

Abstract
Dysregulation of histone modifying enzymes has been associated with numerous psychiatric disorders. Alterations in G9a (Ehmt2), a histone methyltransferase that catalyzes the euchromatic dimethylation of histone H3 at lysine 9 (H3K9me2), has been implicated recently in mediating neural and behavioral plasticity in response to chronic cocaine administration. Here, we show that chronic morphine, like cocaine, decreases G9a expression, and global levels of H3K9me2, in mouse nucleus accumbens (NAc), a key brain reward region. In contrast, levels of other histone methyltransferases or demethylases, or of other methylated histone marks, were not affected in NAc by chronic morphine. Through viral-mediated gene transfer and conditional mutagenesis, we found that overexpression of G9a in NAc opposes morphine reward and locomotor sensitization and concomitantly promotes analgesic tolerance and naloxone-precipitated withdrawal, whereas downregulation of G9a in NAc enhances locomotor sensitization and delays the development of analgesic tolerance. We identified downstream targets of G9a by providing a comprehensive chromatin immunoprecipitation followed by massively parallel sequencing analysis of H3K9me2 distribution in NAc in the absence and presence of chronic morphine. These data provide novel insight into the epigenomic regulation of H3K9me2 by chronic morphine and suggest novel chromatin-based mechanisms through which morphine-induced addictive-like behaviors arise.
Introduction
Increasing evidence suggests that chromatin dysregulation is important in mediating aberrant transcriptional responses in limbic brain regions that contribute to numerous psychiatric disorders, including drug addiction, depression, and schizophrenia (Guidotti et al., 2011; Peter and Akbarian, 2011; Robison and Nestler, 2011; Sun et al., 2012). For example, repeated exposure to drugs of abuse induces persistent changes in gene expression in nucleus accumbens (NAc), a key brain reward region, which are thought to be mediated via chromatin mechanisms (Maze and Nestler, 2011). These transcriptional changes are hypothesized to then contribute to behavioral abnormalities associated with drug addiction.
Chromatin is composed of basic transcriptional units (nucleosomes), in which ∼147 bp of DNA are wrapped around a core histone octamer. These histone proteins have unstructured N-terminal tails that protrude from the core fold of the histone octamer, which can be posttranslationally modified in many ways to alter the structure and transcriptional accessibility of chromatin.
Although most previous studies have focused on how cocaine or related stimulants affect the enzymatic machinery that catalyzes the addition or removal of posttranslation modifications to histone tails, only a few studies have implicated a role for histone acetylation in opiate action (Sanchis-Segura et al., 2009; Wang et al., 2010; Jing et al., 2011; Sheng et al., 2011; Rehni et al., 2012). More recent attention has focused on histone methylation, which has been shown to be important in regulating normal cognitive function and the development of several psychiatric disorders (Day and Sweatt, 2011; Maze and Nestler, 2011; Peter and Akbarian, 2011). G9a is the core subunit of a multimeric repressive histone lysine methyltransferase (KMT) complex and acts primarily to catalyze the dimethylation of histone H3 at lysine 9 (H3K9me2) throughout the genome (Fritsch et al., 2010; Shinkai and Tachibana, 2011). We have established a crucial role for G9a and H3K9me2 in cocaine-induced transcriptional and behavioral plasticity and in the regulation of susceptibility to chronic stress by previous cocaine exposure (Maze et al., 2010; Covington et al., 2011). However, the involvement of such repressive histone methylation in opiate action has not yet been investigated.
Here, we demonstrate that both G9a and H3K9me2 are downregulated in NAc by repeated morphine administration, as observed for cocaine and stress previously (Maze et al., 2010; Covington et al., 2011). We further demonstrate that levels of G9a in NAc exert an important influence on a range of behavioral responses to repeated morphine, including morphine-elicited reward, locomotor sensitization, analgesic tolerance, and physical withdrawal. To investigate putative downstream gene targets that mediate these behavioral abnormalities, we performed a genome-wide analysis, by use of chromatin immunoprecipitation followed by massively parallel sequencing (ChIP-seq), of H3K9me2 binding in NAc after repeated saline or morphine administration. Our results not only confirmed previously identified genes associated with morphine-elicited behaviors (e.g., fosB, grin2a) but also identified numerous novel targets (e.g., metabotropic glutamate receptors, repetitive elements) that shed fundamentally new light on our understanding of the molecular basis of morphine addiction.
Materials and Methods
Animals.
For all experiments, 9- to 11-week-old C57BL/6J male mice (The Jackson Laboratory) or G9afl/fl mice (described in detail by Maze et al., 2010) were used and group housed (five per cage) in a colony room set at constant temperature (23°C) on a 12 h light/dark cycle (lights on from 7:00 A.M. to 7:00 P.M.) with ad libitum access to food and water. All protocols involving mice were approved by the Institutional Animal Care and Use Committee at Mount Sinai School of Medicine.
RNA isolation, reverse transcription, and qPCR.
Bilateral 14-gauge NAc punches were obtained after drug treatment and frozen on dry ice. Samples were then homogenized in TRIzol and processed according to the instructions of the manufacturer (Invitrogen). RNA was purified with RNAeasy Micro columns (Qiagen) and reverse transcribed using an iScript Kit (Bio-Rad). cDNA was quantified by qPCR using SYBR green. Each reaction was run in duplicate and analyzed following the standard ΔΔCt method using glyceraldehyde-3-phosphate dehydrogenase as a normalization control.
Western blotting.
Frozen NAc and dorsal striatum tissue was homogenized in 30 μl of homogenization buffer containing 320 mm sucrose, 5 mm HEPES buffer, 1% SDS, phosphatase inhibitor cocktails I and II (Sigma), and protease inhibitors (Roche) using an ultrasonic processor (Cole Parmer). Protein concentrations were determined using a DC protein assay (Bio-Rad), and 10–30 μg of protein was loaded onto 18% gradient Tris-HCl polyacrylamide gels for electrophoresis fractionation (Bio-Rad). Proteins were transferred to nitrocellulose membranes, blocked with Odyssey blocking buffer (Li-Cor), and incubated overnight at 4°C with primary antibodies in Odyssey blocking buffer. After thorough washing with TBS plus 0.1% Tween 20, membranes were incubated with IRDye secondary antibodies (1:5000 to 1:10,000; Li-Cor) dissolved in Odyssey blocking buffer for 1 h at room temperature. For analysis, the blots were imaged with the Odyssey Infrared Imaging system (Li-Cor) and quantified by densitometry using NIH ImageJ. The amount of protein blotted onto each lane was normalized to levels of actin or tubulin.
Viral-mediated gene transfer.
Expression plasmids for wild-type G9a and Cre were subcloned into herpes simplex virus (HSV) vectors and packaged into high-titer viral particles as described previously (Maze et al., 2010). Viral titers for these experiments are between 3 and 4 × 108 particles/ml. Mice were positioned in small animal stereotaxic instruments, under ketamine (100 mg/kg)/xylazine (10 mg/kg) anesthesia, and their cranial surfaces were exposed. Thirty-three gauge syringe needles were bilaterally lowered into the NAc to infuse 0.5 μl of virus at a 10° angle (anterior/posterior, +1.6 mm; medial/lateral, +1.5 mm; dorsal/ventral, −4.4 mm from bregma). Infusions occurred at a rate of 0.1 μl/min. Animals receiving HSV injections were allowed to recover for at least 24 h after surgery.
Terminal deoxynucleotidyl transferase-dUTP nick end labeling and immunohistochemistry.
Four percent paraformaldehyde-fixed mouse brains were incubated at room temperature overnight in 30% sucrose before being sectioned at 25 μm. Serial NAc sections were then mounted on charged glass slides and subjected to terminal deoxynucleotidyl transferase-dUTP nick end labeling (TUNEL) staining using a Click-iT TUNEL Alexa Fluor Imaging Assay (Invitrogen) according to the instructions of the manufacturer. Briefly, a positive (DNase I incubation) and negative control for apoptosis were prepared using sections obtained from surgery-naive animals. Next, to assess apoptosis specifically in cells infected with HSV–GFP or HSV–G9a–GFP, brain sections were blocked with 3% BSA, followed by incubation with an anti-GFP antibody (1:8000, chicken; Aves Labs) for 24 h at 4°C. After primary antibody incubation, sections were washed with 3% BSA before addition of the secondary antibody (1:400, Alexa Fluor 488, anti-chicken; Invitrogen) for 1 h at room temperature (protected from light). After secondary antibody incubation, sections were once again washed with 3% BSA, followed by Hoechst 33342 staining (1:5000) for 15 min at room temperature. Finally, sections were washed with PBS, submerged in deionized water to remove salts, and coverslipped with an aqueous mounting media (Prolong Gold antifade reagent; Invitrogen). Images were then obtained using standard fluorescent microscopy at 10× (100 μm) and 40× magnification.
Conditioned place preference.
Place conditioning was performed as described previously (Zachariou et al., 2003) with several modifications. Several visual and nonvisual (tactile) cues enabled the animals to distinguish two separate chambers of the apparatus. All conditioning and test sessions were performed under dim illumination. Baseline preference was monitored 1 d after surgery at 12:00 P.M., and the mice did not show a preference for either side of the chamber at baseline. For the next 3 d, mice were conditioned to the saline-paired side in the morning and the morphine-paired side (15 mg/kg, s.c.) in the afternoon. On the final day, mice were placed again in the central compartment at 12:00 P.M. and allowed to move freely between the two chambers for 20 min. Conditioned place preference (CPP) scores were calculated as time spent in the morphine-paired chamber minus time spent in the saline-paired chamber.
Locomotor sensitization.
After recovery from surgery, mice were habituated in the locomotor box the first day for 30 min and then monitored for locomotor activity after a saline injection using the Photobeam Activity System (San Diego Instruments). For the G9a overexpression experiment, morphine (10 mg/kg, s.c) was then administered daily, alternating between locomotor box and home cage for 7 d, and monitored for locomotor activities for 30 min. For the floxed G9a experiment, morphine (5 mg/kg) was administered daily for 5 d, and mice were monitored for locomotor activity each day for 30 min.
Morphine withdrawal.
Mice were injected intraperitoneally with escalating morphine doses (20, 40, 60, 80, 100, and 100 mg/kg) every 8 h for 2.5 d. Three hours after the last morphine injection, 1 mg/kg naloxone hydrochloride (Sigma) was administered subcutaneously. Withdrawal behaviors (jumps, wet dog shakes, tremors, ptosis, diarrhea, and weight loss) were then monitored for 30 min as described (Zachariou et al., 2003).
Morphine analgesia and tolerance.
The hotplate test was performed as described previously (Psifogeorgou et al., 2011). Mice were placed on a platform heated to 52°C with a cutoff of 40 s, and the latency to paw lick or jump was recorded. A control response was determined for each animal before treatment. Morphine sulfate (15 and 20 mg/kg, s.c., for the G9a overexpression and floxed experiments, respectively) was injected daily for 4 d, and analgesia was monitored 30 min after morphine injection. The antinociceptive response was calculated as a percentage of maximal possible effect (MPE), where MPE = (test − control latency)/(cutoff − control) × 100, and then normalized to the responses during day 1.
RNA sequencing and analysis.
Total RNA was isolated from five animals (10 total NAc punches) per replicate as described above with TRIzol according to the protocol of the manufacturer. Libraries were then prepared using the mRNA sequencing kit from Illumina. Briefly, poly(A)-containing mRNAs were purified from total RNA using poly(T) oligo-attached magnetic beads. The mRNA was then fragmented, reverse transcribed, and end repaired. Provided adapters were ligated after the addition of a single A′ base to the end, before they were purified and PCR enriched to create the libraries. Two replicates were sequenced on an Illumina GAII Sequencer with a 36 bp read length. The FASTQ files from the pipeline of Illumina were analyzed by the TopHat software for quality filtering and alignment to the mm9 reference genome (Trapnell et al., 2009). Twenty-two to 24 million short reads alignments were obtained for each replicate. Gene expression levels were then determined by calculating the FPKM (fragments per kilobase of exon model per million mapped fragments) using the software Cufflinks (Trapnell et al., 2010). The four expression groups were defined by their FPKM scores using the following criteria: (1) 0 indicates low expression; (2) >0 but ≤10 indicates medium-low expression; (3) >10 but ≤100 indicates medium-high expression; and (4) >100 indicates high expression.
ChIP, library preparation, and sequencing.
Freshly dissected NAc punches were crosslinked with formaldehyde and prepared for ChIP as described previously (Maze et al., 2011). For each ChIP, bilateral 14 gauge NAc punches were pooled from five mice for quantitative ChIP or 10 mice for ChIP-seq. Tissue was lightly fixed to crosslink DNA with associated proteins, and the material was further sheared and immunoprecipitated using sheep anti-mouse magnetic beads (Invitrogen) conjugated to an antibody that specifically recognizes H3K9me2 or G9a (Abcam). G9a immunoprecipitated DNA was subjected to qPCR analysis, whereas the resulting immunoprecipitated DNA for H3K9me2 and total (input) genomic DNA were prepared for ChIP-seq using an Illumina kit according to the instructions of the manufacturer. Each experimental condition was performed in triplicate. Briefly, each sample underwent end repair followed by addition of an A base to the 3′ end. Proprietary adapters were then ligated to the ends, followed by size selection on a 2% agarose gel. The range of excision was 175–225 bp. After DNA clean up, samples were amplified with 21 cycles of PCR. Amplification and size selection were confirmed with a BioAnalyzer. The resulting libraries were sequenced on an Illumina HiSeq 2000 with 50 bp read length.
ChIP-seq data analysis.
The 50 bp reads were first analyzed by the pipeline software of Illumina for quality filtering and aligned to the reference genome. Additional preprocessing steps were performed as described previously (Maze et al., 2011). On average, 72 million uniquely aligned, nonredundant short reads were obtained for each replicate. The MACS (model-based analysis of ChIP-Seq data) peak finding algorithm was used to identify H3K9me2 peaks in ChIP libraries generated from both saline- and morphine-treated animals (Zhang et al., 2008). Repetitive element alignment and analysis were accomplished as described previously (Maze et al., 2011).
Differential analysis between morphine and saline groups was performed with an in-house program called diffReps. For more details, please refer to http://code.google.com/p/diffreps/. Briefly, diffReps uses a sliding window strategy to identify all genomic regions that display differential levels of histone mark binding between two conditions. The negative binomial distribution is used to model the within-group variation and between-group differences for discrete counting data. Before testing, diffReps obtains global statistics about the ChIP-seq samples under study and filters out the regions with read counts below the mean plus 2 SDs. It then scores all of the windows with fixed sizes and picks up those with a p value <1e-4 as candidate differential windows. Any two overlapping or adjacent candidate windows are merged to make a differential site. Finally, multiple testing corrections are applied to all differential sites on the genome, and an false discovery rate <10% is used as a cutoff. In this study, diffReps was used with a window size of 200 bp and a moving size of 20 bp to obtain a high-resolution profiling of the chromatin regulation by morphine.
Statistical analysis.
Student's t tests were used whenever two groups were being compared, whereas one-way ANOVAs were performed to determine significance for qPCR and Western blotting data comparing saline versus acute versus repeated morphine treatments. Significant main effects (p < 0.05) were further analyzed using Bonferroni's post hoc tests (* in figures) to compare acute and repeated groups with their respective saline controls. For the CPP experiment, Student's t test was used. For locomotor sensitization and analgesic tolerance experiments, two-way ANOVAs followed by Bonferroni's post hoc tests were performed. For withdrawal experiments, Student's t test was used with Bonferroni's correction for multiple comparisons.
Results
Regulation of G9a and H3K9me2 in NAc by chronic morphine administration
Expression changes of all KMTs and lysine demethylases (KDMs) for H3 lysine 9 and lysine 27 methylations, two well-characterized repressive modifications, were examined after acute (6 d, saline; 1 d, 20 mg/kg, i.p.) and repeated (7 d, 20 mg/kg, i.p., once daily) morphine treatments (Fig. 1a). As observed with cocaine, repeated but not acute morphine treatment resulted in an ∼25% reduction in G9a levels in NAc (Fig. 1a). None of the other KMTs and KDMs was significantly altered in this brain region by acute or repeated morphine administration. Time course studies revealed that G9a and its associated mark, H3K9me2, were only downregulated after 5 or 7 d of repeated daily intraperitoneal morphine injections (20 mg/kg) but not after 1 or 3 d (Fig. 1b,c). Furthermore, this effect was dose dependent because seven daily injections of a lower dose of morphine (10 mg/kg) resulted in smaller nonsignificant decreases in G9a and H3K9me2 levels (Fig. 1b,c). The decreases observed in G9a and H3K9me2 were likely not attributable to the number of morphine injections per se, because an escalating morphine injection paradigm (six injections within 2.5 d) did not alter G9a or H3K9me2 levels (Fig. 1d). Corresponding with no expression changes in other KMTs and KDMs, levels of H3K9me1, H3K9me3, and H3K27me3 were not altered by acute or repeated morphine administration (Fig. 1e). The lack of regulation of H3K9me3 by morphine is different from the downregulation that has been observed for cocaine (Maze et al., 2011), but this chromatin mark is catalyzed by other KMTs (e.g., SUV39H1), which were not altered by morphine administration. These data suggest that morphine acts to alter chromatin structure through specific modifications of histone methylation, even at the level of closely related methyl valence states at a particular amino acid residue on H3. Morphine-induced downregulation of H3K9me2 was also brain region specific, because neither acute nor chronic morphine administration altered levels of this mark in dorsal striatum (Fig. 1e).
Figure 1.
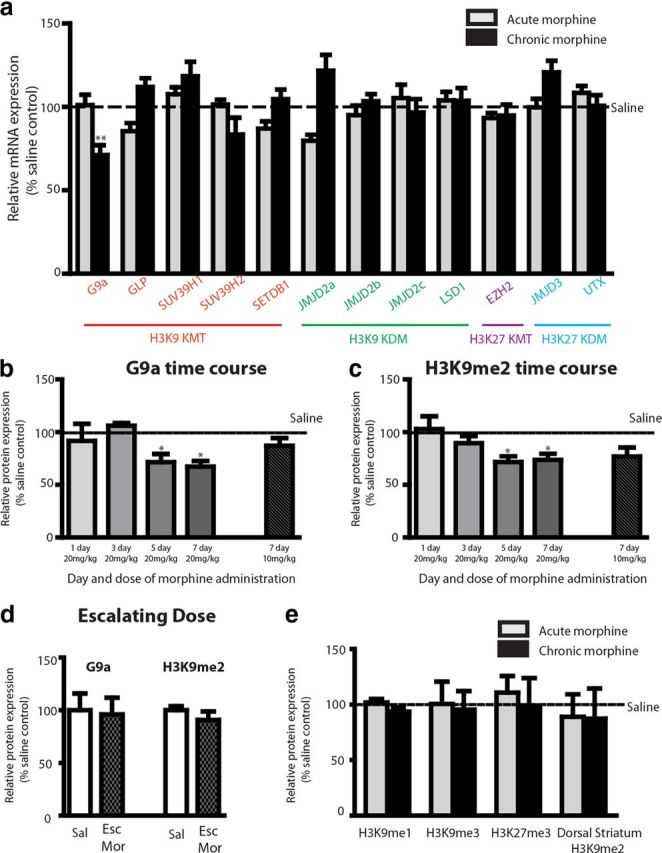
Regulation of repressive H3K9 and H3K27 methylation by morphine administration in the NAc. a, C57BL/6J mice were injected intraperitoneally once daily with either saline or 20 mg/kg morphine for 7 d. Acute morphine-treated animals received six injections of saline, followed by a single intraperitoneal injection of 20 mg/kg morphine. Animals were analyzed 24 h after the last injection, and NAc tissue was collected for qPCR analysis for KMTs and KDMs for H3 lysine 9 and lysine 27. b, c, Animals were injected intraperitoneally once daily (20 mg/kg) for 1, 3, 5, or 7 d or for 7 d with 10 mg/kg morphine. Animals were killed 24 h after the last injection, and NAc tissue was collected for Western blot analysis for G9a and H3K9me2. d, Animals were injected with an escalating morphine (Esc Mor) paradigm (20, 40, 60, 80, 100, 100 mg/kg morphine every 8 h), and killed 24 h after the last injection. NAc tissue was collected for Western blot analysis for G9a and H3K9me2. e, NAc tissue was collected 24 h after 7 d of 20 mg/kg intraperitoneal injections, and Western blot analysis was performed to examine levels of H3K9me1, H3K9me3, and H3K27me3. Dorsal striatum tissue was also collected to examine levels of H3K9me2. Data were expressed as mean ± SEM. Student's t test or one-way ANOVA was performed, and, assuming significance in the main effect, post hoc test were also performed. *p < 0.05, **p < 0.01 compared with saline-treated animals.
Regulation of behavioral responses to morphine by G9a in NAc
We next sought to investigate whether manipulations of G9a levels in the NAc in vivo would alter behavioral responses to morphine. To do so, we began by examining the effect of overexpressing G9a on morphine-elicited behaviors by giving mice bilateral intra-NAc injections of HSV vectors expressing either GFP as a control or G9a plus GFP. Both HSV–GFP and HSV–G9a infusion resulted in minimal cell injury, which was indistinguishable from each other (Fig. 2a) and from vehicle-injected animals (data not shown). Furthermore, compared with HSV–GFP infusion, HSV–G9a selectively increased H3K9me2 levels in NAc, without affecting methylation or acetylation levels at any other valence state on the same residue or at any other residue examined on H3 and H4 (Fig. 2b).
Figure 2.
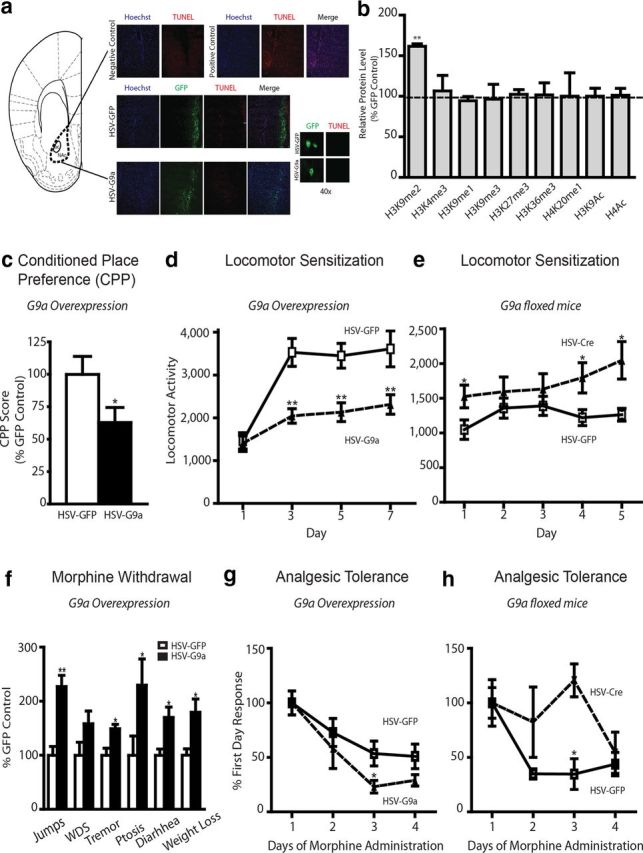
G9a regulates morphine-induced behaviors. a, TUNEL/immunohistochemical analysis of NAc sections after HSV–GFP or HSV–G9a surgery, along with negative and positive controls. b, Western blot analysis for various chromatin modifications after HSV–G9a infusion into the NAc. c, CPP. Animals were trained to pair morphine or saline with two contextually distinct chambers for 3 d after viral-mediated gene transfer with HSV–G9a or HSV–GFP, and CPP scores were calculated as the difference in time spent between the morphine- and saline-paired chambers. Two-tailed Student's t test, *p < 0.05. d, Locomotor sensitization after G9a overexpression. Animals were habituated in a locomotor chamber after saline injections. Morphine (10 mg/kg, s.c.) was then administered daily, alternating between the locomotor box and their home cage for 7 d. Animals (HSV–GFP, white squares; HSV–G9a, black triangles) were monitored for locomotor activity for 30 min on days 1, 3, 5, and 7. A two-way ANOVA was used, followed by Bonferroni's post hoc analysis. Day: F(3,60) = 13.59, p < 0.001; virus: F(1,60) = 28.98, p < 0.001; day × virus: F(3,60) = 2.893, p < 0.05. **p < 0.01, post hoc test. e, Locomotor sensitization after Cre-mediated G9a knockdown in the NAc of G9afl/fl mice. Mice (HSV–GFP, white squares; HSV–Cre, black triangles) were monitored for locomotor activity for 30 min for 5 consecutive days of morphine treatment (5 mg/kg). Two-way ANOVA; day: F(4,76) = 1.004, NS; virus: F(1,76) = 15.54, p < 0.001; day × virus:F(4,76) = 0.7863, NS. *p < 0.05, post hoc test. f, Physical withdrawal. Mice were injected intraperitoneally with escalating doses of morphine (20, 40, 60, 80, 100, and 100 mg/kg) every 8 h for 2.5 d. Two hours after the last morphine injection, naloxone (1 mg/kg) was administered subcutaneously. Withdrawal behaviors (jumps, wet dog shakes, tremors, ptosis, diarrhea, and weight loss) were then monitored for 30 min. Bonferroni's post hoc test, *p < 0.05, **p < 0.01. g, h, Analgesic tolerance. A hotplate test was used, in which the latency for paw lick was recorded. The antinociceptive response was calculated as a percentage of MPE, where MPE = (test − control latency)/(cutoff − control) × 100, and responses were normalized to the first day. Repeated morphine injections (15 and 20 mg/kg, s.c., for g and h, respectively) were given daily for 4 d, and analgesia was measured 30 min after each drug dose. For g, HSV–GFP, white squares; HSV–G9a, black triangles. A two-way ANOVA followed by Bonferroni's post hoc analysis were performed. day: F(3,57) = 12.32, p < 0.001; virus: F(1,57) = 4.17. p < 0.05 *p < 0.05, post hoc test. For h, HSV–GFP, white squares; HSV–Cre, black triangles. A two-way ANOVA followed by Bonferroni's post hoc analysis were performed. virus: F(1,36) = 7.20, p < 0.05. *p < 0.05, post hoc test.
We first investigated the effect of G9a overexpression in NAc on morphine reward through the use of an unbiased morphine CPP paradigm. Animals overexpressing G9a in NAc displayed significantly reduced preferences for morphine compared with those expressing GFP (Fig. 2c). These data indicate the morphine-induced downregulation of G9a and H3K9me2 in NAc serves to increase the rewarding responses to the drug, similar to that observed after repeated cocaine administration (Maze et al., 2010). We next examined the impact of G9a overexpression in NAc on locomotor activity and sensitization elicited by morphine. Although overexpression of G9a did not affect basal (data not shown) locomotor activity or locomotor responses to an initial acute injection of morphine (Fig. 2d, day 1), G9a overexpression did significantly impair locomotor sensitization to repeated morphine administration (Fig. 2d; two-way ANOVA, day, F(3,60) = 13.59, p < 0.001; virus, F(1,60) = 28.98, p < 0.001; day × virus, F(3,60) = 2.893, p < 0.05), with significantly lower locomotor activities observed on days 3, 5, and 7 of morphine treatments (Bonferroni's post hoc test, p < 0.01). To mimic the effect of chronic morphine-induced repression of G9a in NAc, adult G9afl/fl mice were infused with HSV vectors expressing either Cre recombinase plus GFP or GFP alone as a control. Cre expression induces a highly localized knockdown of G9a within the injected region (Maze et al., 2010). Although Cre-mediated G9a knockdown in the NAc did not affect basal locomotor response (data not shown), it enhanced both locomotor responses to an acute dose of morphine (Fig. 2e, Day 1), as well as locomotor sensitization after repeated morphine (Fig. 2e; two-way ANOVA, virus, F(1,76) = 15.54, p < 0.001), with significantly higher locomotor activities observed on days 4 and 5 of morphine treatments (Bonferroni's post hoc test, p < 0.05).
We next turned our attention to morphine physical dependence and withdrawal, another important consequence of chronic opiate exposure. To study the effects of G9a overexpression in NAc on this endpoint, mice were injected with escalating morphine doses every 8 h for 2.5 d (see Materials and Methods) after bilateral intra-NAc injections of either HSV–G9a or HSV–GFP. Three hours after the last morphine injection, naloxone (1 mg/kg), an opioid receptor antagonist, was administered subcutaneously, and a battery of physical signs and symptoms of morphine withdrawal were monitored. Animals receiving HSV–G9a displayed exacerbated physical withdrawal symptoms compared with animals receiving HSV–GFP (Fig. 2f). Thus, G9a overexpression in this brain region had opposite effects on this behavioral adaptation to repeated morphine compared with reward and sensitization, such that the downregulation of G9a after chronic morphine treatment may help to offset the development of physical dependence and withdrawal symptoms.
One of the most important phenomena that limit the clinical use of opiates in the treatment of chronic pain is the development of tolerance to the analgesic effects of these drugs. We therefore examined the effects of G9a overexpression in NAc on the analgesic effects of repeated morphine in the hotplate test. Although no differences were observed for baseline latencies in this test (GFP, 12.2 ± 0.7 s; G9a, 11.0 ± 0.9 s) or for analgesic responses to acute morphine administration (day 1% MPE: GFP, 82.5 ± 9.1%; G9a, 75.0 ± 8.2%), between G9a-overexpressing mice and GFP controls, animals overexpressing G9a in NAc developed tolerance to morphine more rapidly compared with GFP controls (Fig. 2g; two-way ANOVA, day, F(3,57) = 12.32, p < 0.001; virus, F(1,57) = 4.17, p < 0.05). Conversely, Cre-mediated knockdown of G9a in the NAc delayed the development of tolerance compared with control animals receiving HSV–GFP vectors (Fig. 2h; two-way ANOVA, virus, F(1,36) = 7.20, p < 0.05). Together, these experiments show that regulation of G9a and H3K9me2 in NAc exert potent but contrasting effects on various behavioral adaptations to repeated morphine exposure. In the present study, we did not explore possible regulation of G9a/H3K9me2 in NAc during the course of the various behavioral assays used. This would be an interesting focus of future investigations.
Genome-wide regulation of H3K9me2 binding in NAc by chronic morphine administration
To identify the downstream molecular target genes regulated by G9a/H3K9me2 after chronic morphine administration that underlie the role of this repressive histone methylation in regulating morphine-induced behaviors, we turned to ChIP-seq to map the distribution of H3K9me2 binding in NAc of saline- and chronic morphine-treated mice. The overall distribution of H3K9me2 peaks was similar between control and morphine-treated animals (Fig. 3a), with ∼80% of the peaks residing in intergenic regions (gene deserts, pericentromeres, repetitive sequences, etc). The remaining 20% of identified peaks were observed to reside throughout genic regions of the genome, predominantly distributed (>90%) throughout gene bodies, with little enrichment of H3K9me2 associated with gene promoters. This “flat” pattern of H3K9me2 binding across long stretches of genes (Fig. 3b), with slight depletions at transcription start sites (TSSs) and end sites, has also been observed in other tissues (Barski et al., 2007; Wen et al., 2009; Lienert et al., 2011), and contrasts with several other histone methylation marks (e.g., methylation of Lys 4 or Lys 27 on histone H3), which show sharp peaks around gene promoters and other loci.
Figure 3.
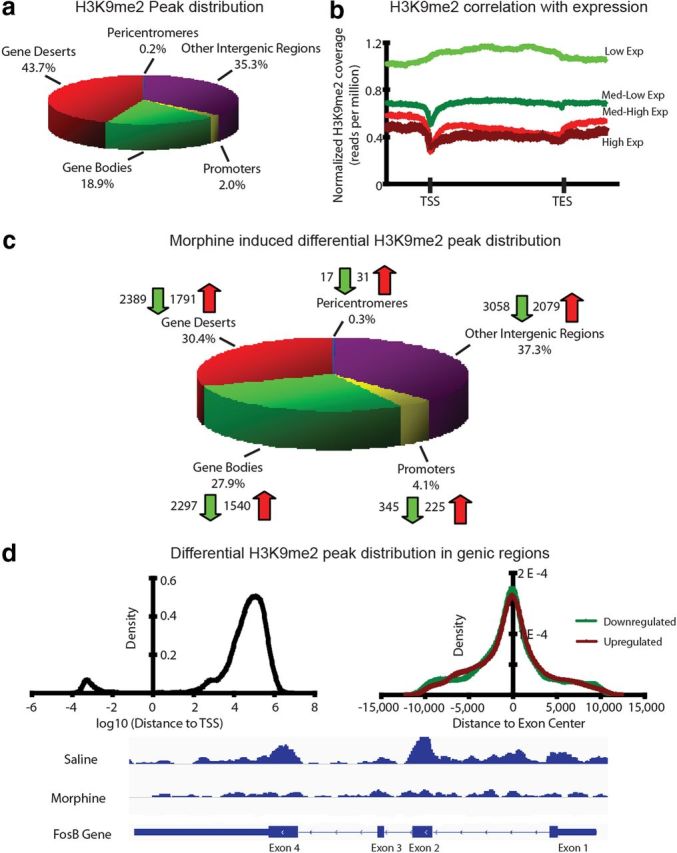
Chronic morphine regulates the genome-wide distribution of H3K9me2 in NAc. a, The global distribution of H3K9me2 peaks was not found to be different between saline- and morphine-treated animals. Combined, ∼80 and 20% of the H3K9me2 peaks were shown to be distributed throughout intergenic and genic regions of the genome, respectively. b, H3K9me2 is inversely correlated with basal gene expression in NAc. c, Morphine treatment results in differential enrichment of H3K9me2 at localized regions throughout the genome, as identified by diffReps analysis (see Materials and Methods). There is a modest enrichment of morphine-regulated peaks in genic regions compared with the overall global distribution, as well as significantly more morphine downregulated H3K9me2 peaks compared with upregulated peaks. d, Distribution within genic regions shows that the morphine-regulated peaks occur distally from TSSs (preferentially in downstream gene bodies) and are centered within exons. An example of this is illustrated for the FosB gene using IGV (Integrative Genomics Viewer) genome browser software, demonstrating that downregulation of H3K9me2 across the gene occurs mostly within exons.
To confirm commonly held assumptions primarily determined from non-neuronal tissues concerning the repressive role of H3K9me2 on basal gene expression, we used RNA sequencing (RNA-seq) data derived from NAc of control animals to divide all detectably expressed genes in NAc into four groups based on endogenous mRNA expression levels: low, medium-low, medium-high, and high. Consistent with a previous report on cultured cells (Barski et al., 2007), gene expression was negatively correlated with H3K9me2 enrichment, with higher H3K9me2 binding associating with more weakly expressed genes and vice versa (Fig. 3b). Using diffReps analysis (for more details, see Materials and Methods) to examine morphine-induced differential regulation of H3K9me2 enrichment throughout the genome, we identified 13,772 differential H3K9me2 sites in NAc between saline- and morphine-treated animals, with 8103 sites downregulated by morphine and 5669 upregulated, thus reflecting our observation of globally downregulated H3K9me2 levels after chronic morphine administration. Two-thirds of these differential sites mapped to intergenic regions of the mouse genome, with one-third of the differential sites mapping to genic loci, displaying a modest enrichment of morphine-induced H3K9me2 changes within genic regions compared with baseline distribution of this mark (Fig. 3c; 28% of differential regions vs 19% total peaks for gene bodies; 4% of differential regions vs 2% total peaks for gene promoters). Within the 5464 morphine-downregulated and 3901 morphine-upregulated H3K9me2 sites found in intergenic regions, there were numerous repetitive elements that displayed significant morphine regulation, with the vast majority (22 of 24) showing decreased H3K9me2 enrichment and de-repression (Table 1). Within genic regions, there were 2642 morphine-downregulated H3K9me2 sites covering ∼590 kb of DNA and 1765 upregulated sites covering ∼396 kb of DNA. Most of these differential sites are centered within exons at some distance from the TSSs, with a preference for downstream gene body associated loci rather than upstream regulatory regions (Fig. 3d, top two panels).
Table 1.
Epigenetic regulation of repetitive elements by chronic morphine administration
| Name | Type | Origin | % H3K9me2 binding relative to saline |
|---|---|---|---|
| HERVFH21I | Endogenous retrovirus | Eutheria | ↓ (29) |
| LTR10A | ERV1 | Eutheria | ↑ (309) |
| MMERVK9C_I | ERV2 | Mus musculus | ↓ (79) |
| ETnERV2 | ERV2 | Mus | ↓ (73) |
| MYSERV6 | ERV2 | Muridae | ↓ (69) |
| RLTR13A | ERV2 | Mus musculus | ↓ (77) |
| ERVB4_1B-I_MM | ERV2 | Mus musculus | ↓ (74) |
| RLTR13A3 | ERV2 | Mus musculus | ↓ (69) |
| ETnERV3 | ERV2 | Mus | ↓ (66) |
| RLTR13A2 | ERV2 | Mus musculus | ↓ (72) |
| MMERVK9E_I | ERV2 | Mus musculus | ↓ (85) |
| RLTR12BD_Mm | ERV2 | Mus | ↓ (61) |
| ORR1A4_LTR | ERV3 | Muridae | ↑ (117) |
| L1_Mur2_orf2 | L1 | Muridae | ↓ (78) |
| MusHAL1 | L1 | Mus | ↓ (73) |
| L1B_MM | L1 | Mus musculus | ↓ (83) |
| L1_Rod_5end | L1 | Muridae | ↓ (26) |
| L1_Mur1_orf2 | L1 | Muridae | ↓ (82) |
| CYRA11_MM | Repeat region | Mus musculus | ↓ (83) |
| (TAAA)n | Simple Repeat | Eukaryota | ↓ (21) |
| (GAAA)n | Simple Repeat | Eukaryota | ↓ (40) |
| MIR | SINE | Mammalia | ↓ (34) |
| MUSID3 | SINE2/tRNA | Rodentia | ↓ (34) |
| tRNA–His–CAY | tRNA | Vertebrata | ↓ (26) |
Repetitive element alignment and analysis were accomplished as described previously (Maze et al., 2011). Briefly, we performed short reads alignment on the canonical repeat sequences and assessed the global quantitative change of H3K9me2 enrichment after chronic morphine administration. The majority of repetitive elements (22 of 24) showed decreased H3K9me2 enrichment after chronic morphine administration. Arrows represent increase or decrease in H3K9me2 binding relative to saline-treated animals, whereas numbers in parentheses represent percentage of H3K9me2 binding relative to saline controls.
We showed previously that ΔFosB, a product of the fosB gene, is important in the development of morphine-related behaviors (Zachariou et al., 2006). Given our previous finding that cocaine-induced alterations in G9a and H3K9me2 in NAc help control fosB gene transcription and ΔFosB accumulation in this brain region (Maze et al., 2010), we looked more closely at the distribution of H3K9me2 before and after morphine treatment at the fosB locus. Interestingly, a general reduction in H3K9me2 enrichment was observed throughout the fosB gene after chronic morphine treatment (Fig. 3d, bottom). Furthermore, in agreement with the global patterns noted above, H3K9me2 reduction by morphine treatment at the fosB gene is primarily centered within its exons. Interestingly, one of the most dramatic downregulation events occurs at the splicing site for fosB transcripts (exon 4), which produces ΔFosB. These analyses offer valuable validation for our genome-wide sequencing data and provide a novel chromatin mechanism by which chronic exposure to morphine releases baseline repression of the fosB gene in the NAc, allowing for the sustained induction of ΔFosB, a key mediator of behavioral adaptations to the drug.
To further investigate the mechanism by which chronic morphine promotes altered patterns of gene expression through regulation of H3K9me2, we next applied ingenuity pathway analysis to the list of genes that displayed differential H3K9me2 peak enrichment as a result of chronic morphine treatment. Glutamatergic signaling presented as a top “hit” pathway showing robust regulation. Three genes (grin2a, grm5, grm8) showed exclusively reduced H3K9me2 binding across the gene after morphine treatment, whereas one gene (gria1) showed exclusively increased H3K9me2 binding (Fig. 4a). All other genes in the list displayed complex H3K9me2 regulation after repeated morphine treatment, with some differential sites along the gene showing upregulation and others showing downregulation. Therefore, to validate that alterations in H3K9me2 enrichment after morphine treatment correlate with transcriptional changes, we studied expression changes of numerous genes in the pathway showing differential H3K9me2 enrichment in our ChIP-seq dataset. Many of the glutamatergic receptor genes (Fig. 4b; e.g., grid2, which encodes a δ subunit of the NMDA receptor) that displayed complex H3K9me2 regulation after repeated morphine treatment were not changed in terms of their mRNA expression levels, with the one exception being grip1, a glutamate receptor interacting protein, which was induced by morphine in the NAc (Fig. 4c). There were five differential H3K9me2 sites along the grip1 gene after morphine treatment, as identified by our ChIP-seq analysis, three of which showed downregulation and two upregulation (two representative downregulated sites and one upregulated site shown in Fig. 4c). Gria1, which encodes the GluA1 (GluR1) subunit of AMPA receptors, was the only gene showing H3K9me2 upregulation after morphine treatment, and it too did not display altered mRNA expression after chronic morphine (Fig. 4b). In contrast, the three genes—grin2a (encoding the NR2a NMDA subunit), grm5 (encoding the metabotropic mGluR5 receptor), and grm8 (encoding the mGluR8 receptor)—that exhibit only downregulation of H3K9me2 at single or multiple sites across their coding regions after morphine treatment displayed corresponding increases in mRNA expression (Fig. 4d–f). Most of these H3K9me2 changes after morphine were correlated with equivalent changes in G9a binding. In addition, many of the differential sites in these glutamatergic genes occurred at introns, rather than coding regions, although some of these changes were in close proximity to neighboring exons. These results thus provide a novel chromatin-based mechanism through which chronic morphine promotes locus-specific redistribution of H3K9me2 throughout the genome, including at genes for specific glutamate receptor subunits, the altered expression of which appears to be a key mechanism of morphine-induced neural and behavioral plasticity (see Discussion).
Figure 4.
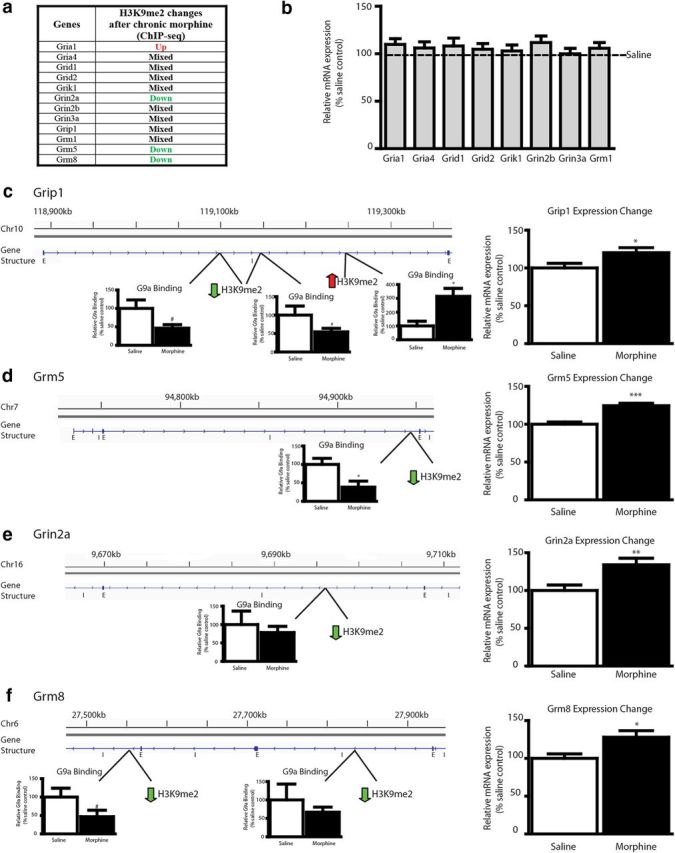
Chronic morphine regulates H3K9me2 and G9a binding at, and expression of, glutamate receptor genes. a, Summary of glutamate signaling genes that were regulated in H3K9me2 binding after chronic morphine treatment, as analyzed by ChIP-seq results. b–f, All glutamatergic-associated genes displaying differential H3K9me2 enrichment after chronic morphine were studied for expression changes after morphine treatment by qPCR in independent tissue samples. b, Several genes in a, which did not show concerted changes in H3K9me2, did not show altered mRNA expression levels. The exception was grip1 (c), which showed an increase in expression after morphine treatment. In contrast, the three genes in a that did display concerted changes in H3K9me2 also show commensurate changes in mRNA expression levels (d–f). Most of these latter genes also exhibited equivalent changes in G9a binding. Representative areas along the gene in which H3K9me2 and corresponding G9a binding changes after morphine treatment are shown in c–f. E, Exons; I, introns. Student's t tests were performed for qPCR validation and G9a ChIP between saline- and morphine-treated animals. #0.05 < p < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001.
The ChIP-seq data show that chronic morphine downregulates H3K9me2 in NAc across several different classes of repetitive elements, including LINE-1 (long interspersed element 1), in this brain region (see Table 1). This is consistent with our recent observation that chronic cocaine decreases enrichment of H3K9me3 at LINE-1 and many other repetitive elements, a modification associated with their induction (Maze et al., 2011). Several recent studies have demonstrated the mobilization, integration, and regulation of LINE-1 retrotransposition in the nervous system, although the exact functional consequences of such events remain essentially unclear (Muotri et al., 2009; Singer et al., 2010). It will be important in the future to elucidate how dysregulation of these elements might contribute to the development of addiction-related behaviors.
Discussion
This study provides novel insight into the regulation of G9a/H3K9me2 in NAc by chronic morphine and point to an integral role for G9a as part of a dynamic repressive machinery in neurons that acts to maintain normal patterns of transcription and prevent aberrant gene expression in response to negative environmental stimuli. The pathological downregulation of this repressive machinery after chronic exposure to morphine, or other emotional stimuli, leads to aberrant transcriptional control that contributes to abnormal behavioral adaptations.
Our data establish a direct contribution of G9a to regulating multiple domains of morphine-related behaviors. Similar to observations with cocaine (Maze et al., 2010), overexpression of G9a in NAc reduced morphine CPP, an indirect measure of morphine reward. Likewise, G9a overexpression and knockdown, respectively, in this brain region reduced and enhanced the development of locomotor sensitization, a phenomenon that is thought to reflect neuroadaptations that underlie addiction (Vanderschuren and Kalivas, 2000). These findings are consistent with our hypothesis that G9a in NAc functions to oppose behavioral maladaptations to drugs of abuse by preventing aberrant gene transcription in response to acute drug exposure. Downregulation of G9a in NAc by repeated drug administration releases this chromatin break and enables pathological behavioral adaptations to occur.
In contrast, overexpression of G9a in the NAc exerted the opposite effect on analgesic tolerance to morphine and on physical morphine dependence and withdrawal. These observations provide additional evidence for the ability of certain molecular adaptations within this “reward” region to also potently influence aspects of chronic morphine action that are primarily mediated via distinct neural and peripheral circuits (Zachariou et al., 2006; Han et al., 2010). The circuit level mechanisms by which the NAc influences analgesic tolerance and physical dependence/withdrawal are poorly understood. Direct infusion of an opioid antagonist into the NAc precipitates modest withdrawal behaviors (Koob et al., 1992). This effect could be mediated by output projections from NAc to regions important in mediating withdrawal behaviors, such as the ventral pallidum (Calvino et al., 1979) and periaqueductal gray (Hu and Jin, 2000), although additional work is needed to study such mechanisms directly. In any event, combined with results from a recent study showing that inhibition of histone deacetylases attenuated morphine withdrawal symptoms (Rehni et al., 2012), our results suggest that chromatin-mediated repression mechanisms may promote opiate dependence and withdrawal. The downregulation of G9a after repeated morphine may help to offset the development of physical dependence and to delay the development of analgesic tolerance, which coupled with increased sensitivity to the rewarding and sensitizing effect of the drug, may enhance the susceptibility of the animal to additional drug exposure.
To investigate downstream mechanisms by which G9a in NAc mediates these morphine-related behavioral changes, we obtained the first genome-wide characterization of H3K9me2 binding in brain after repeated saline and morphine treatments. Similar to previously reported genome-wide characterizations of H3K9me2 binding in mammalian cell lines (Barski et al., 2007; Rosenfeld et al., 2009; Wen et al., 2009; Lienert et al., 2011), we found this mark to be enriched at heterochromatic regions and silent genes. However, appreciable levels of H3K9me2 binding occurred within euchromatic regions and were inversely correlated with basal levels of gene expression. This finding is consistent with the repressive role of H3K9me2 in other systems (Shinkai and Tachibana, 2011). A previous study (Wen et al., 2009), which generated some controversy (Filion and van Steensel, 2010), reported widespread global changes in H3K9me2 in the course of cellular differentiation. However, in accordance with recent reports in the development field (Lienert et al., 2011), we find that H3K9me2 distribution does not change on a global level after repeated morphine but instead occurs in localized regions across the genome. Our observation that ∼40% more H3K9me2 peaks displayed downregulation than upregulation after morphine is in agreement with our biochemical data showing global downregulation in NAc. Together, these findings suggest that de-repression, rather than repression, of gene regulation plays a greater role in neural and behavioral adaptations to chronic morphine. Importantly, insights into the regulation of H3K9me2 throughout genic regions have not been reported previously, and here we demonstrate that morphine-induced changes in H3K9me2 binding indeed occur throughout genic loci, mostly downstream of TSSs within gene bodies, rather than in regulatory promoter regions. This finding suggests a significant influence of chromatin regulation within gene bodies in mediating genomic responses to environmental stimuli and emphasizes the importance for future quantitative ChIP studies to expand investigations to these non-promoter regions of genes.
ΔFosB is induced in NAc in response to repeated morphine (Nye and Nestler, 1996; Kaplan et al., 2011), in which it enhances morphine CPP, leads to more rapid development of analgesic tolerance, and exacerbates physical dependence and withdrawal (Zachariou et al., 2006). Downregulation of G9a and H3K9me2 in NAc has been implicated in the induction of ΔFosB, and subsequent enhancement of reward, after repeated exposure to cocaine (Maze et al., 2010). This previous study used quantitative ChIP and examined fosB promoter regions only. Here we demonstrate decreased H3K9me2 binding throughout the fosB gene body after repeated morphine treatment, with the most dramatic H3K9me2 changes centered at the middle of exons. The robust morphine-induced decrease in H3K9me2 binding at exon 4, which contains the alternative splice site that generates ΔFosB, suggests that alterations in H3K9me2 enrichment at this locus may play a role in the alternative splicing of the fosB gene. Indeed, increasing evidence has implicated H3K9me2 and other chromatin modifications as key determinants of alternative splicing of genes in cultured cells (All ó et al., 2010). In any event, the morphine-induced depletion of H3K9me2 binding at fosB in NAc is associated with ΔFosB induction, which likely contributes to enhancement of morphine reward and locomotor sensitization (Zachariou et al., 2006). The analgesic tolerance and withdrawal behaviors associated with G9a overexpression are opposite to what would be expected assuming that G9a alters morphine-induced behaviors primarily through regulation of ΔFosB; therefore, it is likely that altered G9a activity influences the expression of many other gene targets that differentially affect morphine-associated behaviors.
Numerous studies have investigated the influence of glutamatergic signaling on morphine-related behaviors (Vanderschuren and Kalivas, 2000; Siggins et al., 2003; Noda and Nabeshima, 2004). It has been reported previously that grin2a (NR2a) expression is increased in NAc by repeated morphine (Inoue et al., 2003; Murray et al., 2007), as we demonstrate here, and this adaptation is consistent with electrophysiological observations in this brain region under morphine-treated conditions (Martin et al., 1999, 2004). Because of the absence of NR2a-subtype-selective agonists and antagonists, the functional consequences of altered expression of this subunit are poorly understood. However, it is known that NMDA receptor activation in NAc is important for the establishment of morphine CPP (Wu et al., 2012), locomotor sensitization (Pulvirenti et al., 1991; Bespalov and Zvartau, 1996), analgesic tolerance (Inoue et al., 2003), and physical withdrawal (Ji et al., 2004). Induction of grin2a after repeated morphine through G9a and H3K9me2 is thus consistent with our observations with morphine CPP and locomotor sensitization and supports this chromatin regulatory event as an important molecular mechanism of chronic morphine action. Induction of grip1 and the two identified metabotropic glutamate receptors (mGluR5 and mGluR8) has not been reported previously after repeated morphine. GRIP1 interacts with AMPA receptors and regulates the internalization and recycling of these receptors downstream of mGluR signaling (Casimiro et al., 2011). mGluR5 and mGluR8 are presynaptic group I and III metabotropic glutamate receptors and couple to different G-proteins to increase and decrease NMDA receptor function, respectively (Ambrosini et al., 1995; Lea et al., 2002). Investigations of group I metabotropic glutamate receptor antagonists on morphine-related behaviors agree with its synergistic effect on glutamatergic signaling in partnership with NMDA receptors (Popik and Wróbel, 2002; Kozela et al., 2003; Kotlinska and Bochenski, 2007). Therefore, the induction of grin2a, grip1, and grm5 through de-repression of G9a/H3K9me2 binding at these genes could partially underlie the mechanism by which G9a downregulation leads to the enhanced rewarding and sensitizing effect of morphine, whereas similar induction of grm8 could partially explain how G9a repression could result in reduced physical dependence and delayed development of morphine analgesic tolerance. New tools are now needed to further our understanding of the role played by these genes in opiate addiction. These identified glutamatergic genes demonstrate the utility of ChIP-seq technology, and the database reported here provides the foundation for identifying many additional targets to help understand the molecular basis of drug addiction.
Together, our results demonstrate a unified role for G9a in maintaining gene repression in NAc under basal conditions, whereby chronic exposure to drugs of abuse or severe social stress weakens the ability of G9a to appropriately regulate aberrant patterns of gene expression that may underlie the progression of pathological behaviors that characterize drug addiction or stress-related disorders. Through the first comprehensive characterization of H3K9me2 binding in brain, we also provide novel insight into the location of this chromatin mark and its regulation by morphine, thereby providing a genome-wide template of novel mechanisms through which alterations in repressive chromatin structure mediate maladaptations to drugs of abuse and stress.
Notes
Supplemental material for this article is available at http://neuroscience.mssm.edu/nestler/relatedlinks_morphine.html. The material contains two gene lists from our RNA-seq and ChIP-seq datasets, respectively. Detailed descriptions are as provided in the material. This material has not been peer reviewed.
Footnotes
The authors declare no competing financial interests.
References
- Alló M, Schor IE, Muñoz MJ, de la Mata M, Agirre E, Valcárcel J, Eyras E, Kornblihtt AR. Chromatin and alternative splicing. Cold Spring Harb Symp Quant Biol. 2010;75:103–111. doi: 10.1101/sqb.2010.75.023. [DOI] [PubMed] [Google Scholar]
- Ambrosini A, Bresciani L, Fracchia S, Brunello N, Racagni G. Metabotropic glutamate receptors negatively coupled to adenylate cyclase inhibit N-methyl-d-aspartate receptor activity and prevent neurotoxicity in mesencephalic neurons in vitro. Mol Pharmacol. 1995;47:1057–1064. [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bespalov AY, Zvartau EE. Intraaccumbens administration of NMDA receptor antagonist (+/−)-CPP prevents locomotor activation conditioned by morphine and amphetamine in rats. Pharmacol Biochem Behav. 1996;55:203–207. doi: 10.1016/s0091-3057(96)00065-2. [DOI] [PubMed] [Google Scholar]
- Calvino B, Lagowska J, Ben-Ari Y. Morphine withdrawal syndrome: differential participation of structures located within the amygdaloid complex and striatum of the rat. Brain Res. 1979;177:19–34. doi: 10.1016/0006-8993(79)90915-6. [DOI] [PubMed] [Google Scholar]
- Casimiro TM, Sossa KG, Uzunova G, Beattie JB, Marsden KC, Carroll RC. mGluR and NMDAR activation internalize distinct populations of AMPARs. Mol Cell Neurosci. 2011;48:161–170. doi: 10.1016/j.mcn.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY, Dietz DM, Lobo MK, Ghose S, Mouzon E, Neve RL, Tamminga CA, Nestler EJ. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011;71:656–670. doi: 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70:813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, van Steensel B. Reassessing the abundance of H3K9me2 chromatin domains in embryonic stem cells. Nat Genet. 2010;42 doi: 10.1038/ng0110-4. author reply 5–6. [DOI] [PubMed] [Google Scholar]
- Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C, Harel-Bellan A, Ameyar-Zazoua M, Ait-Si-Ali S. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell. 2010;37:46–56. doi: 10.1016/j.molcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Chen Y, Davis JM, Dong E, Gavin DP, Grayson DR, Matrisciano F, Pinna G, Satta R, Sharma RP, Tremolizzo L, Tueting P. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology. 2011;60:1007–1016. doi: 10.1016/j.neuropharm.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Renthal W, Ring RH, Rahman Z, Psifogeorgou K, Howland D, Birnbaum S, Young K, Neve R, Nestler EJ, Zachariou V. Brain region specific actions of regulator of G protein signaling 4 oppose morphine reward and dependence but promote analgesia. Biol Psychiatry. 2010;67:761–769. doi: 10.1016/j.biopsych.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JY, Jin GZ. Arcuate nucleus of hypothalamus involved in analgesic action of l-THP. Acta Pharmacol Sin. 2000;21:439–444. [PubMed] [Google Scholar]
- Inoue M, Mishina M, Ueda H. Locus-specific rescue of GluRepsilon1 NMDA receptors in mutant mice identifies the brain regions important for morphine tolerance and dependence. J Neurosci. 2003;23:6529–6536. doi: 10.1523/JNEUROSCI.23-16-06529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji D, Sui ZY, Ma YY, Luo F, Cui CL, Han JS. NMDA receptor in nucleus accumbens is implicated in morphine withdrawal in rats. Neurochem Res. 2004;29:2113–2120. doi: 10.1007/s11064-004-6884-y. [DOI] [PubMed] [Google Scholar]
- Jing L, Luo J, Zhang M, Qin WJ, Li YL, Liu Q, Wang YT, Lawrence AJ, Liang JH. Effect of the histone deacetylase inhibitors on behavioural sensitization to a single morphine exposure in mice. Neurosci Lett. 2011;494:169–173. doi: 10.1016/j.neulet.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Kaplan GB, Leite-Morris KA, Fan W, Young AJ, Guy MD. Opiate sensitization induces FosB/DeltaFosB expression in prefrontal cortical, striatal and amygdala brain regions. PLoS One. 2011;6:e23574. doi: 10.1371/journal.pone.0023574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Maldonado R, Stinus L. Neural substrates of opiate withdrawal. Trends Neurosci. 1992;15:186–191. doi: 10.1016/0166-2236(92)90171-4. [DOI] [PubMed] [Google Scholar]
- Kotlinska J, Bochenski M. Comparison of the effects of mGluR1 and mGluR5 antagonists on the expression of behavioral sensitization to the locomotor effect of morphine and the morphine withdrawal jumping in mice. Eur J Pharmacol. 2007;558:113–118. doi: 10.1016/j.ejphar.2006.11.067. [DOI] [PubMed] [Google Scholar]
- Kozela E, Pilc A, Popik P. Inhibitory effects of MPEP, an mGluR5 antagonist, and memantine, an N-methyl-d-aspartate receptor antagonist, on morphine antinociceptive tolerance in mice. Psychopharmacology (Berl) 2003;165:245–251. doi: 10.1007/s00213-002-1287-8. [DOI] [PubMed] [Google Scholar]
- Lea PM, Custer SJ, Vicini S, Faden AI. Neuronal and glial mGluR5 modulation prevents stretch-induced enhancement of NMDA receptor current. Pharmacol Biochem Behav. 2002;73:287–298. doi: 10.1016/s0091-3057(02)00825-0. [DOI] [PubMed] [Google Scholar]
- Lienert F, Mohn F, Tiwari VK, Baubec T, Roloff TC, Gaidatzis D, Stadler MB, Schübeler D. Genomic prevalence of heterochromatic H3K9me2 and transcription do not discriminate pluripotent from terminally differentiated cells. PLoS Genet. 2011;7:e1002090. doi: 10.1371/journal.pgen.1002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Ahmed SH, Blank T, Spiess J, Koob GF, Siggins GR. Chronic morphine treatment alters NMDA receptor-mediated synaptic transmission in the nucleus accumbens. J Neurosci. 1999;19:9081–9089. doi: 10.1523/JNEUROSCI.19-20-09081.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Guadaño-Ferraz A, Morte B, Ahmed S, Koob GF, De Lecea L, Siggins GR. Chronic morphine treatment alters N-methyl-d-aspartate receptors in freshly isolated neurons from nucleus accumbens. J Pharmacol Exp Ther. 2004;311:265–273. doi: 10.1124/jpet.104.067504. [DOI] [PubMed] [Google Scholar]
- Maze I, Nestler EJ. The epigenetic landscape of addiction. Ann N Y Acad Sci. 2011;1216:99–113. doi: 10.1111/j.1749-6632.2010.05893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Covington HE, 3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Feng J, Wilkinson MB, Sun H, Shen L, Nestler EJ. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci U S A. 2011;108:3035–3040. doi: 10.1073/pnas.1015483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri AR, Zhao C, Marchetto MC, Gage FH. Environmental influence on L1 retrotransposons in the adult hippocampus. Hippocampus. 2009;19:1002–1007. doi: 10.1002/hipo.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray F, Harrison NJ, Grimwood S, Bristow LJ, Hutson PH. Nucleus accumbens NMDA receptor subunit expression and function is enhanced in morphine-dependent rats. Eur J Pharmacol. 2007;562:191–197. doi: 10.1016/j.ejphar.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Noda Y, Nabeshima T. Opiate physical dependence and N-methyl-d-aspartate receptors. Eur J Pharmacol. 2004;500:121–128. doi: 10.1016/j.ejphar.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Nye HE, Nestler EJ. Induction of chronic Fos-related antigens in rat brain by chronic morphine administration. Mol Pharmacol. 1996;49:636–645. [PubMed] [Google Scholar]
- Peter CJ, Akbarian S. Balancing histone methylation activities in psychiatric disorders. Trends Mol Med. 2011;17:372–379. doi: 10.1016/j.molmed.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popik P, Wróbel M. Morphine conditioned reward is inhibited by MPEP, the mGluR5 antagonist. Neuropharmacology. 2002;43:1210–1217. doi: 10.1016/s0028-3908(02)00309-x. [DOI] [PubMed] [Google Scholar]
- Psifogeorgou K, Terzi D, Papachatzaki MM, Varidaki A, Ferguson D, Gold SJ, Zachariou V. A unique role of RGS9-2 in the striatum as a positive or negative regulator of opiate analgesia. J Neurosci. 2011;31:5617–5624. doi: 10.1523/JNEUROSCI.4146-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulvirenti L, Swerdlow NR, Koob GF. Nucleus accumbens NMDA antagonist decreases locomotor activity produced by cocaine, heroin or accumbens dopamine, but not caffeine. Pharmacol Biochem Behav. 1991;40:841–845. doi: 10.1016/0091-3057(91)90095-j. [DOI] [PubMed] [Google Scholar]
- Rehni AK, Singh N, Rachamalla M, Tikoo K. Modulation of histone deacetylase attenuates naloxone-precipitated opioid withdrawal syndrome. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:605–619. doi: 10.1007/s00210-012-0739-x. [DOI] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics. 2009;10:143. doi: 10.1186/1471-2164-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis-Segura C, Lopez-Atalaya JP, Barco A. Selective boosting of transcriptional and behavioral responses to drugs of abuse by histone deacetylase inhibition. Neuropsychopharmacology. 2009;34:2642–2654. doi: 10.1038/npp.2009.125. [DOI] [PubMed] [Google Scholar]
- Sheng J, Lv Z, Wang L, Zhou Y, Hui B. Histone H3 phosphoacetylation is critical for heroin-induced place preference. Neuroreport. 2011;22:575–580. doi: 10.1097/WNR.0b013e328348e6aa. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggins GR, Martin G, Roberto M, Nie Z, Madamba S, De Lecea L. Glutamatergic transmission in opiate and alcohol dependence. Ann N Y Acad Sci. 2003;1003:196–211. doi: 10.1196/annals.1300.012. [DOI] [PubMed] [Google Scholar]
- Singer T, McConnell MJ, Marchetto MC, Coufal NG, Gage FH. LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci. 2010;33:345–354. doi: 10.1016/j.tins.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Kennedy PJ, Nestler EJ. Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology. 2012 doi: 10.1038/npp.2012.73. Advance online publication. Retrieved November 4, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Wang R, Zhang Y, Qing H, Liu M, Yang P. The extinction of morphine-induced conditioned place preference by histone deacetylase inhibition. Neurosci Lett. 2010;483:137–142. doi: 10.1016/j.neulet.2010.07.080. [DOI] [PubMed] [Google Scholar]
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Li Y, Gao J, Sui N. Differential effect of NMDA receptor antagonist in the nucleus accumbens on reconsolidation of morphine-related positive and aversive memory in rats. Eur J Pharmacol. 2012;674:321–326. doi: 10.1016/j.ejphar.2011.11.011. [DOI] [PubMed] [Google Scholar]
- Zachariou V, Georgescu D, Sanchez N, Rahman Z, DiLeone R, Berton O, Neve RL, Sim-Selley LJ, Selley DE, Gold SJ, Nestler EJ. Essential role for RGS9 in opiate action. Proc Natl Acad Sci U S A. 2003;100:13656–13661. doi: 10.1073/pnas.2232594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariou V, Bolanos CA, Selley DE, Theobald D, Cassidy MP, Kelz MB, Shaw-Lutchman T, Berton O, Sim-Selley LJ, Dileone RJ, Kumar A, Nestler EJ. An essential role for DeltaFosB in the nucleus accumbens in morphine action. Nat Neurosci. 2006;9:205–211. doi: 10.1038/nn1636. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]