

Abstract
Nitrogen mustard (NM) is a toxic vesicant known to cause damage to the respiratory tract. Injury is associated with increased expression of inducible nitric oxide synthase (iNOS). In these studies we analyzed the effects of transient inhibition of iNOS using aminoguanidine (AG) on NM-induced pulmonary toxicity. Rats were treated intratracheally with 0.125 mg/kg NM or control. Bronchoalveolar lavage fluid (BAL) and lung tissue were collected 1 d - 28 d later and lung injury, oxidative stress and fibrosis assessed. NM exposure resulted in progressive histopathological changes in the lung including multifocal lesions, perivascular and peribronchial edema, inflammatory cell accumulation, alveolar fibrin deposition, bronchiolization of alveolar septal walls, and fibrosis. This was correlated with trichrome staining and expression of proliferating cell nuclear antigen (PCNA). Expression of heme oxygenase (HO)-1 and manganese superoxide dismutase (Mn-SOD) was also increased in the lung following NM exposure, along with levels of protein and inflammatory cells in BAL, consistent with oxidative stress and alveolar-epithelial injury. Both classically activated proinflammatory (iNOS+ and cyclooxygenase-2+) and alternatively activated profibrotic (YM-1+ and galectin-3+) macrophages appeared in the lung following NM administration; this was evident within 1 d, and persisted for 28 d. AG administration (50 mg/kg, 2x/day, 1 d - 3 d) abrogated NM-induced injury, oxidative stress and inflammation at 1 d and 3 d post exposure, with no effects at 7 d or 28 d. These findings indicate that nitric oxide generated via iNOS contributes to acute NM-induced lung toxicity, however, transient inhibition of iNOS is not sufficient to protect against pulmonary fibrosis.
Keywords: vesicants, oxidative stress, iNOS, nitric oxide, pulmonary toxicity, macrophages
INTRODUCTION
Sulfur mustard (SM) and nitrogen mustard (NM) are toxic vesicants initially developed as chemical warfare agents. A major target of both chemicals is the respiratory tract. Following exposure of humans to SM, both acute (e.g., chest tightness, hacking cough, rhinorrhea) and chronic (e.g., bronchiolitis, emphysema, lung fibrosis) effects have been reported, which are major determinants of mortality and long-term morbidity (reviewed in Weinberger et al., 2011). In animal models pulmonary toxicity resulting from exposure to SM and NM is comparable (Anderson et al., 2009; Malaviya et al., 2010; Sunil et al., 2011b). SM and NM are bifunctional alkylating agents. Toxicity results from alkylation and cross linking of nucleic acids, as well as proteins, lipids and other membrane components. This leads to DNA damage, impairment of cellular functioning, apoptosis and autophagy (Kehe et al., 2009; Malaviya et al., 2010; Shakarjian et al., 2010). In addition, both SM and NM deplete cellular glutathione and suppress antioxidant enzymes including catalase, glutathione peroxidase, glutathione reductase and superoxide dismutase (SOD) (Laskin et al., 2010a). These findings, together with reports that antioxidants such as glutathione, catalase, resveratrol, ebselen, trolox, N-acetylcysteine (NAC), or NAC in combination with mixed tocopherols, abrogate vesicant-induced lung injury demonstrate that oxidative stress plays an important role in their pathogenic actions (McClintock et al., 2002; McClintock et al., 2006; Hoesel et al., 2008; Anderson et al., 2009).
Nitric oxide is generated by inflammatory cells via an inducible isoform of nitric oxide synthase (iNOS) (reviewed in Laskin et al., 2010b). Nitric oxide and its oxidation products are highly reactive inducing membrane lipid peroxidation, nitration and hydroxylation of aromatic amino acid residues and sulfhydryl oxidation of proteins. The cytotoxic effects of nitric oxide are tightly related to the production of peroxynitrite, a potent oxidant formed by the interaction of nitric oxide with superoxide anion. The resulting oxidative and nitrosative stress has been reported to exacerbate tissue injury in response to various pulmonary toxicants (Laskin et al., 2011; Yao and Rahman, 2011). Exposure of rodents to SM or NM results in increased expression of iNOS in the lung within 24 h (Ucar et al., 2007; Yaren et al., 2007; Malaviya et al., 2010; Sunil et al., 2011b). This is associated with endothelial cell damage, alveolar hemorrhagic edema and inflammatory cell infiltration into the lung (Yaren et al., 2007; Anderson et al., 2009; Sunil et al., 2011b). Pretreatment of rodents with aminoguanidine (AG), a specific iNOS inhibitor (Southan and Szabo, 1996), has previously been shown to reduce acute histopathological changes in the lung induced by NM and increases in malonaldehyde levels (Yaren et al., 2007). The effects of transient inhibition of iNOS on the long term pulmonary effects of NM are unknown, and were investigated in the present studies.
MATERIALS AND METHODS
Animals and Treatments
Male Wistar rats (225 250 g) were purchased from Harlan Laboratories (Indianapolis IN) and maintained in an AALAC approved animal care facility. Animals were housed in filter top microisolation cages and provided food and water ad libitum. Animals received humane care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Rats were treated with PBS or NM (0.125 mg/kg) by intratracheal instillation as previously described (Sunil et al., 2011b). NM (mechlorethamine hydrochloride, Sigma-Aldrich, St. Louis, MO) was prepared immediately before administration. Preparation and instillation of NM, which included the use of double gloves, safety glasses, and masks, were performed in a designated room under a chemical hood following Rutgers University Environmental Health and Safety guidelines. In some experiments, animals were treated (i.p.) twice a day for 1 d or 3 d beginning one hour prior to NM with 50 mg/kg AG (Sigma-Aldrich, St. Louis, MO). This treatment protocol was based on the IC50 (5.4 μM) and plasma half-life (1.9 h) of AG (Alderton et al., 2001), and has previously been reported to be effective in inhibiting iNOS in vivo (Yaren et al., 2007).
Bronchoalveolar Lavage (BAL) and Tissue Collection
Animals were euthanized by i.p. injection of Nembutal (250 mg/kg) 1 d, 3 d, 7 d or 28 d after administration of NM or PBS control. BAL was collected by slowly instilling and withdrawing PBS (10 ml) into the lung through a cannula in the trachea. BAL was centrifuged (300×g, 8 min), and cell-free supernatants stored at -80 °C until analysis. Cell pellets were resuspended in 1 ml PBS and cells enumerated using a hemocytometer. Differential analysis was performed on Giemsa-stained cytospin preparations. Protein was quantified in cell-free BAL using a BCA protein assay kit (Pierce Biotechnologies Inc., Rockford, IL) with bovine serum albumin as the standard. For preparation of histological sections, the left lobe of the lung was perfused with 3% paraformaldehyde in PBS, removed and fixed in 3% paraformaldehyde overnight at 4°C, and then transferred to 50% ethanol. The remaining lung lobes were removed and stored at -80°C until analysis.
Histology
Sections (5 μm) were stained with hematoxylin and eosin or Masson’s Trichrome stain, and examined by light microscopy. Images were acquired using DP controller software (Ver. 3.3.1.292) from Olympus Corporation (Center Valley, PA). The extent of inflammatory changes including macrophage and neutrophil localization, alterations in alveolar epithelial barriers, edema, thickening of alveolar septa and fibrosis were assessed blindly by a board certified veterinary pathologist (LeRoy Hall, PhD, DVM). Semiquantitative grades (0 to 3) were assigned to specimens, with grade 0 indicating no changes; grade 1, minimal or small changes; grade 2, medium changes, and grade 3, moderate to extensive changes, relative to PBS controls. Images were also acquired at high resolution using a NanoZoomer 2.0-RS system (Hamamatsu, Japan).
Antibodies
Rabbit polyclonal anti-heme oxygenase (HO)-1 and anti-Mn-SOD antibodies were from Assay Designs (Ann Arbor, MI). Rabbit polyclonal anti-proliferating cell nuclear antigen (PCNA), anti-cyclooxygenase (COX)-2 and anti-iNOS antibodies were from Abcam Inc. (Cambridge, MA). Primary goat polyclonal anti-galectin-3 antibody was from R & D Systems (Minneapolis, MN), and rabbit polyclonal anti-YM-1 antibody from Stemcell Technologies (Vancouver, Canada).
Immunohistochemistry
Paraffin sections (5 μm) were deparaffinized and endogenous peroxidase quenched using 3% hydrogen peroxide diluted in methanol. Antigen retrieval was performed by boiling the specimens for 10 min in 10 mM sodium citrate buffer (pH 6.0) containing 0.05% Tween-20. To block nonspecific binding, sections were incubated for 1- 2 h at room temperature in buffer (Tris-buffered saline/Tween 20 containing goat or rabbit serum). Sections were then incubated overnight at 4°C in a humidified chamber with primary antibody or the appropriate IgG control diluted in blocking buffer, followed by incubation at room temperature for 30 min with biotinylated secondary antibody (Vectastain Elite ABC kit, Vector Labs, Burlingame, CA). Binding was visualized using an avidin-biotyinylated enzyme complex (Vectastain Elite ABC kit) with 3, 3’-diaminobenzidine (DAB) as the substrate (Vector Labs).
Statistical Analysis
Experimental treatment groups consisted of a minimum of 3-4 animals. Data were analyzed using two-way ANOVA. The two independent variables in our studies were: treatment (Control ± AG and NM ± AG) and time (1 d, 3 d, 7 d, 28 d). A p value of ≤ 0.05 was considered statistically significant.
RESULTS
Effects of AG on NM-induced Lung Histopathology
Treatment of rats with NM resulted in multifocal lesions characterized by perivascular and peribronchial edema, blood vessel hemorrhage, inflammatory cell infiltrates, and luminal accumulation of cellular debris, which was evident within 1 d (Fig. 1, panels D - F). Increased numbers of alveolar macrophages and neutrophils (Fig. 1, panels D - O), extravasation of red blood cells and fibrin deposits were also observed, which persisted for at least 28 days (Fig. 1, panels H and L). The bronchial epithelium became hyperplastic and exhibited plying of epithelial cells (Fig 1, panel I). NM also induced multifocal hyperplasia in the lung parenchyma, as well as hyperplasia and hypertrophy of goblet cells in the bronchial wall, an effect that was most prominent 3 d after exposure (Fig. 1, panels G and H). Patchy mild thickening of alveolar septa was visible at 3 d and 7 d post-exposure, characterized by increased numbers of mononuclear cells, neutrophils and macrophages (Fig. 1, panels D - O). At these times prominent trichrome staining in the alveolar septal wall and peribronchiolar region was apparent (Fig. 2). These deposits were mainly localized within inflammatory lesions with a few areas exhibiting organized fibrin deposits. At 28 d post NM exposure, multiple areas of fibrosis containing collagen fibers were observed around airways and bronchioles (Fig. 1, panels M and N). This was correlated with the appearance of foamy macrophages and neutrophils in the lung which occluded the alveoli. Neutrophils, eosinophils, fibroblasts and lymphocytes were also present within the thickened alveolar septa (Fig. 1, panels N and O). Bronchiolization, a phenomenon of ingrowth of cuboidal cells lining adjacent bronchioles to alveoli which form a tube like alveolar structure, was observed in lungs of NM treated animals beginning at 3 d and persisting for 28 d (Fig. 1, panels H, I and N). Erythrophagocytosis, fibroplasia, squamous metaplasia of the bronchial wall and emphysema-like changes in the alveolar tissue were also evident 3 d, 7 d and 28 d after NM exposure (Fig. 1, panels K - O). Multifocal hyperplasia and bronchiolization were correlated with increased expression of the proliferation marker, PCNA, which was most notable 3 d post exposure; PCNA expression in epithelial cells was mainly identified within bronchiolized alveolar wall (Fig. 3).
Figure 1. Histopathological changes in rat lung following NM exposure.
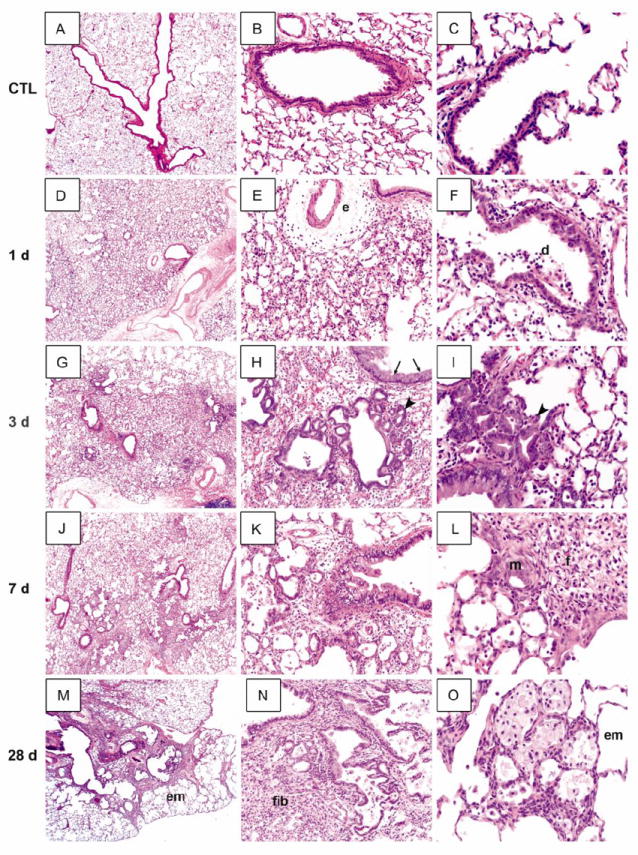
Lung sections, prepared 1 d (D-F), 3 d (G-I), 7 d (J-L) and 28 d (M-O) after exposure of rats to NM (0.125 mg/kg) or PBS control (CTL; A-C), were stained with H & E. Arrowheads, bronchiolization of alveolar septal wall; arrows, goblet cell hyperplasia and hyperproliferation; e, perivascular edema; d, cell debris; em, emphysema; m, metaplasia; f, fibrin deposits; fib, fibrosis. Original magnification, 2x (A, D, G, J, M); 10x (B, E, H, K, N); 20x (C, F, I, L, O). Representative sections from 3-4 rats/treatment group are shown.
Figure 2. Effects of AG on NM-induced collagen deposition in the lungs.
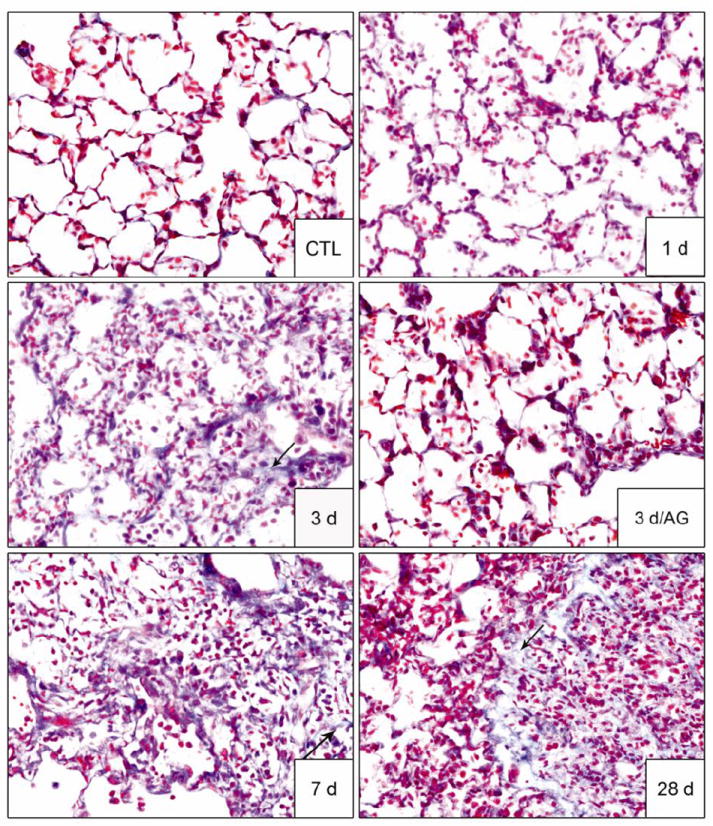
Lung sections, prepared 1 d, 3 d, 7 d and 28 d after exposure of rats to PBS control (CTL) or NM, or 3 d after NM + AG, were stained with Masson’s trichrome stain. Arrows indicate collagen deposits in alveolar wall. Original magnification, 40x. Representative sections from 3-4 rats/treatment group are shown.
Figure 3. Effects of AG on NM-induced cell proliferation in the lung.

Lung sections, prepared 1 d, 3 d, 7 d and 28 d after exposure of rats to PBS control (CTL) or NM, or 3 d after NM + AG, were stained with antibody to PCNA. Binding was visualized using a Vectastain kit. Arrowheads, epithelial cells; arrows, alveolar macrophages. Original magnification, 60x. Representative sections from 3 rats/treatment group are shown.
Treatment of animals with AG significantly reduced NM-induced structural alterations in the lung; however, these effects were only noted 1 d and 3 d post NM exposure during the course of AG treatment (Fig. 4 and not shown). Thus, at these post-exposure times, NM-induced thickening of the alveolar septal and bronchial walls and inflammatory cell infiltration were blunted by AG; fewer deposits of plasma proteins including fibrin were also noted in lung parenchyma, along with reduced edema, bronchoalveolar hyperplasia, hypertrophy, bronchial degeneration, necrosis, and numbers of goblet cells (Fig. 4). AG treatment of rats also mitigated NM-induced fibroplasia and squamous metaplasia. This was correlated with reduced trichrome staining (Fig. 2) and decreased PCNA expression (Fig. 3). In contrast, AG had no effect on NM-induced histopathologic alterations, trichrome staining or PCNA expression in the lung at 7 d or 28 d post exposure (data not shown). AG by itself also had no effect on lung histology.
Figure 4. Effects of AG on NM-induced structural alternations in the lung.
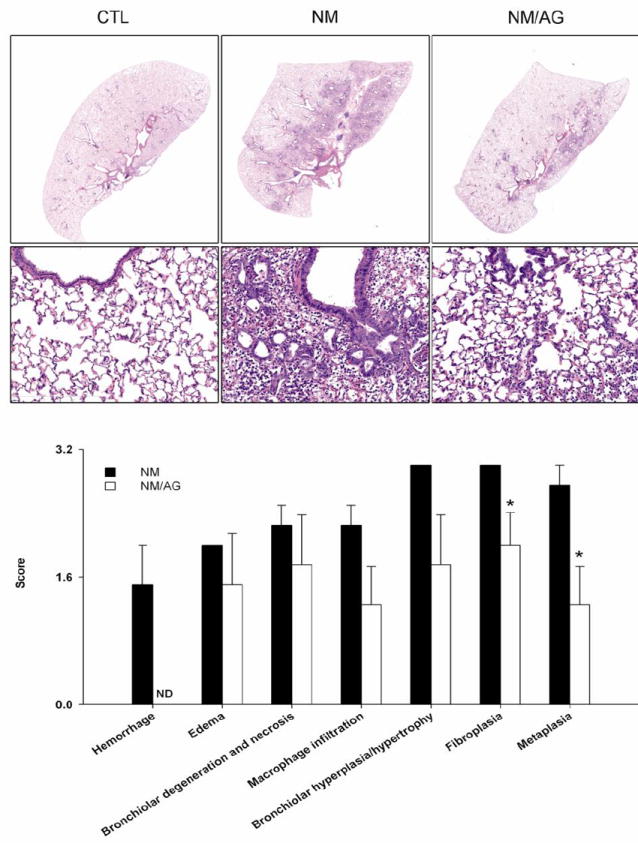
Upper and middle panels. Lung sections, prepared 3 d after treatment of rats with PBS control (CTL), NM, or NM + AG (NM/AG), were stained with H & E. Images were acquired using a NanoZoomer 2.0-RS scanner. Original magnification, 0.65x (top panel); 20x (middle panel). Lower panel. Lung sections were scored for histopathologic changes. PBS control (CTL) = zero for all parameters assessed. Each bar is the mean ± SE (n = 4 rats/treatment group). *Significantly (p ≤ 0.05) different from NM; ND, not detectable.
To further assess NM-induced alveolar epithelial damage, we measured protein accumulation in BAL. In accord with our histologic findings of structural damage and inflammation, NM exposure resulted in significant increases in BAL protein which peaked at 3 d (Fig. 5, upper panel). BAL cell number also increased significantly after NM. As observed with BAL protein, this was most notable 3 d post exposure (Fig. 5, lower panel). Differential analyses revealed that in control animals, BAL cells were >98% macrophages. Following NM exposure, the percentage of BAL neutrophils increased from 0.3 ± 0.2 to 14.7 ± 2.1 (mean ± SE) at 3 d. Treatment of animals with AG inhibited NM-induced increases in BAL protein at 1 d, whereas a reduction in BAL cell number was observed at 1 d and 3 d post NM exposure (Fig. 5, lower panel). AG also suppressed BAL protein and cell number in control animals.
Figure 5. Effects of AG on BAL cell and protein content.
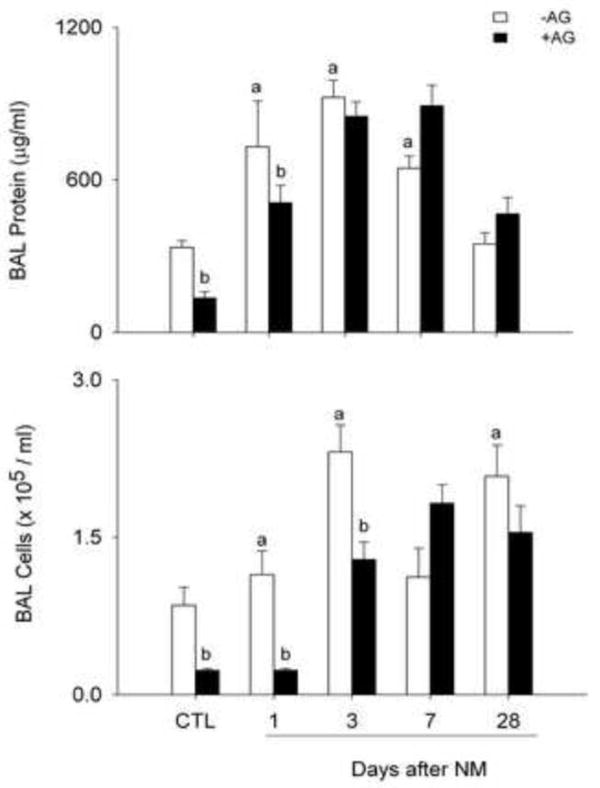
BAL was collected 1 d, 3 d, 7 d and 28 d after treatment of rats with PBS control (CTL), NM, or NM + AG. Each bar is the mean ± SE (n = 3-16 rats/treatment group). aSignificantly (p ≤ 0.05) different from CTL; bSignificantly (p ≤ 0.05) different from NM.
Effects of AG on NM-induced Oxidative Stress, Lung Macrophages and Inflammatory Mediator Expression
In further studies we analyzed the effects of AG on NM-induced expression of enzymes important in protecting the lungs from oxidative stress including HO-1 and Mn-SOD (Rahman et al., 2006). In control rats, HO-1 and Mn-SOD were detected in alveolar macrophages; some type II cells also expressed Mn-SOD (Figs. 6 and 7). Treatment of rats with NM resulted in increased numbers of alveolar macrophages expressing HO-1. This was observed 3 d, 7 d and 28 d post exposure (Fig. 6). NM also upregulated Mn-SOD in alveolar macrophages and type II cells at 7 d and 28 d post exposure (Fig. 7). AG treatment reduced the effects of NM on macrophage expression of HO-1 at 3 d post exposure, with no effects at 7 d or 28 d, or on constitutive HO-1 expression (Fig. 6). AG also had no effect on NM-induced expression of Mn-SOD at 7 d and 28 d post exposure.
Figure 6. Effects of AG on NM-induced HO-1 expression.
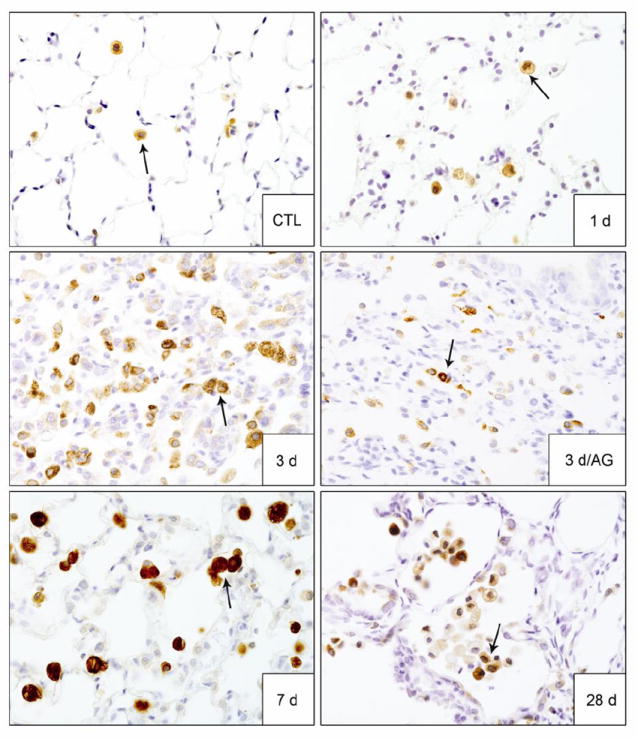
Lung sections, prepared 1 d, 3 d, 7 d and 28 d after exposure of rats to PBS control (CTL) or NM, or 3 d after NM + AG, were stained with antibody to HO-1. Binding was visualized using a Vectastain kit. Arrows indicate alveolar macrophages. Original magnification, 60x. Representative sections from 3 rats/treatment group are shown.
Figure 7. Effects of AG on NM-induced Mn-SOD expression.
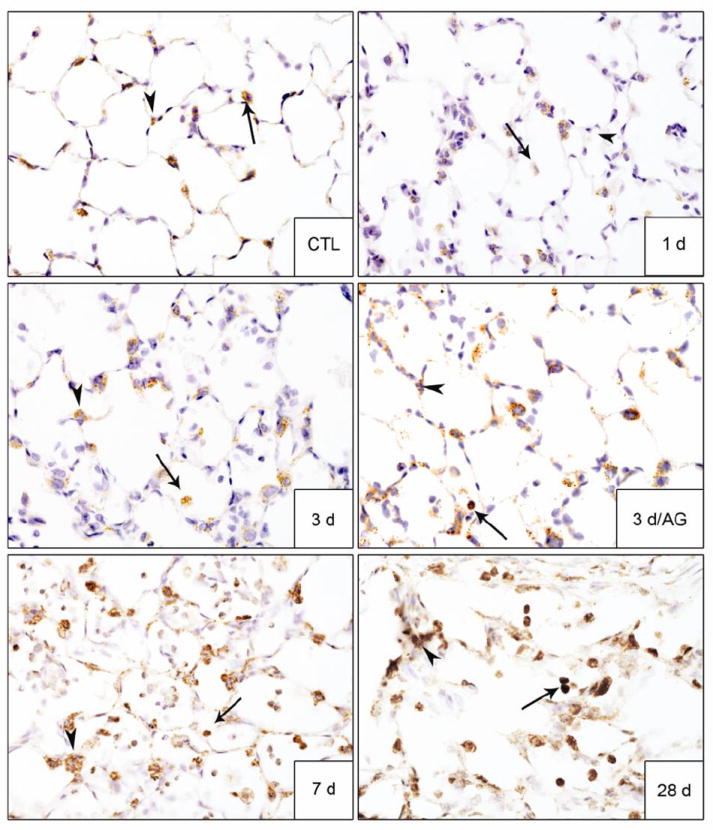
Lung sections, prepared 1 d, 3 d, 7 d and 28 d after exposure of rats to PBS control (CTL) or NM, or 3 d after NM + AG, were stained with antibody to Mn-SOD. Binding was visualized using a Vectastain kit. Arrowheads, epithelial cells; arrows, alveolar macrophages. Original magnification, 60x. Representative sections from 3 rats/treatment group are shown.
We next examined the effects of AG on NM-induced expression of iNOS and COX-2, proinflammatory mediators implicated in lung injury and markers of classically activated macrophages (Laskin et al., 2011). In control rats, low level expression of both iNOS and COX-2 was noted in alveolar macrophages, as well as type II cells (Figs. 8 and 9). NM exposure resulted in a marked increase in iNOS+ macrophages in the lung beginning at 1 d and persisting for 28 d (Fig. 8). NM also upregulated iNOS expression in type II cells. A time related increase in COX-2+ macrophages was also observed in the lung which was most prominent 7 d post NM exposure (Fig. 9). Treatment of the animals with AG suppressed the accumulation of iNOS+ and COX-2+ macrophages in the lung at 3 d and 7 d post NM exposure (Figs. 8, 9 and not shown); no effects were observed in PBS exposed controls or at 28 d post NM (data not shown).
Figure 8. Effects of AG on NM-induced iNOS expression.
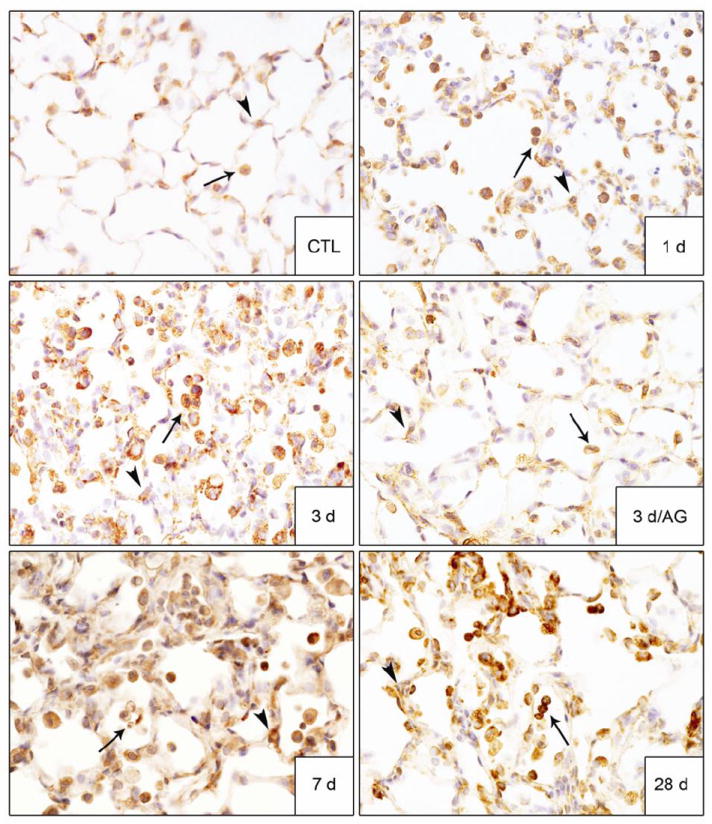
Lung sections, prepared 1 d, 3 d, 7 d and 28 d after exposure of rats to PBS control (CTL) or NM, or 3 d after NM + AG, were stained with antibody to iNOS. Binding was visualized using a Vectastain kit. Arrowheads, epithelial cells; arrows, alveolar macrophages. Original magnification, 60x. Representative sections from 3 rats/treatment group are shown.
Figure 9. Effects of AG on NM-induced COX-2 expression.
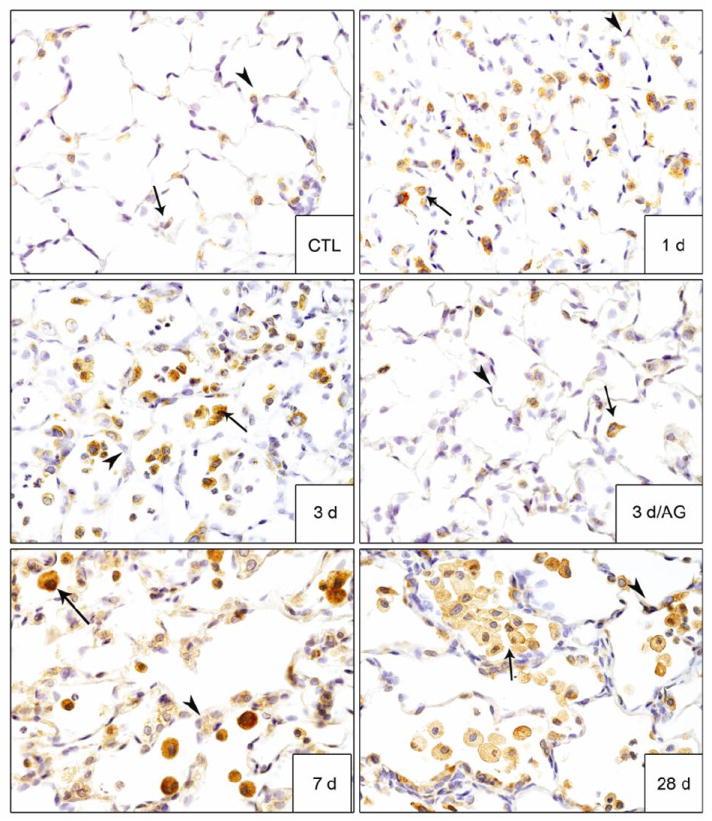
Lung sections, prepared 1 d, 3 d, 7 d and 28 d after exposure of rats to PBS control (CTL) or NM, or 3 d after NM + AG, were stained with antibody to COX-2. Binding was visualized using a Vectastain kit. Arrowheads, epithelial cells; arrows, alveolar macrophages. Original magnification, 60x. Representative sections from 3 rats/treatment group are shown.
Accumulating evidence suggests that alternatively activated macrophages contribute to fibrogenesis (reviewed in Laskin et al., 2011). In further studies we determined if NM exposure was associated with an accumulation of these macrophages in the lung. YM-1 and galectin-3 have been identified as markers of alternatively activated macrophages (Misson et al., 2004; Henderson et al., 2008; Lopez-Navarrete et al., 2011). Following NM exposure, increases in YM-1+ and galectin-3+ macrophages were observed in the lung (Figs. 10 and 11). While galactin-3+ macrophages appeared within 1 d of NM exposure, YM-1+ macrophages were noted after 3 d. Both macrophage populations persisted in the lung up to 28 d. Macrophages staining for YM-1 or galectin-3 were larger in size relative to macrophages in control lungs; this became more pronounced with time. By 28 days post NM, YM-1+ and galectin-3+ macrophages also exhibited a foamy appearance. Treatment of rats with AG resulted in a significant decrease in YM-1+ and galectin-3+ macrophages in the lung at 3 d post NM exposure (Figs. 10 and 11), with no effect at 7 d or 28 d (not shown). AG alone also had no effect on expression of YM1 or galectin-3 in lung macrophages (not shown).
Figure 10. Effects of AG on NM-induced YM-1 expression in the lung.
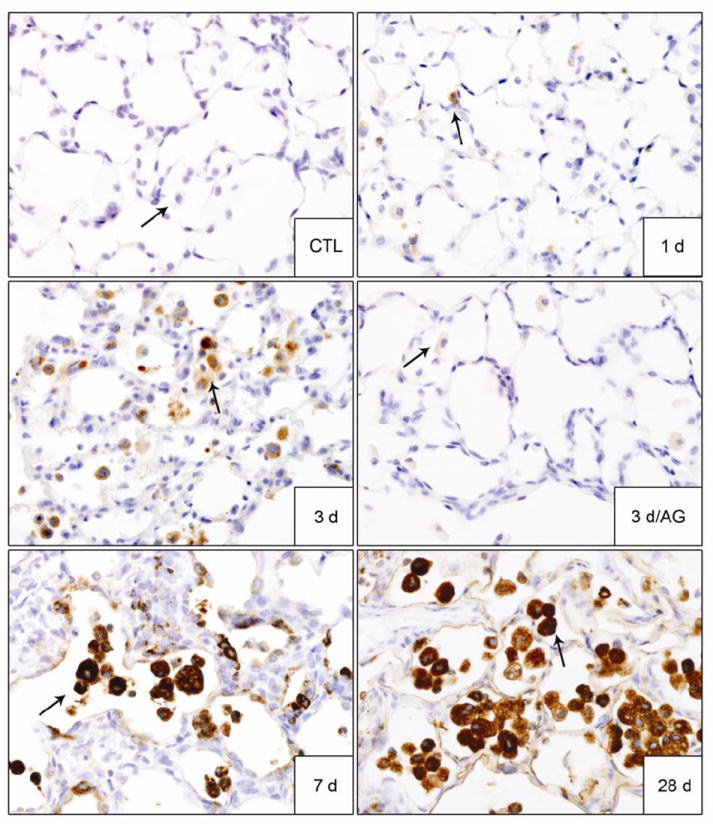
Lung sections, prepared 1 d, 3 d, 7 d and 28 d after exposure of rats to PBS control (CTL) or NM, or 3 d after NM + AG, were stained with antibody to YM-1. Binding was visualized using a Vectastain kit. Arrows indicate alveolar macrophages. Original magnification, 60x. Representative sections from 3 rats/treatment group are shown.
Figure 11. Effects of AG on NM-induced galectin-3 expression in the lung.

Lung sections, prepared 1 d, 3 d, 7 d and 28 d after exposure of rats to PBS control (CTL) or NM, or 3 d after NM + AG, were stained with antibody to galectin-3. Binding was visualized using a Vectastain kit. Arrows indicate alveolar macrophages. Original magnification, 60x. Representative sections from 3 rats/treatment group are shown.
DISCUSSION
RNS generated via iNOS have been implicated in numerous pulmonary inflammatory pathologies (Laskin et al., 2011; Sugiura and Ichinose, 2011). iNOS is rapidly upregulated in the lung following vesicant exposure (Ucar et al., 2007; Yaren et al., 2007; Gao et al., 2008; Malaviya et al., 2010; Sunil et al., 2011a; Sunil et al., 2011b), suggesting that inhibition of this enzyme and the production of RNS may be effective in mitigating acute injury and inflammation. In this regard, previous studies have shown that transient inhibition of nitric oxide or peroxynitrite production attenuates the acute effects of NM on lung oxidative stress and histopathologic changes (Ucar et al., 2007; Yaren et al., 2007). In the present studies, we extended these findings, assessing the effects of transient iNOS inhibition on lung inflammation, macrophage activation and fibrosis.
NM exposure was associated with progressive histopathologic changes in the lung which were evident within 1 d and persisted for at least 28 d. Similar persistent histological changes have been described in the lung up to 14 d after exposure of rodents to NM, as well as SM (Calvet et al., 1994; Ucar et al., 2007; Yaren et al., 2007; Sunil et al., 2011b). A unique finding in our studies was bronchiolization of the alveolar septal wall. This pathogenic response has also been noted in the lung after exposure of animals to fibrogenic agents such as bleomycin or nickel (Wehner et al., 1984; Kawamoto and Fukuda, 1990; Kim et al., 2009), suggesting that it may be important in the development of fibrosis. Disruption of the alveolar epithelial barrier results in leakage of albumin, fibrin and plasma proteins into alveolar regions of the lung (Bhalla, 1999). In accord with previous reports (Sunil et al., 2011b), we observed a rapid (within 1 d) increase in BAL protein in the lung following NM exposure confirming alveolar epithelial cell injury. The observation that protein levels remained elevated for up to 7 d suggests that repair processes are delayed. This is supported by our findings of a persistent increase in inflammatory cells in BAL and in histologic sections, as well as fibrin and plasma deposits in alveolar epithelial regions of the lung after NM exposure.
Consistent with NM-induced fibroplasia and bronchiolization, an increase in expression of the proliferation marker, PCNA was noted in epithelial cells 3 d post exposure; some subepithelial cells also expressed PCNA. Comparable increases in PCNA staining have been described in rodents exposed to the half mustard, 2-chloroethyl ethyl sulfide (CEES) (Mukhopadhyay et al., 2009). Interestingly, increased PCNA staining was also detected in epithelial cells and some alveolar macrophages 28 d post NM, a time coordinate with significant thickening of the alveolar septae, along with multiple fibrotic foci. At this time, morphologically flat squamous appearing PCNA+ epithelial cells were evident mainly lining fibrotic foci. A similar staining pattern of squamous epithelial cells has been reported in rat lung during the pathogenesis of bleomycin-induced fibrosis (Kim et al., 2009). The flat squamous PCNA positive cells surrounding fibrotic foci may be important in growth of fibrotic tissue in the lung.
Evidence suggests that oxidative stress plays a role in vesicant-induced pulmonary toxicity (Mukhopadhyay et al., 2006; Hoesel et al., 2008; Anderson et al., 2009; Gould et al., 2009; Shohrati et al., 2010; Weinberger et al., 2011). A characteristic response of cells and tissues to oxidative stress is upregulation of HO-1, a phase II stress response enzyme with antioxidant and antiinflammatory activity (Otterbein et al., 1999; Rahman et al., 2006). We observed a persistent induction of HO-1 in lung macrophages beginning 3 d after NM exposure. Analogous increases in HO-1 have been described in lung macrophages after SM inhalation (Malaviya et al., 2010). Our findings that HO-1 expression was predominantly localized in lung macrophages suggest that these cells may be a significant source of cytotoxic oxidants. NM intoxication was also associated with increases in Mn-SOD expression in lung macrophages, as well as epithelial cells; however this was delayed relative to HO-1 becoming most pronounced 28 d post exposure. These data indicate that NM-induced oxidative stress is persistent.
Classically activated proinflammatory macrophages have been implicated in lung injury induced by pulmonary toxicants (reviewed in Laskin et al., 2011). These cells release mediators such as nitric oxide and prostaglandin E2 which promote oxidative stress and inflammation. We found that iNOS and COX-2, enzymes regulating the synthesis of RNS and eicosanoids, respectively, were upregulated in macrophages within 1 d of NM intoxication, suggesting that they are classically activated. Interestingly, macrophages expressing these markers persisted in the lung for at least 28 d post exposure, indicating that NM-induced injury and oxidative stress involves prolonged proinflammatory macrophage activity. Lung epithelial cells also stained positively for iNOS and COX-2 following NM exposure. As observed with macrophages, this was evident at all post-exposure time points. These data are consistent with findings that both cell types have the capacity to the generate RNS and eicosanoids in the lung following injury (Punjabi et al., 1994; Tasaka et al., 2008; Laskin et al., 2011). Increases in expression of iNOS and COX-2 have been described in lungs of patients with idiopathic pulmonary fibrosis (Saleh et al., 1997; Lappi-Blanco et al., 2006). These findings, together with reports that suppressing iNOS or COX-2 mitigates the development of silica- or bleomycin-induced lung fibrosis in rodents, suggest that products of these enzymes contribute to the fibrogenic process (Giri et al., 2002; Arafa et al., 2007; Kalayarasan et al., 2008). Our observation that AG treatment of rats reduced NM-induced PCNA expression, fibroplasia and collagen deposition at 3 d post exposure are in accord with this idea.
Macrophages are also known to play an important role in the resolution of inflammatory responses. This activity is mediated by a distinct subpopulation of macrophages that are alternatively activated (reviewed in Laskin et al., 2011). Exaggerated responses of alternatively activated macrophages are thought to contribute to the development of fibrosis. Following NM intoxication, we observed rapid increases (within 1-3 d) in the number of lung macrophages expressing YM-1 and galectin-3, markers of alternatively activated macrophages, suggesting that the process of fibrosis begins early in the pathogenic process. This is supported by our findings that the appearance of YM-1+ and galectin-3+ macrophages in the lung correlated with increases in fibroplasia at 3 d post NM, and collagen deposits around bronchioles and alveolar septae after 7 d; by 28 d multiple fibrotic foci with mature collagen were evident around the airways distorting the normal parenchymal structure. At this time, macrophages expressing YM-1 and galectin-3 occluding the alveoli were prominent. Additionally, these cells were markedly increased in size and exhibited a foamy appearance, which is consistent with the morphology of profibrotic macrophages (Wang and Lyerla, 2010). Galectin-3 has been linked to the promotion of collagen synthesis by fibroblasts, and the development of fibrosis in liver, kidney and lung (Misson et al., 2004; Nishi et al., 2007; Henderson et al., 2008; Lopez-Navarrete et al., 2011). High levels of galectin-3 have also been detected in BAL from patients with idiopathic pulmonary fibrosis and interstitial pneumonia (Nishi et al., 2007). Further studies are required to assess whether galactin-3 plays is also involved in NM-induced fibrosis.
AG is an analog of L-arginine that competitively binds to iNOS, inhibiting the synthesis of nitric oxide (Southan and Szabo, 1996). AG administration has been reported to mitigate the severity of pathological changes attributed to excess production of RNS in a number of inflammatory conditions including allergic asthma, ischemia/reperfusion induced lung injury and fibrosis (Giri et al., 2002; Landgraf et al., 2005; Yeh et al., 2008). We found that transient inhibition of iNOS using AG reduced histopathological changes in the lung induced by NM at 1 d and 3 d post exposure. Thus, decreases in both the extent and the severity of early lung injury were observed. We also noted less edema, alveolar hemorrhage and inflammatory cell infiltration into the lung in AG-treated rats which is in accord with previous studies (Ucar et al., 2007; Yaren et al., 2007). This was correlated with attenuation of NM-induced increases in BAL cell number and protein. These data, together with recent reports that iNOS-/- mice are protected from lung injury induced by the half mustard, CEES (Sunil et al., 2012), provide support for the idea that RNS participate in the early pathogenic response to vesicants. Interestingly, AG was also found to reduce BAL protein and cell content in control animals. This may be due to the ability of AG to scavenge free radicals and consequently, stabilize the alveolar epithelial barrier (Courderot-Masuyer et al., 1999).
AG treatment has been reported to attenuate NM-induced oxidative stress in the lung, as measured by decreases in lung malonaldehyde and urinary nitrite and nitrate levels (Yaren et al., 2007). Consistent with these findings, we observed that NM-induced HO-1 expression was suppressed in lung macrophages following AG administration. We also found that the number of iNOS+ lung macrophages was decreased, a response which persisted for 7 d. These data confirm that early oxidative stress induced by NM involves RNS. Our findings that AG treatment suppressed iNOS may also be due to feedback inhibition (Cooper, 1999). In contrast, treatment of rats with AG had no effect on expression of Mn-SOD. This is most likely due to the fact that expression of Mn-SOD was delayed for 7 d post NM, a time when AG administration had already been discontinued. AG treatment was also found to suppress NM-induced increases in COX-2+ macrophages in the lung at 3 d and 7 d post exposure. Decreases in both COX-2+ and iNOS+ proinflammatory macrophages in lungs of AG treated mice are in accord with reduced tissue injury in these animals. Previous reports demonstrated that AG inhibits COX-2 expression in rat models of lung infection and injury (Md et al., 2005; Natarajan et al., 2007). It remains to be determined if nitric oxide generated via iNOS plays a similar role in regulating COX-2 expression following NM exposure.
Early increases in hyperplasia, as measured by PCNA and fibrogenesis, were also suppressed by transient iNOS inhibition with AG. This was correlated with reduced numbers of YM-1+ and galectin-3+ profibrotic macrophages in the lung. Antioxidants have been reported to inhibit proliferation of cells within hyperplastic lesions, collagen accumulation, and the development of fibrotic lesions in the lung following exposure to bleomycin or to CEES (Ikezaki et al., 1996; Giri et al., 2002; Hoesel et al., 2008). Our findings provide additional support for a role of oxidative stress in fibroplasia and the development of lung fibrosis (Kliment and Oury, 2010). Reduced levels of PCNA expression in AG-treated animals indicates that RNS contribute to proliferative responses in the lungs of NM-exposed rats.
Vesicant exposure involves both acute and long term pathologic consequences. Our studies provide evidence of persistent activation of macrophages, epithelial cells and fibroblasts in the lungs of rats following a single acute NM exposure, which is in accord with findings in humans exposed to SM (Weinberger et al., 2011). Moreover, the acute pulmonary effects of NM intoxication can be mitigated by transient inhibition of iNOS; in contrast the longer term consequences were unaffected. Further studies are required to determine if continuous inhibition of iNOS is effective in mitigating lung fibrosis induced by NM.
Highlights.
Nitrogen Mustard (NM) induces acute lung injury and fibrosis.
Pulmonary toxicity is associated with increased expression of iNOS.
Transient inhibition of iNOS attenuates acute lung injury induced by NM.
Acknowledgments
This work was supported by NIH Grants AR055073 HL096426, GM034310, ES004738, CA132624 and ES005022. We would like to thank Dr. David Reimer, DVM, MBA from the Laboratory Animal Services, Rutgers University for intratracheal instillations.
Footnotes
Conflict of Interest: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DR, Taylor SL, Fetterer DP, Holmes WW. Evaluation of protease inhibitors and an antioxidant for treatment of sulfur mustard-induced toxic lung injury. Toxicology. 2009;263:41–46. doi: 10.1016/j.tox.2008.08.025. [DOI] [PubMed] [Google Scholar]
- Arafa HM, Abdel-Wahab MH, El-Shafeey MF, Badary OA, Hamada FM. Anti-fibrotic effect of meloxicam in a murine lung fibrosis model. Eur J Pharmacol. 2007;564:181–189. doi: 10.1016/j.ejphar.2007.02.065. [DOI] [PubMed] [Google Scholar]
- Bhalla DK. Ozone-induced lung inflammation and mucosal barrier disruption: toxicology, mechanisms, and implications. J Toxicol Environ Health B Crit Rev. 1999;2:31–86. doi: 10.1080/109374099281232. [DOI] [PubMed] [Google Scholar]
- Calvet JH, Jarreau PH, Levame M, D’Ortho MP, Lorino H, Harf A, Macquin-Mavier I. Acute and chronic respiratory effects of sulfur mustard intoxication in guinea pig. J Appl Physiol. 1994;76:681–688. doi: 10.1152/jappl.1994.76.2.681. [DOI] [PubMed] [Google Scholar]
- Cooper CE. Nitric oxide and iron proteins. Biochim Biophys Acta. 1999;1411:290–309. doi: 10.1016/s0005-2728(99)00021-3. [DOI] [PubMed] [Google Scholar]
- Courderot-Masuyer C, Dalloz F, Maupoil V, Rochette L. Antioxidant properties of aminoguanidine. Fundam Clin Pharmacol. 1999;13:535–540. doi: 10.1111/j.1472-8206.1999.tb00358.x. [DOI] [PubMed] [Google Scholar]
- Gao X, Ray R, Xiao Y, Ray P. Suppression of inducible nitric oxide synthase expression and nitric oxide production by macrolide antibiotics in sulfur mustard-exposed airway epithelial cells. Basic Clin Pharmacol Toxicol. 2008;103:255–261. doi: 10.1111/j.1742-7843.2008.00255.x. [DOI] [PubMed] [Google Scholar]
- Giri SN, Biring I, Nguyen T, Wang Q, Hyde DM. Abrogation of bleomycin-induced lung fibrosis by nitric oxide synthase inhibitor, aminoguanidine in mice. Nitric Oxide. 2002;7:109–118. doi: 10.1016/s1089-8603(02)00102-7. [DOI] [PubMed] [Google Scholar]
- Gould NS, White CW, Day BJ. A role for mitochondrial oxidative stress in sulfur mustard analog 2-chloroethyl ethyl sulfide-induced lung cell injury and antioxidant protection. J Pharmacol Exp Ther. 2009;328:732–739. doi: 10.1124/jpet.108.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson NC, Mackinnon AC, Farnworth SL, Kipari T, Haslett C, Iredale JP, Liu FT, Hughes J, Sethi T. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol. 2008;172:288–298. doi: 10.2353/ajpath.2008.070726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesel LM, Flierl MA, Niederbichler AD, Rittirsch D, McClintock SD, Reuben JS, Pianko MJ, Stone W, Yang H, Smith M, Sarma JV, Ward PA. Ability of antioxidant liposomes to prevent acute and progressive pulmonary injury. Antioxid Redox Signal. 2008;10:973–981. doi: 10.1089/ars.2007.1878. [DOI] [PubMed] [Google Scholar]
- Ikezaki S, Nishikawa A, Enami T, Furukawa F, Imazawa T, Uneyama C, Fukushima S, Takahashi M. Inhibitory effects of the dietary antioxidants butylated hydroxyanisole and butylated hydroxytoluene on bronchioloalveolar cell proliferation during the bleomycin-induced pulmonary fibrosing process in hamsters. Food Chem Toxicol. 1996;34:327–335. doi: 10.1016/0278-6915(96)00127-5. [DOI] [PubMed] [Google Scholar]
- Kalayarasan S, Sriram N, Sudhandiran G. Diallyl sulfide attenuates bleomycin-induced pulmonary fibrosis: critical role of iNOS, NF-kappaB, TNF-alpha and IL-1beta. Life Sci. 2008;82:1142–1153. doi: 10.1016/j.lfs.2008.03.018. [DOI] [PubMed] [Google Scholar]
- Kawamoto M, Fukuda Y. Cell proliferation during the process of bleomycin-induced pulmonary fibrosis in rats. Acta Pathol Jpn. 1990;40:227–238. doi: 10.1111/j.1440-1827.1990.tb01556.x. [DOI] [PubMed] [Google Scholar]
- Kehe K, Balszuweit F, Steinritz D, Thiermann H. Molecular toxicology of sulfur mustard-induced cutaneous inflammation and blistering. Toxicology. 2009;263:12–19. doi: 10.1016/j.tox.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Kim JY, Choeng HC, Ahn C, Cho SH. Early and late changes of MMP-2 and MMP-9 in bleomycin-induced pulmonary fibrosis. Yonsei Med J. 2009;50:68–77. doi: 10.3349/ymj.2009.50.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliment CR, Oury TD. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic Biol Med. 2010;49:707–717. doi: 10.1016/j.freeradbiomed.2010.04.036. [DOI] [PubMed] [Google Scholar]
- Landgraf RG, Russo M, Jancar S. Acute inhibition of inducible nitric oxide synthase but not its absence suppresses asthma-like responses. Eur J Pharmacol. 2005;518:212–220. doi: 10.1016/j.ejphar.2005.04.047. [DOI] [PubMed] [Google Scholar]
- Lappi-Blanco E, Kaarteenaho-Wiik R, Maasilta PK, Anttila S, Paakko P, Wolff HJ. COX-2 is widely expressed in metaplastic epithelium in pulmonary fibrous disorders. Am J Clin Pathol. 2006;126:717–724. doi: 10.1309/PFGX-CLNG-2N17-PJX9. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol. 2011;51:267–288. doi: 10.1146/annurev.pharmtox.010909.105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin JD, Black AT, Jan YH, Sinko PJ, Heindel ND, Sunil V, Heck DE, Laskin DL. Oxidants and antioxidants in sulfur mustard-induced injury. Ann N Y Acad Sci. 2010a;1203:92–100. doi: 10.1111/j.1749-6632.2010.05605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin JD, Heck DE, Laskin DL. Nitric oxide pathways in toxic responses. In: Ballantine B, Marrs T, Syversen T, editors. General and Applied Toxicology. Wiley-Blackwell; UK: 2010b. pp. 425–438. [Google Scholar]
- Lopez-Navarrete G, Ramos-Martinez E, Suarez-Alvarez K, Aguirre-Garcia J, Ledezma-Soto Y, Leon-Cabrera S, Gudino-Zayas M, Guzman C, Gutierrez-Reyes G, Hernandez-Ruiz J, Camacho-Arroyo I, Robles-Diaz G, Kershenobich D, Terrazas LI, Escobedo G. Th2-associated alternative Kupffer cell activation promotes liver fibrosis without inducing local inflammation. Int J Biol Sci. 2011;7:1273–1286. doi: 10.7150/ijbs.7.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Sunil VR, Cervelli J, Anderson DR, Holmes WW, Conti ML, Gordon RE, Laskin JD, Laskin DL. Inflammatory effects of inhaled sulfur mustard in rat lung. Toxicol Appl Pharmacol. 2010;248:89–99. doi: 10.1016/j.taap.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock SD, Hoesel LM, Das SK, Till GO, Neff T, Kunkel RG, Smith MG, Ward PA. Attenuation of half sulfur mustard gas-induced acute lung injury in rats. J Appl Toxicol. 2006;26:126–131. doi: 10.1002/jat.1115. [DOI] [PubMed] [Google Scholar]
- McClintock SD, Till GO, Smith MG, Ward PA. Protection from half-mustard-gas-induced acute lung injury in the rat. J Appl Toxicol. 2002;22:257–262. doi: 10.1002/jat.856. [DOI] [PubMed] [Google Scholar]
- Md S, Moochhala SM, Siew Yang KL, Lu J, Anuar F, Mok P, Ng KC. The role of selective nitric oxide synthase inhibitor on nitric oxide and PGE2 levels in refractory hemorrhagic-shocked rats. J Surg Res. 2005;123:206–214. doi: 10.1016/j.jss.2004.07.243. [DOI] [PubMed] [Google Scholar]
- Misson P, van den Brule S, Barbarin V, Lison D, Huaux F. Markers of macrophage differentiation in experimental silicosis. J Leukoc Biol. 2004;76:926–932. doi: 10.1189/jlb.0104019. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Mukherjee S, Stone WL, Smith M, Das SK. Role of MAPK/AP-1 signaling pathway in the protection of CEES-induced lung injury by antioxidant liposome. Toxicology. 2009;261:143–151. doi: 10.1016/j.tox.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Rajaratnam V, Mukherjee S, Smith M, Das SK. Modulation of the expression of superoxide dismutase gene in lung injury by 2-chloroethyl ethyl sulfide, a mustard analog. J Biochem Mol Toxicol. 2006;20:142–149. doi: 10.1002/jbt.20128. [DOI] [PubMed] [Google Scholar]
- Natarajan G, Glibetic M, Raykova V, Ofenstein JP, Thomas RL, Aranda JV. Nitric oxide and prostaglandin response to group B streptococcal infection in the lung. Ann Clin Lab Sci. 2007;37:170–176. [PubMed] [Google Scholar]
- Nishi Y, Sano H, Kawashima T, Okada T, Kuroda T, Kikkawa K, Kawashima S, Tanabe M, Goto T, Matsuzawa Y, Matsumura R, Tomioka H, Liu FT, Shirai K. Role of galectin-3 in human pulmonary fibrosis. Allergol Int. 2007;56:57–65. doi: 10.2332/allergolint.O-06-449. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Kolls JK, Mantell LL, Cook JL, Alam J, Choi AM. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest. 1999;103:1047–1054. doi: 10.1172/JCI5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjabi CJ, Laskin JD, Pendino KJ, Goller NL, Durham SK, Laskin DL. Production of nitric oxide by rat type II pneumocytes: increased expression of inducible nitric oxide synthase following inhalation of a pulmonary irritant. Am J Respir Cell Mol Biol. 1994;11:165–172. doi: 10.1165/ajrcmb.11.2.7519435. [DOI] [PubMed] [Google Scholar]
- Rahman I, Biswas SK, Kode A. Oxidant and antioxidant balance in the airways and airway diseases. Eur J Pharmacol. 2006;533:222–239. doi: 10.1016/j.ejphar.2005.12.087. [DOI] [PubMed] [Google Scholar]
- Saleh D, Barnes PJ, Giaid A. Increased production of the potent oxidant peroxynitrite in the lungs of patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:1763–1769. doi: 10.1164/ajrccm.155.5.9154889. [DOI] [PubMed] [Google Scholar]
- Shakarjian MP, Heck DE, Gray JP, Sinko PJ, Gordon MK, Casillas RP, Heindel ND, Gerecke DR, Laskin DL, Laskin JD. Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol Sci. 2010;114:5–19. doi: 10.1093/toxsci/kfp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohrati M, Ghanei M, Shamspour N, Babaei F, Abadi MN, Jafari M, Harandi AA. Glutathione and malondialdehyde levels in late pulmonary complications of sulfur mustard intoxication. Lung. 2010;188:77–83. doi: 10.1007/s00408-009-9178-y. [DOI] [PubMed] [Google Scholar]
- Southan GJ, Szabo C. Selective pharmacological inhibition of distinct nitric oxide synthase isoforms. Biochem Pharmacol. 1996;51:383–394. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]
- Sugiura H, Ichinose M. Nitrative stress in inflammatory lung diseases. Nitric Oxide. 2011;25:138–144. doi: 10.1016/j.niox.2011.03.079. [DOI] [PubMed] [Google Scholar]
- Sunil VR, Patel-Vayas K, Shen J, Gow AJ, Laskin JD, Laskin DL. Role of TNFR1 in lung injury and altered lung function induced by the model sulfur mustard vesicant, 2-chloroethyl ethyl sulfide. Toxicol Appl Pharmacol. 2011a;250:245–255. doi: 10.1016/j.taap.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil VR, Patel KJ, Shen J, Reimer D, Gow AJ, Laskin JD, Laskin DL. Functional and inflammatory alterations in the lung following exposure of rats to nitrogen mustard. Toxicol Appl Pharmacol. 2011b;250:10–18. doi: 10.1016/j.taap.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil VR, Shen J, Patel-Vayas K, Gow AJ, Laskin JD, Laskin DL. Role of reactive nitrogen species generated via inducible nitric oxide synthase in vesicant-induced lung injury, inflammation and altered lung functioning. Toxicol Appl Pharmacol. 2012;261:22–30. doi: 10.1016/j.taap.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaka S, Amaya F, Hashimoto S, Ishizaka A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid Redox Signal. 2008;10:739–753. doi: 10.1089/ars.2007.1940. [DOI] [PubMed] [Google Scholar]
- Ucar M, Korkmaz A, Reiter RJ, Yaren H, Oter S, Kurt B, Topal T. Melatonin alleviates lung damage induced by the chemical warfare agent nitrogen mustard. Toxicol Lett. 2007;173:124–131. doi: 10.1016/j.toxlet.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Wang L, Lyerla T. Histochemical and cellular changes accompanying the appearance of lung fibrosis in an experimental mouse model for Hermansky Pudlak syndrome. Histochem Cell Biol. 2010;134:205–213. doi: 10.1007/s00418-010-0724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner AP, Dagle GE, Busch RH. Pathogenicity of inhaled nickel compounds in hamsters. IARC Sci Publ. 1984:143–151. [PubMed] [Google Scholar]
- Weinberger B, Laskin JD, Sunil VR, Sinko PJ, Heck DE, Laskin DL. Sulfur mustard-induced pulmonary injury: therapeutic approaches to mitigating toxicity. Pulm Pharmacol Ther. 2011;24:92–99. doi: 10.1016/j.pupt.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol. 2011;254:72–85. doi: 10.1016/j.taap.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaren H, Mollaoglu H, Kurt B, Korkmaz A, Oter S, Topal T, Karayilanoglu T. Lung toxicity of nitrogen mustard may be mediated by nitric oxide and peroxynitrite in rats. Res Vet Sci. 2007;83:116–122. doi: 10.1016/j.rvsc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Yeh DY, Feng NH, Chen CF, Lin HI, Wang D. Inducible nitric oxide synthase expressions in different lung injury models and the protective effect of aminoguanidine. Transplant Proc. 2008;40:2178–2181. doi: 10.1016/j.transproceed.2008.07.087. [DOI] [PubMed] [Google Scholar]