with rTGF-β1 reduced macrophage count and neutrophils influx in lungs of infected mice.
Keywords: Inflammatory response, influenza A virus, rTGF-β, viral pathogenesis
Influenza virus is a major human pathogen that causes epidemics and pandemics of significant morbidity1–5. In the elderly and those with pre-existing medical conditions, it can cause increased mortality. Influenza A virus infects and replicates in mucosal epithelial cells of respiratory tract. Replication in the respiratory tract leads to cell damage and liberation of cytokines and chemokines, which further leads to inflammation and respiratory symptoms3,6. The systemic and bronchealveolar production of pro-inflammatory cytokines is a well established feature of influenza virus infections7–9.
Several studies in laboratory from influenza infected cases and animal model confirmed that the immunopathological responses are significant factor of morbidity and mortality10,11. The inflammatory response in the upper respiratory tract after intranasal infection with influenza virus in ferrets showed an increase of neutrophils one day after infection. Thus, neutrophil infiltration during the early phase of infection is considered to be one of the characteristic features of influenza virus pathogenesis12. Virus infection induces the production of pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) which plays a significant role in both protective immunity and pathology of the infection, and the inflammatory response may be regulated by interleukin-10 (IL-10) and transforming growth factor- β1 (TGF-β1).
TGF-β1 is a potent immunomodulator and regulates the inflammatory process in a complex biphasic fashion. The immune response to influenza A virus is characterized by an influx of both macrophages and lymphocytes into the lungs of the infected host. In general, the pathogenesis of influenza infection can be divided into two phases, the cellular events that precede the lymphocyte invasion and those events that follow it13. We hypothesize that the TGF-β negatively affects the inflammatory response by regulating the lymphocyte influx to the airway and by further modulating release of pro-inflammatory and anti-inflammatory cytokines. The production of cytokine is strongly cell type-dependent, and the use of different cell types may account for discrepancies in in vitro studies.
So far, the in vivo cytokine induction and modulation by recombinant TGF-β1 (rTGF-β1) during influenza infection has not been characterized. It will, therefore, be important to elucidate the host and pathogen molecules that are involved in immune response. In this study we investigated the effect of TGF-β in resolution of inflammatory response on infection with influenza virus in mice.
Material & Methods
Mouse infection: The experiments were performed with BALB/c mice at the Department of Respiratory Virology at the V.P. Chest Institute, Delhi, India. Mice were purchased from National Institute of Nutrition, Hyderabad (A.P.) and V.P. Chest Institute, University of Delhi (Delhi), India. All animals used for the experiments were between 6 and 8 wk of age and were housed in experimental animal facility of V.P. Chest Institute. Also, ethical clearance was obtained from the Institutional Animal Ethical Committee. The influenza virus A/Udorn/317/72(H3N2), obtained from Centres for Disease Control (CDC), Atlanta was grown in the allantoic fluid of 10 day old embryonated eggs.
Experimental groups: Mice were distributed in four groups, each group consisted of 15 mice, and all the groups were anaesthetized with chloroform before treatment, (I) Control group: mice were mock infected by instillation of 50μl of allantoic fluid, (II) Virus group: mice were intranasally instilled with sublethal dose of influenza A virus (4.1×103 plaque forming unit (pfu)/mouse), (III) rTGF-β1 treated virus group: infected mice were administered intravenously with 200 μl rTGF-β1 (Peprotech Inc., USA), through the tail vein (5.0 μg/kg body weight of mice), (IV) rTGF-β1 treated control group: mock infected mice were administered intravenously with 200μl rTGF-β1 through the tail vein (5.0 μg/kg body weight of mice). In all the experiments virus and rTGF-β1 were given on day 0 but rTGF-β1 was inoculated after 10 min of virus instillation.
Survival experiments: Survival against experimental infection was evaluated in three experimental groups of 15 mice each i.e. virus group (II), rTGF-β1 treated virus group (III), and rTGF-β1 treated control group (IV) by instilling a lethal dose of influenza A virus (4.1×104 pfu/mouse). The virulence of influenza virus was expressed on the 5th to 6th day after virus infection. The survival of rTGF-β1treated and untreated animals was observed during the next 7 days.
Experimental infection and pfu enumeration: Viral load was done by pfu enumeration, 36 mice were intranasally instilled with sub-lethal dose of influenza A virus (4.1×103 pfu/mouse) in 50 μl of allantoic fluid. Of the 36 infected mice, a group of 18 infected mice administered, intravenously 5.0 μg/kg body weight of mouse per 200 μl rTGF-β1. At 2 to 7 days after infection, three mice were sacrificed from both infected and infected and rTGF- β1 treated groups, by cervical dislocation, their lungs with trachea were aseptically removed and homogenized in 2 ml of phosphate buffered saline (PBS), virus yield was quantified by the plaque-forming assay as described by Kurokawa et al14. Briefly, confluent monolayers of Madin-Darby canine kidney cells were incubated with homogenate serially diluted in PBS containing 1 per cent bovine serum albumin for 1 h at room temperature. The cells were then overlaid with Eagle's minimal essential medium supplemented with 0.2 per cent bovine serum albumin, 0.1 per cent DEAE dextran, 1 μg trypsin/ml and 0.8 per cent agar, and maintained in a humidified atmosphere containing 5 per cent CO2 for 3 days. The agar media were then removed and the cells were fixed with 5 per cent formalin solution and stained with 0.03 per cent methylene blue solution. Visualized plaques were counted and the virus titre was expressed as log10 pfu/ml14.
Collection of bronchoalveolar lavage (BAL) fluid: The mice were anaesthetized by chloroform, bronchoalveolar lavage (BAL) was performed as described earlier15,16. In brief, the lungs were lavaged twice with a total volume of 2.0 ml of saline (4°C) inserted through an endotracheal tube. The rate of recovery of BAL fluid (BALF) was greater than 90 per cent for all the animals tested. After the amount of fluid recovered was recorded, the BALF was centrifuged (1500 rpm for 15 min at 4°C) and the supernatant was collected and stored at -70°C until measurement of cytokines IL-1β, IL-10 and IFN-γ. The cells infiltrated in the BALF were resuspended in the original volume of PBS, and the total number was determined after erythrocyte lysis by trypan blue dye exclusion with a Neubauer hemocytometer under a microscope. Cell differential counts were determined by Wright staining of sample, on the basis of morphologic criteria, under a light microscope with evaluation of at least 200 cells per slide in triplicate.
Histopathology of the lungs: The lungs were fixed in 10 per cent neutral buffered formalin solution, sectioned, and stained with haematoxylin and eosin (H & E). Paraffin embedded tissue was sectioned (3 to 4) and stained with H & E for general morphology.
Cytokine assay in BAL fluid: After the collection of BAL, levels of IL-1β, IL-10 and IFN-γ were measured using ELISA kits (BD OptEIA, CA, USA) according to the instructions.
Statistical analysis: All experiments were repeated thrice to ensure reproducibility. Statistical analysis was performed with computer based software ‘PRISM’ for one way and two way ANOVA, followed by Bonferroni's multiple comparison tests and P<0.05 was considered as significant.
Results
Alterations in BALF cellularity: Influenza infection significantly induced cellular alterations in the BALF of infected animals. Comparison of the BALF cell counts and differentials of virus (Group II) with control (Group I) animals showed impressive increase in the levels of BALF cellularity post-infection (p.i.) (Fig. 1). BALF total cell counts increased by about 270 and 600 per cent in group II mice on 3rd and 5th day p.i. This increase in BALF cellularity was significantly (P<0.001) attenuated in the, virus +rTGF-β1 (Group III) animals (Fig. 1a). The BALF cellular differential counts were significantly increased on 3rd day and 5th day p.i. in group II with substantially high lymphocyte on the 3rdand 5thday post-infection, administration of rTGF-β1 with virus instillation reduced macrophage count in BALF (Fig. 1b), lymphocyte infiltration was decreased in alveolar lavage 5th and 7th day post infection by rTGF-β1 treatment (Fig. 1c). Virus infection also increased recruitment of polymorphonuclear neutrophils in lungs that was observed on 3rd and 5th day p.i. Treatment with rTGF-β1 diminished neutrophils influx in lungs (Fig. 1d).
Fig. 1.
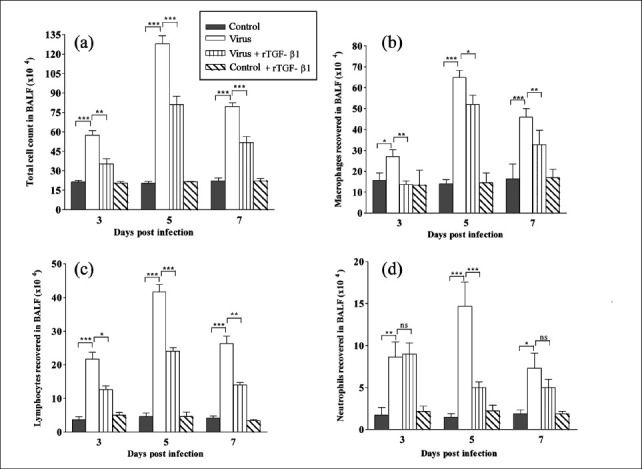
(a) Total cell count in bronchoalveolar lavage fluid (BALF), (b) Macrophages recovered in BALF, (c) Lymphocytes recovered in BALF, (d) Neutrophils recovered in BALF. Values are means ± SD; n =5 mice/group. *P<0.05 between groups.
Effect of rTGF-β1 on influenza virus clearance: To determine effect of rTGF-β1 on virus clearance in vivo, the virus titre of lung homogenates was measured from virus instilled mice and infected mice with rTGF-β1 simultaneously, administered once over the course of infection. The infectious virus was detected in the lungs after 48 h, and was generally cleared from the lung in 6 days (Fig. 2). When the pulmonary virus titre of lung homogenates from infected mice with simultaneous rTGF-β1 administration was measured at 2-6 days post infection, influenza virus titres were consistently lower than titres observed in the only virus instilled group. It was observed that in virus instilled group, titre peaked on day 4 of post-infection and rTGF-β1 treated mice showed 51 per cent lower pulmonary virus titer 4th day after infection. However, when the pulmonary virus titres of lung homogenates obtained on days 5, 6 and 7 post infections were compared, only a small amount of infectious virus was detected in lungs from both the groups.
Fig. 2.
Maximum virus titre observed on 4th day p.i. and administration of rTGF-β1 kept virus titre lower than the only virus instilled group. Administration of rTGF-β1 (group III) decreased virus titre (P*<0.05; **P<0.01 and ***P<0.001) on 3rd, 4th and 5th day p.i.
Effect of rTGF-β on survival rate of mice: Kaplan-Meier method was used to investigate whether intravenous administration of rTGF-β1 affected survival of mice intranasally instilled with influenza A virus. The survival of rTGF- β1 treated and untreated animals were observed during the next 7 days. Mock infected mice served as controls. It was observed that survival rate of mice in the control group was 100 per cent but the survival rate of the mice in the infected group was 13.3 per cent (Fig. 3). It was found that the survival rate of the mice in the rTGF-β1 administered group was 60 per cent higher than that of the mice in the virus infected group. Intravenous administration of rTGF-β1 protected mice against death due to influenza virus infection.
Fig. 3.
Virus instillation with lethal dose resulted increased mortality from 2nd day p.i. to 7th day p.i. (group II) compared with control (group I). Administration for TGF-β1 increased survival to 60% in Group III (rTGF-β1 + virus).
Effect of rTGF-β on lung morphology: Histological examination revealed that showed that on day 3 p.i. there was infiltration of inflammatory cells into alveolar walls with focal areas of consolidation the lungs of influenza virus instilled mice (group II), (Fig. 4a). The alveoli in the lungs of the control mice (group I) were normal in size and appearance. The bronchioles were affected, with necrosis of individual epithelial cells and focal intra-luminal aggregates of necrotic cellular debris in the influenza virus infected group (group II) 5th day after infection (Fig. 4b). The clear progression of the inflammation, significant alveolitis, with necrosis of epithelial cells was observed in the lungs of mice on 7th day after virus infection. The alveoli, interstitial septa and perivascular spaces were extensively infiltrated by a mixture of inflammatory cells, predominantly the mononuclear cells (Fig. 4c). Mice inoculated with influenza virus and administered with rTGF-β1 (group III) had minimal lung involvement on 3rd day post infection. Only a few small areas of lung showed an inflammatory mononuclear cell infiltrate in the alveoli (Fig. 4d). Lungs of mice infected with influenza administered with TGF-β1, showed minimal increase in the cellularity of the interstitial septa and no inflammatory infiltration of the alveoli and perivascular spaces 5th and 7th day post infection (Fig. 4e) and normal parenchyma (Fig. 4f).
Fig. 4.
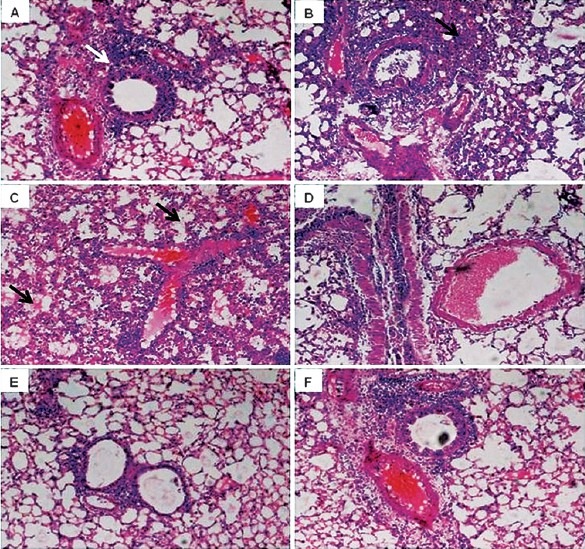
Lung histopathology of mice after infection with influenza virus. Lungs were fixed, sectioned, and stained with haematoxylin and eosin. A-C: Tissue from mice infected with influenza virus alone showing increase in infiltration of inflammatory cells into alveolar walls (white arrow head in A) with focal areas of consolidation (black arrow head in B) and necrosis of epithelial cells (arrows in C) on day 3, 5 and 7 p.i.; D-F: Tissue from mice simultaneously administered with rTGF-β1 showing less involvement of bronchioles and infiltration of inflammatory cells and normal parenchyma on day 3, 5 and 7.
Alterations in pro-inflammatory and anti-inflammatory cytokines: Highest levels of IFN-γ were observed on 5th day post infection. Treatment with rTGF-β1 decreased IFN-γ significantly (P<0.05) on 3rd day and (P<0.001) on 5th and 7th day p.i (Fig. 5a). The level of IL-1β in BALF of infected mice increased by 16-fold (from 9.33 to 151 pg/ml; P<0.001) (Fig. 5b) on 3rd day p.i. and by 22-fold on 5th day p.i. The level of IL-1β remained consistently low in rTGF-β1 treated group. IL-10 remained unaffected in BALF on day 3 and 5 p.i. with respect to control group and increased significantly to 7.8 fold (P<0.001) on day 7 p.i. (Fig. 5c) relative to uninfected mice. IL-10 concentration in rTGF-β1 treated group was increased significantly (P<0.001) compared to rest of the groups on days 3, 5 and 7 post-infection.
Fig.

5a. The IFN-γ level in BALF. The level observed was seen to be significantly increased in virus group (II) (P<0.001) on 3rd, 5th and 7th day p.i as compared to control group I (control mice) and group IV (rTGF- β1 treated control mice). Administration of rTGF-β1 down regulated IFN-γ significantly in group III (influenza virus with rTGF- β1) P<0.05 on 3rd day and P<0.001 on 5th and 7th day p.i. 5b. Level of IL-1 β in BALF of infected mice group II increased an average of 16-fold (from 9.33 to 151 pg/ml; P<0.001) on 3rd day p.i. from control group I and maximum increase 22 fold, observed on 5th day p.i. Level of IL-1β remained consistently lower in rTGF-β treated group III and rTGF-β control group IV. 5c. IL-10 level in BALF showing no significant change at day 3 and 5 p.i. and increased significantly (P<0.001) at day 7 p.i. in virus group (II) relative to control group (I) mice. IL-10 concentration in rTGF-β treated group (III) mice increased significantly (P<0.001) from rest of the groups including group IV (rTGF-β control mice) at day 3, 5 and 7 post-infection.
Discussion
Our findings demonstrate that rTGF-β1 plays a significant role in resolution of inflammatory response in the mouse model. It was observed that immunomodulation of mice infected with influenza virus by rTGF-β1 resulted in a reduced inflammatory infiltrate in the lungs, decreased concentrations of cytokines in the BAL fluid, and decreased mortality. A previous study17 has reported that rTGF-β1 impedes virus clearance, however, our findings did not corroborate. This indicates that generalized pulmonary inflammation does not contribute to viral clearance but represents an immunological background within which antiviral immunity operates or that the inflammatory response is magnified several fold over what is necessary and sufficient for effective clearance.
Investigation of immunopathological response induced by influenza virus revealed alterations in BAL cellularity: a local immune response initiated to support virus clearance. The increase in BAL cellularity was significantly attenuated in the infected rTGF-β1 administered animals over the period of infection. Treatment with rTGF-β1 reduced macrophage count and neutrophils influx in lungs. The significant morbidity and mortality associated with influenza virus infection is because of the involvement of lungs18 and local inflammatory response exhibited by influx of immune cells in an order to suppress the virus replication and to clear the virus from the site of infection19.
Elevated polymorphonuclear neutrophils (PMNs) are associated with pulmonary pathology after influenza infection20. The histopathlogical study of the lungs after experimental instillation of influenza virus A/Udorn/317/72(H3N2) suggest that in the mouse model, there is progressive damage of alveolar cells with acute inflammatory reaction and development of pneumonitis and bronchitis. There was high recruitment of neutrophils and macrophages along with epithelial damage. The massive infiltration of leukocytes into alveolar walls and spaces was due to the pathogenic effects of the virus. Mice inoculated with influenza virus and administered with rTGF-β1 had minimal lung involvement after infection with small areas of lung which showing inflammatory mononuclear cell infiltrate in the alveoli.
Our results showed increased survival of animals on administration of rTGF-β1, but to study its implications on the course of natural virus clearance, pfu enumeration in lung homogenate over the period of infection was assayed. Administration of rTGF-β1 reduced the number of infective virus particles in the lung homogenate. The maximum decline of infective virus particles was on 5th day p.i., where virus exerted maximum deleterious effects on the pathology of mouse lungs. Perhaps this could have contributed to the increased survival rate in mice on treatment with rTGF-β1, during the virus infection.
It has been reported that pathological changes associated with influenza virus infection are the consequence of cytokine response displayed by immune system and replication of virus in mice lungs21. Studies revealed protective roles of IFN-γ in animal models of infection with herpes simplex virus, cytomegalovirus, murine hepatitis virus, lymphocytic choriomeningitis virus, and adenovirus22. Mice lacking IFN-γ did not display a reduced ability to recover from infection with the A/JAP/57 (H2N2) strain of influenza virus and mounted cytotoxic T lymphocyte (CTL) activity comparable to that of their wild-type counterparts23. It has been reported that IFN-γ and influenza A virus synergistically induce inducible nitric oxide synthase (iNOS) which stimulates NO mediated inflammation and causes mortality24. In our study, attempts were made to minimize the inflammatory response without obliterating virus clearance mechanism of host immune system. rTGF-β1 is an immunomodulatory cytokine which has the ability to downregulate the IFN-γ and consequently the molecules having deleterious effects in an overshooting immune response.
The present data showed that the treatment with rTGF-β1 significantly decreased IFN-γ post-infection which correlated with a decrease in infiltration of inflammatory cells in lungs. rTGF-β1 showed a noticeable effect on local response over the period of infection as also demonstrated earlier25. IL-1 is also a primary inflammatory cytokine26. Its two forms, IL-1α and IL-1β, are products of adjacent but highly divergent genes. Our study showed an increase in level of IL-1 β in BALF of infected mice on 3rd day p.i. and maximum increase was observed on day 5 post infection. rTGF-β1 treatment kept level of IL-1β consistently lower. This pattern of inflammatory cytokine release would predict an early, marked inflammatory reaction in the lungs of untreated animals after influenza infection and is entirely consistent with the histopathology results and bronchial lavage cytology. This explains the interference of rTGF-β1 in early stages of IL-1β and IFN-γ induced inflammation during experimental influenza virus infection.
IL-10 is a potent anti-inflammatory cytokine which operates on many levels, including the inhibition of lymphocyte activation and macrophage function. There is no change observed in IL-10 concentration in BALF at early stage of infection. The administration of rTGF-β1 significantly reduced the levels of inflammatory cytokines, and induced a higher level of IL-10 in infected mouse lungs as compared to the only virus instilled group of animals. As in the group III, level of IL-10 increased significantly compared to rest of the groups. IL-10 is known to suppress the release of virus induced inflammatory cytokine27,28. This observation is important to add to our understanding that cytokine response plays an important role in preventing tissue damage at the site of infection, i.e. the lungs. Thus, the lungs of mice infected with virus demonstrated elevated levels of pro-inflammatory cytokines, particularly IL-1β, IFN-γ, and a low level of the anti-inflammatory cytokine IL-10. The high level of inflammatory cytokines was also consistent with the histological findings, the pulmonary lesions, inflammation and more intense cellular infiltration in mice infected with virus.
Several studies have shown that the TGF-β1 plays a critical role in inflammation and resolution of tissue injury in multiple organs, including the lungs. The TGF-β1 null mice [TGF-β1 (-/-)] show severe and generalized inflammatory disorders29,30.
Inhibition of inflammatory cell infiltration by rTGF-β1 administration was apparent upon analysis of the cells in BALF obtained from the virus-infected mice. Therefore, it is possible that suppression of IFN-γ and IL-1β is caused by the anti-inflammatory effect of rTGF-β1 on virus-induced acute inflammatory responses in the host. Of relative importance is our finding that rTGF-β1 administration significantly influences the virus yield in the lung. This result may substantiate the notion that rTGF-β1 exhibits a potent anti-inflammatory action31 against influenza-virus-induced lung injury by ameliorating virus-induced inflammation and modulating the pathogenesis of influenza virus infection. This supports our hypothesis that TGF- β1 may have a role in resolution of inflammation.
Acknowledgment
The authors acknowledge the Indian Council of Medical Research (ICMR), New Delhi, for providing the Senior Research Fellowship to the first author (VS).
References
- 1.Khanna M, Srivastava V, Kumar P. Influenza: A serious global threat. J Infect Dis Antimicrob Agents. 2002;19:25–39. [Google Scholar]
- 2.Khanna M, Akhtar N, Srivastava V, Kumar P, Vijayan VK. Biological and epidemiological aspects of influenza virus H5N1 in context of India. Indian J Exp Biol. 2006;44:265–78. [PubMed] [Google Scholar]
- 3.Khanna M, Kumar P, Singh V, Nazir R, Vijayan VK. Expression of macrophage inflammatory protein-1α and eosinophilic derived neurotoxin into upper respiratory secretions during influenza virus infection in children. Indian J Chest Dis. 2007;49:144–6. [Google Scholar]
- 4.Khanna M, Kumar P, Choudhary K, Kumar B, Vijayan VK. Emerging influenza: A global threat. J Biosci. 2008;33:475–82. doi: 10.1007/s12038-008-0066-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khanna M, Gupta N, Gupta A, Vijayan VK. Influenza A (H1N1) 2009: a pandemic alarm. J Biosci. 2009;34:481–9. doi: 10.1007/s12038-009-0053-z. [DOI] [PubMed] [Google Scholar]
- 6.Julkunen I, Melen K, Nyqvist M, Pirhonen J, Sareneva T, Matikainen S. Inflammatory responses in influenza A virus infection. Vaccine. 2000;19:S32–7. doi: 10.1016/s0264-410x(00)00275-9. [DOI] [PubMed] [Google Scholar]
- 7.Julkunen I, Sareneva T, Pirhonen J, Ronni T, Melen K, Matikainen S. Molecular pathogenesis of influenza A virus infection and virus-induced regulation of cytokine gene expression. Cytokine Growth Factor Rev. 2001;12:171–80. doi: 10.1016/s1359-6101(00)00026-5. [DOI] [PubMed] [Google Scholar]
- 8.Gern JE, Martin MS, Anklam A, Shen K, Roberg KA, Carlson-Dakes KT, et al. Relationships among specific viral pathogens, virus-induced interleukin-8, and respiratory symptoms in infancy. Pediatric Allergy Immun. 2002;13:386–93. doi: 10.1034/j.1399-3038.2002.01093.x. [DOI] [PubMed] [Google Scholar]
- 9.Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol. 2001;64:262–8. doi: 10.1002/jmv.1045. [DOI] [PubMed] [Google Scholar]
- 10.Kumar P, Sood V, Vyas R, Gupta N, Banerjea AC, Khanna M. Potent inhibition of influenza virus replication with novel siRNA-chimeric-ribozyme constructs. Antiviral Res. 2010;87:204–12. doi: 10.1016/j.antiviral.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Choi AM, Jacoby DB. Influenza virus A infection induces interleukin-8 gene expression in human airway epithelial cells. FEBS Lett. 1992;309:327–9. doi: 10.1016/0014-5793(92)80799-m. [DOI] [PubMed] [Google Scholar]
- 12.Sweet C, Smith H. Pathogenicity of influenza virus. Microbiol Mol Bio Rev. 1980;44:303–30. doi: 10.1128/mr.44.2.303-330.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol. 2000;156:1951–9. doi: 10.1016/S0002-9440(10)65068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurokawa M, Ochiai H, Nakajima K, Niwayama S. Inhibitory effect of protein kinase C inhibitor on the replication of influenza A virus. J Gen Virol. 1990;71:2149–55. doi: 10.1099/0022-1317-71-9-2149. [DOI] [PubMed] [Google Scholar]
- 15.Mejeski EI, Pa MK, Lopez AD, Harley RA, London SD, London L. Respiratory reovirus 1/L induction of intraluminal fibrosis, a model of bronchiolitis obliterans organizing pneumonia, is dependent on T lymphocytes. Am J Pathol. 2003;163:1467–79. doi: 10.1016/S0002-9440(10)63504-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nick JA, Young SK, Brown KK, Avdi NJ, Arndt PG, Suratt BT, et al. Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammation. J Immunol. 2000;164:2151–9. doi: 10.4049/jimmunol.164.4.2151. [DOI] [PubMed] [Google Scholar]
- 17.Schultz-Cherry S, Hinshaw VS. Influenza virus neuraminidase activates latent transforming growth factor beta. J Virol. 1996;70:8624–9. doi: 10.1128/jvi.70.12.8624-8629.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brody JA, Brock DB. Epidemiologic and statistical characteristics of the United States elderly population. In: Finch CE, Schneider EL, editors. Handbook of the biology of aging. 2nd ed. 1985. pp. 3–26. [Google Scholar]
- 19.Netea MG, Fantuzzi G, Kullberg BJ, Stuyt RL, Pulido EJ, McIntyre RC, et al. Neutralization of IL-18 reduces neutrophil tissue accumulation and protects mice against lethal Escherichia coli and Salmonella typhimurium endotoxemia. J Immunol. 2000;164:2644–9. doi: 10.4049/jimmunol.164.5.2644. [DOI] [PubMed] [Google Scholar]
- 20.Suliman HB, Ryan LK, Bishop L, Folz RJ. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am J Physiol Lung Cell Mol Physiol. 2001;280:L69–78. doi: 10.1152/ajplung.2001.280.1.L69. [DOI] [PubMed] [Google Scholar]
- 21.Ryan LK, Copeland LR, Daneils MJ, Costa ER, Selgrade MJ. Proinflammatory and Th1cytokine alterations following ultraviolet radiation enhancement of disease due to influenza infection in mice. Toxicol Sci. 2002;67:88–97. doi: 10.1093/toxsci/67.1.88. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, Xiang Z, Ertl HC, Wilson JM. Upregulation of class I major histocompatibility complex antigens by interferon-gamma is necessary for T cell mediated elimination of recombinant adenovirus infected hepatocytes in vivo. Proc Natl Acad Sci USA. 1995;92:7257–61. doi: 10.1073/pnas.92.16.7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham BM, Dalton DK, Giltinan D, Braciale VL, Stewart TA, Braciale TJ. Response to influenza infection in mice with a targeted disruption in the interferon γ gene. J Exp Med. 1993;178:1725–32. doi: 10.1084/jem.178.5.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kacergius T, Ambrozaitis A, Deng Y, Kuhn NS, Gravenstein S. Interferon-gamma augments cytotoxicity in influenza virus-infected RAW 264.7 and AMJ2-C11 macrophages by upregulating the expression of inducible nitric oxide synthase. Acta Medica Lituanica. 2008;15:76–80. [Google Scholar]
- 25.Carlson CM, Turpin EA, Moser LA, O’Brien KB, Cline TD, Jones JC, et al. Transforming growth factor-β: Activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 2010;6:e1001136. doi: 10.1371/journal.ppat.1001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinarello CA. Biologic basis for interleukin1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 27.Moore KW, De Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 28.Hamilton TA, Ohmori Y, Tebo J. Regulation of chemokine expression by anti inflammatory cytokines. Immunol Res. 2002;25:229–45. doi: 10.1385/IR:25:3:229. [DOI] [PubMed] [Google Scholar]
- 29.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulkarni AB, Huh G, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor-beta l null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–4. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Massague J. The transforming growth factor-beta family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]