

Abstract
Background & objectives:
Mutations in the oncogene and tumour suppressor genes play an important role in carcinogenesis. We investigated the association of p53 and K-ras gene mutation and Helicobacter pylori infection in patients with gastric cancer (GC) and peptic ulcer disease (PUD) attending a tertiary care hospital in north India.
Methods:
In total, 348 adult patients [62 GC, 45 PUD and 241 non-ulcer dyspepsia (NUD)] who underwent an upper gastrointestinal endoscopy were enrolled. H. pylori infection was diagnosed by rapid urease test, culture, histopathology and PCR. Mutation in the exon 5-8 of p53 gene was analyzed by PCR-single stranded conformational polymorphism (SSCP) and confirmed by sequence analysis. K-ras gene codon 12 mutation was analyzed by PCR-based restriction fragment length polymorphism.
Results:
Overall p53 gene mutation was found in 4.6 per cent of the study population, and its distribution in GC, PUD and NUD was 21, 4.4 and 0.4 per cent, respectively. p53 gene mutation was significantly higher in patients with GC than PUD (P<0.05) and NUD (P<0.001). No difference in p53 gene mutation was observed between H. pylori infected and non-infected individuals. K-ras gene mutation was absent in all the patients.
Interpretation & conclusions:
Our results show that p53 gene mutation may be associated with gastric carcinogenesis independent to H. pylori infection and absence of K-ras gene mutation questions its role in the pathogenesis of GC and PUD in Indian patients.
Keywords: Gastric cancer, gene mutations, Helicobacter pylori, peptic ulcer disease
Helicobacter pylori has been classified as a major cause of peptic ulcer disease (PUD) and a risk factor for gastric adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma1–3. On a global scale, gastric cancer is the second commonest cancer in the world. There is substantial international variation in gastric cancer incidence with the highest rates reported from China, Japan and other Eastern Asian countries. Epidemiological studies have proved that H. pylori infection is considered as a risk factor for gastric cancer and the International Agency for Research on Cancer (IARC) has classified this bacterium as a definite carcinogen2. While the majority of the infected individuals develop no significant clinical disease, others develop two kinds of divergent clinical outcomes – PUD and gastric cancer4. The reasons for developing these two extreme phenotypes remain poorly understood and are not explained by bacterial virulence factors alone4,5. This highlights the need to explore potential candidate genes of the host involved in the H. pylori- associated gastric carcinogenesis.
p53 protein plays an important role in the maintenance of genomic integrity through the induction of cell growth arrest or apoptosis following DNA damage6. Alterations in p53 gene, leading to a loss of tumour-suprressor function of p53 protein have been implicated in the aetiology and progression of a variety of human cancers7,8. In October 2006, the p53 database of IARC listed 31.2 per cent gastric cancers with point mutation in the p53 gene9. K-ras oncogene encodes a membrane-associated protein, p21RAS, with intrinsic GTPase activity involved in cellular signal transduction10. It is well known that K-ras plays an important role in the pathogenesis of various types of human cancer11. Point mutations at codons 12, 13 and 61 of K-ras result in a shift of K-ras protein toward the activated state, which constitutively activates the mitogenic signal transduction pathway12. Frequency of mutated K-ras varies among the different tumour types13. Point mutations of the K-ras are found predominantly in adenocarcinomas. The highest incidence is found in adenocarcinomas of the pancreas, in which approximately 90 per cent of the tumours harbour mutated K-ras10,11. There were inconclusive data available on p53 and K-ras gene mutational pattern in gastric cancer. It remains unclear that whether mutations in the above mentioned tumour suppressor p53 gene and ras-oncogene are associated with H. pylori infection and tumourigenesis. Therefore, this study was undertaken to investigate p53 and K-ras gene mutation in patients with gastroduodenal diseases in addition to H. pylori infection attending a tertiary care hospital in north India.
Material & Methods
Study population: A total of 348 consecutive adult patients [62 gastric adenocarcinoma (GC), 45 PUD and 241 non-ulcer dyspepsia (NUD)] who underwent upper gastrointestinal endoscopy at a tertiary referral center in northern India between September 2002 and May 2007 were enrolled in the study. The diagnosis of gastroduodenal diseases was based on clinical, endoscopical and histopathological examinations. Patients with NUD were considered as controls. The ethics committee of the institute granted approval for the study and the written consent was obtained from all the patients. Subjects who had received anti-microbial therapy, H2 receptor blockers, proton pump inhibitors and non-steroidal anti-inflammatory drugs in the preceeding 30 days prior to endoscopy or anti-H. pylori treatment in the past were excluded from this study.
DNA extraction: For PCR and mutation detection of p53 and K-ras gene, genomic DNA was isolated from gastric tissues using the QIAamp DNA mini kit (QIAGEN, Hilden, Germany) as per the manufacturer's instructions.
Detection of H. pylori infection: During each endoscopic examination antral biopsies were obtained and subjected to the following tests: rapid urease test (RUT), culture, histopathology and H. pylori specific ureA PCR following the standard protocol as described earlier12–15. H. pylori infection was diagnosed if any of the above tests was positive.
Detection of p53 gene mutation: Mutations of the p53 gene in exon 5-8 were identified by PCR- single stranded conformation polymorphism (PCR-SSCP). Mutations obtained by SSCP in the p53 gene were finally confirmed by sequence analysis. In brief, PCR was used to amplify exons 5-8 of p53 gene which are known to be mutational hot spots16. PCR was performed in a 50 μl reaction volume containing 100 ng of genomic DNA, 1× PCR buffer, 1.5 mM MgCl2, 0.2 mM each deoxynucleotide, 0.5 μM each specific primers (Table I) and 1.25 U of Taq polymerase (Bangalore Genei, India). The conditions of PCR were as follows: 35 cycles at 94°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec and final extension at 72°C for 10 min were carried out in a thermal cycler (MJ Research, USA) as described previously16. A negative control (no DNA template) and positive control (mutated DNA for each exon 5-8 of p53 gene obtained from Dr Pierre Hainaut, Head, Molecular Carcinogenesis group, IARC, WHO, France, as generous gifts) were run in parallel for each amplification reaction.
Table I.
Primer sequences used for p53 and K-ras gene

Before carrying out SSCP, 10 μl of PCR product was electrophoresed in 2 per cent agarose and visualized with ethidium bromide stain (0.5 μg/ml) to confirm the absence of contamination and to ensure that the PCR product was a single band of the appropriate size. Ten microlitre of each PCR product was mixed with 10 μl of SSCP dye containing 95 per cent (v/v) formamide, 20 mmol/l di-sodium EDTA, 0.05 per cent (w/v) xylene cyanol and 0.05 per cent (w/v) bromophenol blue. Before electrophoresis, the PCR products were heated at 95°C for 5 min in a water bath. After heat denaturation, the products were immediately placed on ice to prevent renaturation. An aliquot of 20 μl of each denatured sample was loaded onto 0.75 mm thick, 12 per cent (w/v) polyacrlylamide gel (29:1 ratio of acrylamide to bisacrylamide) with 5x TBE and EDTA. Electrophoresis of gel was carried out in TBE for 12 h at 18°C with constant current of 200 V. Single stands of DNA were visualized with silver staining. Any extra band(s) and/or change of mobility of bands present in the sample were considered as positive for a mutation. PCR-SSCP was run for all the samples in duplicates to reduce the possibility of false positive results due to polymerase errors or contamination. The samples that showed consistently aberrant patterns in replicate PCR-SSCP assays were subjected to direct sequencing. Sequencing of the PCR-SSCP positive samples was performed by commercial available sequencing services of Chromous Biotech Pvt. Limited, India.
Detection of K-ras gene mutation: Mutation of K-ras gene codon 12 was screened by using PCR-restriction fragment length polymorphism (PCR-RFLP) as described previously17. In brief, PCR was used to amplify K-ras codon 12 region using sequence specific forward and reverse primers (Table I). PCR was performed in a 50 μl reaction volume containing 100 ng of genomic DNA, 1x PCR buffer, 1.5 mM MgCl2, 0.2 mM each deoxynucleotide, 0.5 μM each specific primer and 1.25 U of Taq polymerase. The conditions of PCR were as follows: 35 cycles at 94°C for 1 min, 55°C for 1 min, 72°C for 1 min and final extension at 72°C for 10 min were carried out in a thermal cycler (MJ Research, USA). A negative control containing no DNA template was run in parallel for each amplification reaction. Before carrying out RFLP, 8 μl of PCR product was electrophoresed in 3 per cent agarose and visualized with ethidium bromide stain (0.5 μg/ml) to confirm the absence of contamination and to ensure that the PCR product was a single band of the appropriate size (106 bp).
Amplified PCR products were digested with Mva I (MBI, Fermentas, Vilnius, Lithuania) to distinguish the mutant allele from the wild type allele. Digestion of PCR products with the restriction enzyme was performed at 42°C for 8 h. After digestion, PCR products were electrophoresed on 2.5 per cent agarose gels, followed by ethidium bromide staining. If K-ras gene codon 12 was normal, wild type fragments cleaved to yield 77 and 29 bp products. If codon 12 contained a mutation, mutant type fragment yielded a single 106 bp product. The DNA with K-ras codon 12 mutation (kindly gifted by Dr Angelina Quintero, University of Mexico city, Mexico) was used as positive control in the study.
Statistical analysis: The data were analyzed by Chi square test. The SPSS 12.0 statistical package (Chicago, IL, USA) was used for data management and analysis.
Results
A total of 348 patients (mean age: 46.78 ± 15.96 yr; 216 male) were enrolled in the study and their distributions were as follows: gastric adenocarcinoma 62 (mean age: 56.60 ± 15.42 yr; 47 male), PUD 45 (mean age: 49.47 ± 17.22 yr; 31 male) and NUD 241 (mean age: 43.75 ± 14.76 yr; 138 male) (Table II). Presence of H. pylori infection was seen in 58.6 per cent patients. H. pylori infection was significantly higher in patients with PUD than with gastric adenocarcinoma (80 vs 56.5%, P<0.01) and NUD (80 vs 55.2%, P<0.01) (Table II).
Table II.
Demography of the study populations, p53 gene mutation and Helicobacter pylori infection
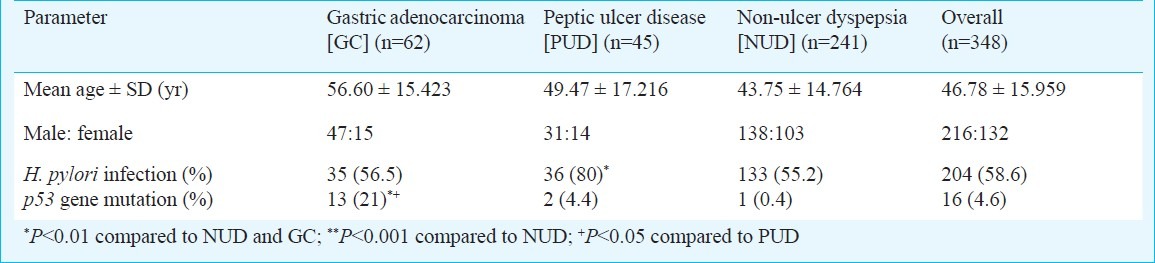
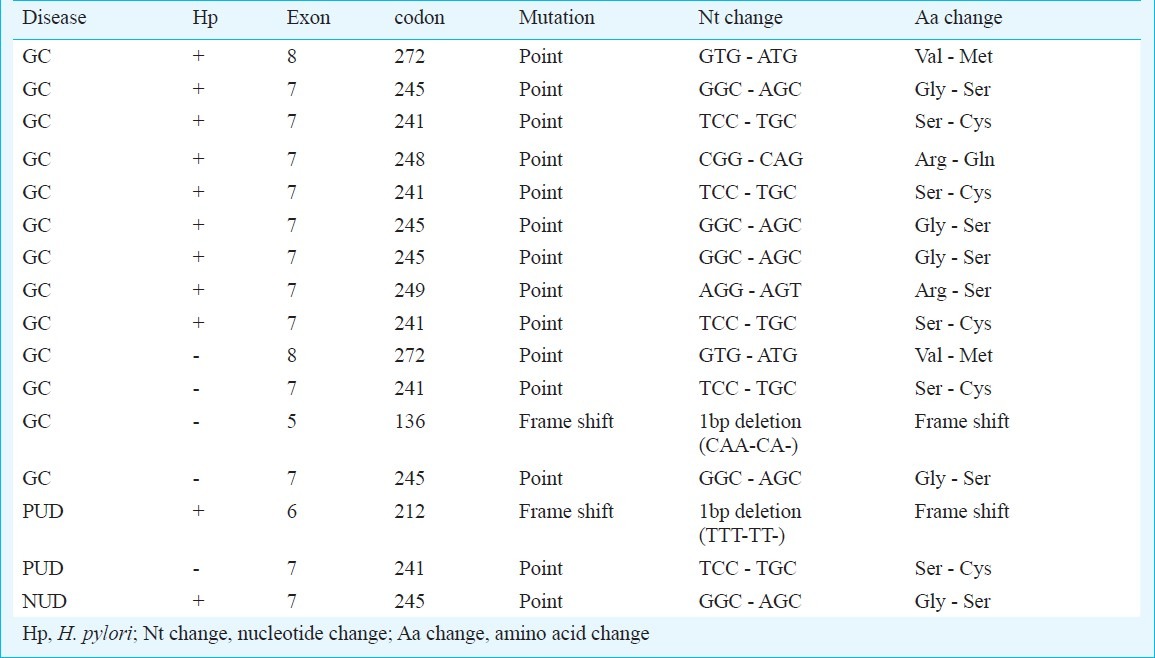
PCR-SSCP analysis detected alterations in the p53 gene in 16 (4.6%) of 348 patients. There were 1, 1, 12 and 2 alterations in exons 5, 6, 7 and 8, of p53 gene, respectively. p53 gene mutation in patients with GC, PUD and NUD was 21 per cent (13/62), 4.4 per cent (2/45) and 0.4 per cent (1/241), respectively. p53 gene mutation was significantly higher in patients with GC than PUD (P<0.05) and NUD (P<0.001). No difference in p53 gene mutation was observed between H. pylori infected and non-infected individuals (Table II). p53 gene mutation was significantly higher in males compared to females [87.5% (14/16) vs 12.5% (2/16), P<0.05]. All the mutations obtained by PCR-SSCP were confirmed by sequence analysis (Table III). Amplified products from three tumour samples that yielded normal mobility patterns and from three corresponding adjacent normal tissue samples were sequenced, and no mutations were found in the sequence of the exon 5-8 regions. K-ras gene mutation was absent in all the patients with GC, PUD and NUD in our study.
Table III.
Details and distribution of p53 gene mutation (exon 5-8) in patients with gastric adenocarcinoma (GC), peptic ulcer disease (PUD) and non-ulcer dyspepsia (NUD)
Discussion
Tumourigenesis occurs when a cell loses its regulated growth cycle and clonally expands beyond control. This requires a series of critical molecular events that cause the cell to divide and escape from normal proliferative control18. GC may follow this model. The product of the p53 gene plays an important role in the negative regulation of cell growth. The wild-type p53 protein binds to specific DNA sequences as a transcriptional factor that regulates the expression of particular genes in the cell. Consequently, it blocks cell progression through the late G1 phase of the cell cycle19. Some mutant proteins fail to block this progression, whereas others can gain a novel function and actually promote cellular proliferation20. There are significant geographic differences in the p53 gene mutation rates in GC. Several investigators have reported p53 gene mutations in GC around the world. In our study, 21 per cent patients with gastric carcinoma showed p53 mutations. This falls within a wide range of p53 mutation frequency of 0 to 82 per cent21,22 in GC from previous studies that used similar methods for mutation detection.
There is incessant debate on the association of p53 gene mutation and H. pylori infection with GC. Some investigators have reported that H. pylori infection induces p53 gene mutation in gastric carcinogenesis23,24. However, others25,26 and the results of the present study showed that p53 gene mutations occurred independent to H. pylori infection in GC. Hongyo et al26 reported that H. pylori infection was present in 55.8 per cent gastric cancer cases and was somewhat more common in cases lacking p53 gene mutation. In our study H. pylori infection was found in 56.5 per cent patients with gastric cancer and p53 gene mutation was higher in H. pylori infected individuals when compared with H. pylori uninfected individuals, however, the difference was not statistically significant (69.2%, 9/13 vs 30.8%, 4/13). The role of p53 gene mutation has not been studied in patients with PUD and NUD. In the present study, 4.4 and 0.4 per cent p53 gene mutation was seen in 4.4 per cent patients with peptic ulcer and in 0.4 per cent with non-ulcer dyspepsia. The occurrence of p53 mutations was higher in H. pylori infected individuals than in H. pylori non-infected individuals but the difference was not significant [5.4% (11/204) vs 3.5% (5/144)]. Thus, our study provides no evidence that H. pylori infection directly induces p53 gene alterations.
The K-ras encodes a membrane-associated protein, p21RAS, with intrinsic GTPase activity involved in cellular signal transduction9. Point mutations of K-ras at specific codons lead to activated oncoprotein, GTP-RAS, with reduced GTPase activity. A mutation at codon 12 alone is sufficient for oncogenic activation9. These findings suggest that mutations at codon 12 of K-ras may play an important role in gastric carcinogenesis. Of the 348 adult patients enrolled in the study, none had K-ras gene mutation.
In literature, variable incidence of K-ras gene mutation has been reported in gastric cancer27,28. However, some investigators found no evidence for mutation in codon 12 of K-ras gene in GC29,30, which are concordant to the current finding. Mutations in K-ras oncogene do not appear to have any role in gastric carcinogenesis, at least in our population.
In conclusion, we observed p53 gene mutation in 4.6 per cent of our study population. This mutation was significantly higher in GC when compared with PUD and NUD and it was independent to H. pylori infection indicating a role of p53 gene mutation in gastric carcinogenesis, independent to H. pylori infection. Absence of K-ras gene mutation in our population questions its role in the pathogenesis of GC and PUD in Indian patients.
We analyzed p53, K-ras gene mutation and H. pylori infection in patients with gastric cancer and peptic ulcer disease and compared it with non-ulcer dyspepsia which is considered as disease control group. Ideally we should have taken asymptomatic healthy controls instead of disease controls but practically that was not possible on ethical ground. Healthy individuals cannot be subjected to endoscopy.
Acknowledgment
Authors acknowledge Dr Pierre Hainaut, Head, Molecular Carcinogenesis group, IARC, WHO, France and Dr Angelina Quintero, University of Meixco city, Mexico for providing the mutated DNA samples for p53 gene exon 5-8 and K-ras gene codon 12, respectively as a generous gift. The study was supported by Council of Science and Technology, Government of Uttar Pradesh, India, through grant no. CST/SERPD/D-3402. The first author (AS) acknowledges the financial assistance as senior research fellowship from the Indian Council of Medical Research (ICMR) and the second author (ss) acknowledges the Department of Biotechnology (DBT), Government of India, New Delhi (India), for providing junior research fellowship.
References
- 1.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–9. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 2.International Agency for Research on Cancer. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risk Hum. 1994;61:177–241. [PMC free article] [PubMed] [Google Scholar]
- 3.Saxena A, Prasad KN, Ghoshal UC, Bhagat MR, Krishnani N, Husain N. Polymorphism of -765G > C COX-2 is a risk factor for gastric adenocarcinoma and peptic ulcer disease in addition to H. pylori infection: a study from northern India. World J Gastroenterol. 2008;14:1498–503. doi: 10.3748/wjg.14.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Omar EM, Chow WH, Rabkin CS. Gastric cancer and H. pylori: Host genetics open the way. Gastroenterology. 2001;121:1002–4. [PubMed] [Google Scholar]
- 5.Weinberg RA. Tumor suppressor genes. Science. 1991;254:1138–46. doi: 10.1126/science.1659741. [DOI] [PubMed] [Google Scholar]
- 6.Sarbia M, Porschen R, Borchard F, Horstmann O, Willirs R, Gobbert HE. p53 protein expression and prognosis in squamous cell carcinoma of the esophagus. Cancer. 1994;74:2218–23. doi: 10.1002/1097-0142(19941015)74:8<2218::aid-cncr2820740803>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 7.Renault B, Van den Broek M, Fodde R, Wijmen J, Pellegata NS, Amadori D, et al. Base transitions are the most frequent genetic changes at p53 in gastric cancer. Cancer Res. 1993;53:2614–7. [PubMed] [Google Scholar]
- 8.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–9. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 9.Soh K, Yanagisawa A, Hiratsuka H, Sugano H, Kato Y. Variation in K-ras codon 12 point mutation rate with histological atypia within individual colorectal tumors. Jpn J Cancer Res. 1993;84:388–93. doi: 10.1111/j.1349-7006.1993.tb00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee KH, Lee JS, Suh C, Kim SW, Kim SB, Lee JH, et al. Clinicopathological significance of the K-ras gene codon 12 point mutation in stomach cancer. An analysis of 140 cases. Cancer. 1995;75:2794–801. doi: 10.1002/1097-0142(19950615)75:12<2794::aid-cncr2820751203>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 11.Capon DJ, Seeburg PH, McGrath JP, Hayflick JS, Edman U, Levinson AD, et al. Activation of Ki-ras 2 gene in human colon and lung carcinoma by two different point mutations. Nature. 1983;304:507–13. doi: 10.1038/304507a0. [DOI] [PubMed] [Google Scholar]
- 12.Thillainayagam AV, Arvind AS, Cook RS, Harrison IG, Tabaqchali S, Farthing MJ. Diagnostic efficiency of an ultra rapid endoscopy room test for Helicobacter pylori. Gut. 1991;32:467–9. doi: 10.1136/gut.32.5.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma S, Prasad KN, Chamoli D, Ayyagari A. Antimicrobial susceptibility and biotyping pattern of Helicobacter pylori isolates from patients with peptic ulcer diseases. Indian J Med Res. 1995;102:261–6. [PubMed] [Google Scholar]
- 14.Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996;20:1161–81. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Clayton CL, Kleanthous H, Coates PJ, Morgan DD, Tabaqchali S. Sensitive detection of Helicobacter pylori by using polymerase chain reaction. J Clin Microbiol. 1992;30:192–200. doi: 10.1128/jcm.30.1.192-200.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiao YH, Rugge M, Correa P, Lehmann HP, Scheer WD. p53 alteration in gastric precancerous lesions. Am J Pathol. 1994;144:511–7. [PMC free article] [PubMed] [Google Scholar]
- 17.Singh MK, Chetri K, Pandey UB, Kapoor VK, Mittal B, Choudhuri G. Mutational spectrum of K-ras oncogene among Indian patients with gallbladder cancer. J Gastroenterol Hepatol. 2004;19:916–21. doi: 10.1111/j.1440-1746.2004.03355.x. [DOI] [PubMed] [Google Scholar]
- 18.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 19.May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene. 1999;18:7621–36. doi: 10.1038/sj.onc.1203285. [DOI] [PubMed] [Google Scholar]
- 20.Chen PL, Chen YM, Bookstein R, Lee WH. Genetic mechanisms of tumor suppression by the human p53 gene. Science. 1990;250:1576–80. doi: 10.1126/science.2274789. [DOI] [PubMed] [Google Scholar]
- 21.Mattar R, Nonogaki S, Silva C, Alves V, Gama-Rodrigues JJ. P53 and Rb tumor suppressor gene alterations in gastric cancer. Rev Hosp Clin Fac Med Sao Paulo. 2004;59:172–80. doi: 10.1590/s0041-87812004000400004. [DOI] [PubMed] [Google Scholar]
- 22.Fenoglio-Preiser CM, Wang J, Stemmermann GN, Noffsinger A. TP53 and gastric carcinoma: a review. Hum Mutat. 2003;21:258–70. doi: 10.1002/humu.10180. [DOI] [PubMed] [Google Scholar]
- 23.Murakami K, Fujioka T, Okimoto T, Mitsuishi Y, Oda T, Nishizono A, et al. Analysis of p53 gene mutations in Helicobacter pylori-associated gastritis mucosa in endoscopic biopsy specimens. Scand J Gastroenterol. 1999;34:474–7. doi: 10.1080/003655299750026191. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Chi DS, Kalin GB, Sosinski C, Miller LE, Burja I, et al. Helicobacter pylori infection and oncogene expressions in gastric carcinoma and its precursor lesions. Dig Dis Sci. 2002;47:107–13. doi: 10.1023/a:1013223722331. [DOI] [PubMed] [Google Scholar]
- 25.Lima VP, de Lima MA, André AR, Ferreira MV, Barros MA, Rabenhorst SH. H. pylori (CagA) and Epstein-Barr virus infection in gastric carcinomas: correlation with p53 mutation and c-Myc, Bcl-2 and Bax expression. World J Gastroenterol. 2008;14:884–91. doi: 10.3748/wjg.14.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hongyo T, Buzard GS, Palli D, Weghorst CM, Amorosi A, Galli M, et al. Mutations of the K-ras and p53 genes in gastric adenocarcinomas from a high-incidence region around Florence, Italy. Cancer Res. 1995;55:2665–72. [PubMed] [Google Scholar]
- 27.Watari J, Tanaka A, Tanabe H, Sato R, Moriichi K, Zaky A, et al. K-ras mutations and cell kinetics in Helicobacter pylori associated gastric intestinal metaplasia: a comparison before and after eradication in patients with chronic gastritis and gastric cancer. J Clin Pathol. 2007;60:921–6. doi: 10.1136/jcp.2006.041939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hiyama T, Haruma K, Kitadai Y, Masuda H, Miyamota M, Tanaka S, et al. K-ras mutation in Helicobacter pylori associated chronic gasstritis in patients with and without gastric cancer. Int J Cancer. 2002;97:562–6. doi: 10.1002/ijc.1644. [DOI] [PubMed] [Google Scholar]
- 29.Kasper HU, Schneider-Stock R, Mellin W, Roessner A. P21 protein expression and ras-oncogene mutations in gastric carcinoma: correlation with clinical data. Int J Oncol. 1998;12:69–74. doi: 10.3892/ijo.12.1.69. [DOI] [PubMed] [Google Scholar]
- 30.Craanen ME, Blok P, Top B, Boerrigter L, Dekker W, Offerhaus GJ, et al. Absence of ras gene mutations in early gastric carcinomas. Gut. 1995;37:758–62. doi: 10.1136/gut.37.6.758. [DOI] [PMC free article] [PubMed] [Google Scholar]