

Abstract
The World Health Organization recommends that the daily intake of added sugars should make up no more than 10% of total energy. The consumption of sugar-sweetened beverages is the main source of added sugars. Fructose, together with glucose, as a component of high fructose corn syrups or as a component of the sucrose molecule, is one of the main sweeteners present in this kind of beverages. Data from prospective and intervention studies clearly point to high fructose consumption, mainly in the form of sweetened beverages, as a risk factor for several metabolic diseases in humans. The incidence of hypertension, nonalcoholic fatty liver disease (NAFLD), dyslipidemia (mainly hypertriglyceridemia), insulin resistance, type 2 diabetes mellitus, obesity, and the cluster of many of these pathologies in the form of metabolic syndrome is higher in human population segments that show high intake of fructose. Adolescent and young adults from low-income families are especially at risk. We recently reviewed evidence from experimental animals and human data that confirms the deleterious effect of fructose on lipid and glucose metabolism. In this present review we update the information generated in the past 2 years about high consumption of fructose-enriched beverages and the occurrence of metabolic disturbances, especially NAFLD, type 2 diabetes mellitus, and metabolic syndrome. We have explored recent data from observational and experimental human studies, as well as experimental data from animal and cell models. Finally, using information generated in our laboratory and others, we provide a view of the molecular mechanisms that may be specifically involved in the development of liver lipid and glucose metabolic alterations after fructose consumption in liquid form.
Keywords: Obesity, Metabolic syndrome, Hypertension, Dyslipidemia, Nonalcoholic fatty liver disease, Clinical studies, Experimental studies, Sweetened beverages
INTRODUCTION
At the end of 2011, the United Nations declared that, for the first time in the history of humanity, non-communicable diseases had outpaced infectious diseases as the main threat to human health globally. Among them, cardiovascular diseases associated with metabolic disorders, such as obesity, metabolic syndrome, and type 2 diabetes mellitus, are of paramount importance. Changes in human dietary habits in recent decades have led to the consumption of hypercaloric diets that are rich in saturated fats and simple sugars (sucrose, glucose and fructose). This, combined with decreased physical activity, is one of the key factors contributing to the ever-increasing prevalence of metabolic disorders. This situation recently prompted Lustig et al[1] to request the legal regulation of foodstuffs containing added sugars in a way similar to the control of tobacco and alcohol.
The World Health Organization recommends that the daily intake of added sugars should make up no more than 10% of total energy[2]. The consumption of sugar-sweetened beverages is the main source of added sugars[3]. Fructose, together with glucose, as a component of high fructose corn syrups (HFCSs) or as a component of the sucrose molecule, is mainly responsible for the metabolic disturbances associated with excessive consumption of added sugars. We recently reviewed evidence from experimental animals and human data that confirms the deleterious effect of fructose on lipid and glucose metabolism[4]. Given the relevance of this issue to public health policies, in this review we update information on the effects of fructose on human health. We focus also on new experimental data from our laboratory and others on molecular mechanisms involved in the disturbance of liver metabolism by fructose.
FRUCTOSE: THE BENCH SIDE
Data from prospective and intervention studies clearly point to high fructose consumption, mainly in the form of sweetened beverages, as a risk factor for several metabolic diseases in humans. The incidence of hypertension, nonalcoholic fatty liver disease (NAFLD), dyslipidemia (mainly hypertriglyceridemia), insulin resistance, type 2 diabetes mellitus, obesity, and the cluster of many of these pathologies in the form of metabolic syndrome is higher in human population segments that show high intake of fructose. Adolescent and young adults from low-income families are especially at risk. We and others have recently reviewed the evidence of this relationship[4-8]. In the present review, we provide an overview of recent data, from 2011 onwards that has not been discussed previously (Table 1). For readers interested in recent reviews on this subject, particularly regarding fructose consumption, uric acid metabolism and hypertension, we refer to two excellent reviews published in 2011[9,10].
Table 1.
Overview of fructose-related human studies
| Authors | Subjects | Study characteristics | Sugar | Main results |
| Browning et al[11] | 18 NAFLD (5 men, 13 women), BMI: 35 ± 7 kg/m2 | Intervention study 2 wk dietary carbohydrate and calorie restriction | Reductions in body weight (-4.6 ± 1.5 kg vs -4.0 ± 1.5 kg) and hepatic triglycerides (-55% ± 14% vs -28% ± 23%) were significantly greater with dietary carbohydrate restriction than with calorie restriction | |
| Maersk et al[12] | 60 overweight/obese nondiabetic subjects | Randomized intervention study Ingestion of 4 different drinks (1 L/d, SSB, isocaloric semiskim milk, aspartame-sweetened and water) for 6 mo | S | Daily intake of SSB with sucrose increased ectopic fat accumulation (liver, skeletal muscle) and lipids (blood cholesterol and triglycerides) compared with the other beverages |
| Silbernagel et al[13] | Healthy male (12) and female (8) adults | Dietary intervention study 150 g/d for 4 wk | F and G | Visceral and liver fat content associated to cholesterol synthesis Cholesterol synthesis appeared to be dependent on fructose/glucose intake |
| Stanhope et al[14] | 48 adults, BMI 18-35 kg/m2 | Dietary intervention study Consumption of simple sugars at 25% of energy requirements for 2 wk | F and G | F consumption increased cardiovascular risk factors (AUC-Tg, fasting LDL and apo B) more than G |
| de Koning et al[15] | 40 389 healthy men | Prospective cohort study 20 yr of follow-up of SSB and artificially sweetened beverages consumption | F, G and S SSB | After adjustment for several confounders, the hazard ratio for the association of SSB with incident type 2 diabetes was 1.24 for the comparison of the top with the bottom quartile of SSB intake |
| Silbernagel et al[16] | Healthy male (12) and female (8) adults | Dietary intervention study 150 g/d for 4 wk | F and G | Insulin sensitivity decreased in both intervention groups, while plasma triglycerides were increased in the F group |
| Cox et al[17] | Overweight/obese male (16) and female (15) adults | Intervention study 10 wk supplementation with SSB at 25% of energy requirements | F and G SSB | F-consuming subjects had a significant reduction in net postprandial fat oxidation and resting energy expenditure |
| Maier et al[18] | 15 overweight/obese children (5-8 yr) | Dietary intervention study parental training to reduce dietary sugar content (–50% from baseline, 12 wk) and 12 wk of follow-up | F, G and S | Reductions in sugar intake were related to significant reductions in BMI and BMI standard deviation scores |
| Pollock et al[19] | 559 adolescents (14-18 yr) | Association study of F intake and cardiometabolic risk factors | F | After adjustment, higher F consumption directly associated to BP, fasting glucose, HOMA-IR and C-reactive protein, and inversely to HDL-cholesterol and adiponectin. The introduction of visceral fat as a covariate attenuated these trends |
| Cox et al[21] | Overweight/obese male (16) and female (15) adults | Intervention study 10 wk supplementation with SSB at 25% of energy requirements | F and G SSB | Fasting concentrations of MCP-1, PAI-1 and E-selectin as well as postprandial concentrations of PAI-1 increased in subjects consuming F but not in those consuming G |
| Brown et al[22] | 2696 people | Cross-sectional association study | F, G and S SSB | Direct and independent associations of SSB intake and BP |
| Greater sugar-BP differences for persons with higher sodium excretion | ||||
| Friberg et al[25] | 61 226 women | Population-based cohort study 18.4 yr of follow-up of total sucrose, high-sugar-foods | F, G and S | Total sucrose intake and consumption of sweet buns and cookies was associated with increased risk of endometrial cancer |
| Ye et al[26] | 737 non diabetic adults | Association study of sugar intake and cognitive function | F, G and S | Greater intakes of total sugars, added sugars and SSB beverages, but not of sugar sweetened solid foods, were significantly associated with lower MMSE scores, after adjusting for covariates |
F: Fructose; G: Glucose; S: Sucrose; SSB: Sugar-sweetened beverages; BP: Blood pressure; NAFLD: Nonalcoholic fatty liver disease; BMI: Body mass index; MCP-1: Monocyte chemoattractant protein-1; PAI-1: Plasminogen activator inhibitor-1; HOMA-IR: Homeostasis model assessment-estimated insulin resistance; HDL: High-density lipoprotein; AUC-Tg: 24 h area under the curve for plasma triglycerides; LDL: Low-density lipoprotein; MMSE: Mini-mental state examination.
One of the ongoing controversies about fructose consumption in humans is related to the difficulty in identifying effects that are not strictly related to the simple consumption of an excess of daily calories. In a short (2 wk) dietary intervention study in NAFLD subjects, Browning et al[11] showed that carbohydrate restriction (< 20 g/d) was significantly more effective in reducing hepatic triglyceride content than the restriction of calories to 1200-1500 kcal/d (55% vs 28%, respectively), despite the fact that both interventions similarly reduced body weight (by about 4.3%). In a randomized intervention study comparing the consumption of sucrose-sweetened beverages (1 L/d for 6 mo) with other isocaloric beverages in obese subjects, Maersk et al[12] demonstrated that sucrose significantly increased triglyceride deposition, not only in liver, but also in skeletal muscle and visceral adipose tissue.
In another intervention study in healthy people who consumed a balanced diet supplemented with 150 g/d fructose or glucose, Silbernagel et al[13] showed that endogenous cholesterol synthesis was associated with visceral and liver fat content. However, in this study the strongest association was observed in glucose-consuming individuals. Nevertheless, in a well-conducted interventional study by Stanhope et al[14], subjects who consumed fructose (at 25% of energy requirements), either as such or as HFCS, but not glucose, showed an increased fasting concentration of low density lipoprotein (LDL) cholesterol. Fructose consumption also increased the 24-h triglyceride area under the curve and the fasting apolipoprotein (apo)B concentration.
In a prospective cohort study that analyzed 40 389 healthy men over 20 years of follow up, de Koning et al[15] clearly found an association between sugar-sweetened beverage consumption and an elevated risk of type 2 diabetes mellitus. Although it was suggested that fructose was mainly responsible for this association, Silbernagel et al[16] did not find any differences between fructose and glucose in the reduction of insulin sensitivity when these sugars were administered to 20 healthy subjects in a small intervention study. However, plasma triacylglycerol concentrations only increased significantly in the fructose group.
Fructose-induced obesity is closely related to type 2 diabetes mellitus. In a well- conducted intervention study by Cox et al[17] in overweight/obese male and female subjects, consumption of fructose (at 25% of energy requirements for 10 wk), but not glucose, clearly led to significant decreases in net postprandial fat oxidation and resting energy expenditure, thus contributing to the build-up of excess energy substrates. Furthermore, in one of the population segments at high risk of fructose-related obesity, Maier et al[18] demonstrated that a significant reduction in fructose and/or general sugar intake over a short period of time (3 mo) in overweight and obese children may reduce the body mass index. Mainly through increases in visceral fat, fructose-induced obesity is positively associated in adolescents with cardiometabolic risk markers, such as systolic blood pressure, fasting glucose, homeostasis model assessment-estimated insulin resistance index, and C-reactive protein[19].
Cardiovascular accidents originate as thrombi deposits on atheromatous plaques, which obstruct blood circulation[20]. Atherosclerosis is promoted by dyslipidemia, hypertension, and chronic low-grade inflammation. Besides increasing plasma triglycerides and LDL cholesterol[14], fructose seems to promote a proinflammatory milieu that favors atherosclerosis development. In an intervention study in overweight/obese subjects, Cox et al[21] demonstrated that fructose supplementation in liquid form (at 25% of energy requirements for 10 wk), but not glucose, clearly increases proinflammatory and prothrombotic mediators, such as monocyte chemoattractant protein-1, plasminogen activator inhibitor-1, and E-selectin. Furthermore, in a cross-sectional study including 2696 participants in the International Study of Macro/Micronutrients and Blood Pressure, Brown et al[22] found a direct association between sugar-sweetened beverage intake and systolic and diastolic blood pressure increases. Thus, fructose seems to contribute directly to increased prevalence in the three main risk factors for atherosclerosis-related cardiovascular diseases.
Besides the association between fructose consumption and common metabolic diseases, there is growing evidence of a relationship with other diseases, such as cancer and Alzheimer’s disease, that are also closely connected to the cellular metabolic status[23,24]. Very recently, Friberg et al[25] analyzed data on total sucrose and high-sugar food consumption during 18.4 years of follow-up in 61 226 women. They found a direct association with increased risk of endometrial cancer. In addition, high sugar intake has recently been associated with lower cognitive function among middle-aged and older Puerto Ricans without diabetes, in an analysis of data from a substudy of the Boston Puerto Rican Health Study 2004-9[26]. Although a high fructose diet does not affect spatial water maze learning and memory in female rats[27], the presence of NAFLD, which is one of the main consequences of fructose consumption in men and experimental animals, seems to somehow impair hippocampal-dependent memory in male rats[28].
Thus, overall, it seems that a high intake of sugar-sweetened beverages containing fructose places a metabolic burden on humans that facilitates the development of metabolic and cardiovascular diseases. What molecular mechanisms are involved in the production of these effects by fructose?
FRUCTOSE: MOLECULAR INSIGHTS FROM ANIMAL STUDIES
Fructose administration, mainly in drinking water, to laboratory rats and mice reproduces almost all of the features of metabolic syndrome and associated diseases in humans. These include left ventricular hypertrophy[29,30], insulin resistance[30-33], hypertension and related hyperuricemia[34-36], NAFLD[37,38], and metabolic syndrome itself[39].
London et al[40] have investigated the role of increased 11-hydroxysteroid dehydrogenase type 1 in liver and visceral adipose tissue in rats after fructose, but not glucose, consumption. Their results indicate that deregulated local glucocorticoid production plays a role at the onset of fructose-induced obesity[40]. Morris et al[41] put forward the hypothesis that the timing of fructose intake, mainly during the daylight period, could induce a mismatch in caloric consumption that favors the development of obesity and other metabolic alterations, at least in C57BL mice. Furthermore, several possible hypotheses related to the development of NALFD by fructose consumption have been pursued, including increased oxidative and inflammatory stress through nitric oxide synthase induction[42] and tumor necrosis factor α production[43]. A very concise and interesting review on the issue of possible molecular mechanisms involved in fructose induced lipogenesis was published in 2011[44].
In the past few years, our laboratory has researched three main issues regarding the molecular effects of fructose on liver fat and glucose metabolism: (1) possible drug therapies for the prevention and/or correction of fructose-induced metabolic pathologies; (2) molecular mechanisms that are responsible for early induction of glucose intolerance in female rats, as a previous step to developing insulin resistance and type 2 diabetes mellitus; and (3) molecular mechanisms leading to reduced peroxisome proliferator-activated receptor (PPAR) expression and activity in livers of female rats.
NAFLD is by far the most common cause of liver dysfunction. It is a spectrum of diseases ranging from fatty liver (steatosis) to steatohepatitis[45]. To date, the only effective treatment for NAFLD is modest calorie restriction and gradual weight loss[46]. Statins, hypolipidemic drugs that act by inhibiting the hydroxymethyl-glutaryl-CoA reductase enzyme, can be safely used in NAFLD patients[47], and there is evidence of improved liver histology in NAFLD patients treated with atorvastatin[48,49]. In a recently published study, we proposed a possible molecular mechanism for the therapeutic effect of atorvastatin on NAFLD[50]. Besides its well-known anti-inflammatory effect[51,52], atorvastatin reduced the liver expression of fructokinase in male rats supplemented with a 10% w/v solution of fructose for 14 d. Fructose consumption induces the expression of liver fructokinase in experimental animals[53,54] and in NAFLD patients[55]. As fructokinase is essential in controlling fructose metabolism, its induction establishes a vicious circle that progressively increases the deleterious effect of fructose on liver metabolism. Atorvastatin effectively facilitates the breaking of this circle. It contributes to an increase in fatty acid metabolism[56] and to a reduction in fatty acid synthesis that is driven by increased carbohydrate response element binding protein (ChREBP) transcriptional activity[57,58], which are necessary to revert the deposition of triglycerides in liver tissue.
We used the same experimental model of rats supplemented with a 10% w/v solution of fructose for 14 d, to show that female rats were more sensitive to the deleterious effect of fructose on glucose homeostasis than male rats, as only females showed signs of glucose intolerance[54]. In the same study, we found a marked reduction in insulin receptor substrate (IRS)-2 in the livers of fructose-supplemented female rats. IRS-2 is the main transducer of insulin signaling in hepatic tissue[59]. We have further pursued research of molecular changes related to fructose consumption in liver. We have confirmed that female rats supplemented with liquid fructose for 14 d, but not 7 d, are glucose intolerant (as shown by glucose tolerance test; GTT). This situation correlates with a decrease in the amount of IRS-2 protein expressed in liver. The same animals showed a marked increase in mammalian target of rapamycin (mTOR) activity and mitogen-activated protein kinase (p38-MAPK) activity.
p38-MAPK is a stress-related kinase[60] whose activity can be increased by the metabolic burden imposed by fructose metabolism in hepatocytes through two mechanisms: increased activity of protein phosphatase A2[54,61]; and the presence of bacterial toxins in blood, as a result of fructose-related alteration of the intestinal barrier permeability[43,62]. Furthermore, increased p38-MAPK activity, by phosphorylating the tuberous sclerosis 2 gene product or tuberin, could release its inhibitory activity on mTOR complex 1 (mTORC1)[63]. This would explain the observed increase in mTOR activity. The mTOR signaling pathway transduces information from different signals, such as growth factors, amino acids and energy overload of the cell[64]. Finally, as Guo et al[65] have shown that mTOR activation causes IRS-2 degradation, the increase in mTOR activity could be the final molecular factor resulting in a decreased liver expression of liver IRS-2 protein, as we have found[54].
Surprisingly, although female rats supplemented with liquid fructose for 14 d, had reduced liver expression of IRS-2, were hyperinsulinemic and showed an altered GTT, they were normoglycemic and their liver expression of gluconeogenic genes was unchanged (glucose-6-phosphatase) or even decreased (phosphoenolpyruvate carboxykinase). An explanation for this discrepancy can be found in a recent report indicating that X-box-binding protein (XBP)-1, an endoplasmic reticulum stress transcription factor, plays an essential role in maintaining plasma glucose concentration and glucose tolerance[66]. It has been described that mTORC1 activity increases the splicing of XBP-1[67], while p38-MAPK phosphorylates the spliced-derived protein, facilitating its nuclear localization and activity[68]. Indeed, in the liver samples from the fructose-fed rats used in our study, there was a marked increase in the spliced form of XBP-1 mRNA and nuclear protein, in accordance with the increased activity of mTOR and p38-MAPK. Thus, although the decreased expression of IRS-2 in liver represents an impairment of insulin signaling, the increased expression and activity of XBP-1 could compensate for this deficit and maintain appropriate gluconeogenesis (Figure 1). Data from skeletal muscle that indicate a deficit in adiponectin receptor and signaling in 14-d fructose-supplemented rats, could explain the fact that these animals do not have increased liver gluconeogenesis, but do have significant glucose tolerance impairment, as evaluated by an GTT.
Figure 1.
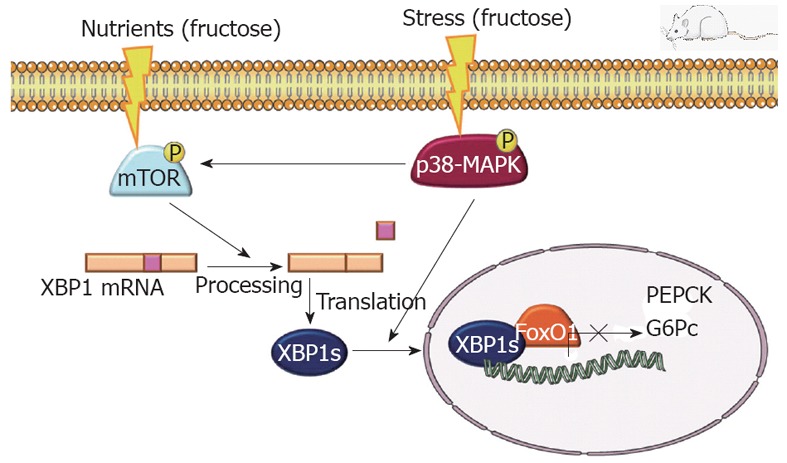
X-box-binding protein-1, an endoplasmic reticulum stress transcription factor, plays an essential role in maintaining plasma glucose concentration and glucose tolerance. Indeed, in the liver samples from the fructose-fed rats used in the study, there was a marked increase in the spliced form of X-box-binding protein (XBP)-1 mRNA and nuclear protein, in accordance with the increased activity of mammalian target of rapamycin (mTOR) activity and mitogen-activated protein kinase (p38-MAPK). Thus, although the decreased expression of insulin receptor substrate-2 in liver represents an impairment of insulin signaling, the increased expression and activity of XBP-1 could compensate for this deficit and maintain appropriate gluconeogenesis. PEPCK G6Pc: Phosphoenolpyruvate carboxykinase and glucose-6-phosphatase; FoxO1: Forkhead box protein O1.
We have previously shown that there is a state of leptin resistance in livers of male rats supplemented with liquid fructose. This results in increased binding of unphosphorylated active forkhead box protein (Fox)O1 to the transcription factor PPARα, which causes the inhibition of PPARα transcriptional activity and, as a consequence, reduces the liver capacity to oxidize fatty acids[57,58]. FoxO-1 is a transcription factor that is regulated by insulin and deeply involved in the control of liver gluconeogenesis[65]. Female rats equally supplemented with liquid fructose respond similarly with a reduction in liver PPARα activity and fatty acid oxidation. However, there is no involvement of leptin resistance and FoxO-1 interaction[54]. Thus, we have pursued the search for a possible molecular mechanism involved in the downregulation of the PPARα system in the liver of fructose-supplemented female rats.
ChREBP is a transcription factor responsible for inducing liver lipogenesis after carbohydrate ingestion[69]. We have previously reported that ChREBP is the main factor responsible for the increase in rat liver lipogenesis following fructose supplementation[50,54,57,58,70]. Unpublished results from our group indicate that there is also a close relationship between ChREBP activation and PPARα downregulation across different experimental settings (in vivo studies in female rats, cultured FaO and HepG2 hepatoma cells, primary cultures of human hepatocytes). It has been described that ChREBP controls the expression of regulator of G protein signaling (RGS) 16, a regulator of G protein signaling that inhibits hepatic fatty acid oxidation[71]. Although fructose markedly increased the mRNA level of RGS16 in livers of female rats, there was no change in the amount of the expressed protein. This suggests that increased expression of RGS16 is not involved in downregulation of the PPARα system. In rat hepatoma FaO cells cultured in the presence of a high concentration of fructose (25 mmol/L), we are performing knock-down experiments with siRNA against ChREBP to demonstrate clearly the direct involvement of ChREBP in the production of the fructose effect on the PPAR system. Confirmation of this hypothesis will indicate that fructose can simultaneously switch on liver fatty acid synthesis and switch off liver fatty acid catabolism by a single molecular mechanism: the intense activation of ChREBP. This would explain the effectiveness of fructose in inducing fatty liver and hypertriglyceridemia. We are also exploring possible mechanisms to explain why fructose stimulates the activity of ChREBP with such intensity. We have found that fructose supplementation markedly reduces the amount of the NAD-dependent deacetylase sirtuin 1 protein in livers of female rats, but not males. This reduction increases the amount of acetylated ChREBP. As it has been shown that ChREBP hyperacetylation increases its transcriptional activity[72], the reduction of sirtuin 1 expression could be one mechanism involved in the intense activation of ChREBP by fructose in the liver of female rats.
Footnotes
Supported by Fundació Privada Catalana de Nutrició i Lípids; and Grant SAF2010-15664 from the Spanish Ministry of Science and Innovation
Peer reviewer: Jian Wu, Associate Professor of Medicine, Internal Medicine/Transplant Research Program, University of California, Davis Medical Center, 4635 2nd Ave. Suite 1001, Sacramento, CA 95817, United States
S- Editor Gou SX L- Editor Kerr C E- Editor Li JY
References
- 1.Lustig RH, Schmidt LA, Brindis CD. Public health: The toxic truth about sugar. Nature. 2012;482:27–29. doi: 10.1038/482027a. [DOI] [PubMed] [Google Scholar]
- 2.Aller EE, Abete I, Astrup A, Martinez JA, van Baak MA. Starches, sugars and obesity. Nutrients. 2011;3:341–369. doi: 10.3390/nu3030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dallongeville J, Charbonnel B, Desprès JP. Sugar-sweetened beverages and cardiometabolic risk. Presse Med. 2011;40:910–915. doi: 10.1016/j.lpm.2011.02.042. [DOI] [PubMed] [Google Scholar]
- 4.Alegret M, Roglans N, Laguna JC. Fructose consumption and leptin resistance: What have we learnt from animal studies? In: Hemling RM, Belkin AT, editors. Leptin: Hormonal Functions, dysfunctions and clinical uses. Hauppauge, NY, USA: Nova Science Publishers Inc; 2011. pp. 210–230. [Google Scholar]
- 5.Hu FB, Malik VS. Sugar-sweetened beverages and risk of obesity and type 2 diabetes: epidemiologic evidence. Physiol Behav. 2010;100:47–54. doi: 10.1016/j.physbeh.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malik VS, Popkin BM, Bray GA, Després JP, Hu FB. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation. 2010;121:1356–1364. doi: 10.1161/CIRCULATIONAHA.109.876185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–264. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 8.Tappy L, Lê KA. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev. 2010;90:23–46. doi: 10.1152/physrev.00019.2009. [DOI] [PubMed] [Google Scholar]
- 9.Lanaspa MA, Tapia E, Soto V, Sautin Y, Sánchez-Lozada LG. Uric acid and fructose: potential biological mechanisms. Semin Nephrol. 2011;31:426–432. doi: 10.1016/j.semnephrol.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Soleimani M. Dietary fructose, salt absorption and hypertension in metabolic syndrome: towards a new paradigm. Acta Physiol (Oxf) 2011;201:55–62. doi: 10.1111/j.1748-1716.2010.02167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Browning JD, Baker JA, Rogers T, Davis J, Satapati S, Burgess SC. Short-term weight loss and hepatic triglyceride reduction: evidence of a metabolic advantage with dietary carbohydrate restriction. Am J Clin Nutr. 2011;93:1048–1052. doi: 10.3945/ajcn.110.007674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maersk M, Belza A, Stødkilde-Jørgensen H, Ringgaard S, Chabanova E, Thomsen H, Pedersen SB, Astrup A, Richelsen B. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: a 6-mo randomized intervention study. Am J Clin Nutr. 2012;95:283–289. doi: 10.3945/ajcn.111.022533. [DOI] [PubMed] [Google Scholar]
- 13.Silbernagel G, Lütjohann D, Machann J, Meichsner S, Kantartzis K, Schick F, Häring HU, Stefan N, Fritsche A. Cholesterol synthesis is associated with hepatic lipid content and dependent on fructose/glucose intake in healthy humans. Exp Diabetes Res. 2012;2012:361863. doi: 10.1155/2012/361863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanhope KL, Bremer AA, Medici V, Nakajima K, Ito Y, Nakano T, Chen G, Fong TH, Lee V, Menorca RI, et al. Consumption of fructose and high fructose corn syrup increase postprandial triglycerides, LDL-cholesterol, and apolipoprotein-B in young men and women. J Clin Endocrinol Metab. 2011;96:E1596–E1605. doi: 10.1210/jc.2011-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Koning L, Malik VS, Rimm EB, Willett WC, Hu FB. Sugar-sweetened and artificially sweetened beverage consumption and risk of type 2 diabetes in men. Am J Clin Nutr. 2011;93:1321–1327. doi: 10.3945/ajcn.110.007922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silbernagel G, Machann J, Unmuth S, Schick F, Stefan N, Häring HU, Fritsche A. Effects of 4-week very-high-fructose/glucose diets on insulin sensitivity, visceral fat and intrahepatic lipids: an exploratory trial. Br J Nutr. 2011;106:79–86. doi: 10.1017/S000711451000574X. [DOI] [PubMed] [Google Scholar]
- 17.Cox CL, Stanhope KL, Schwarz JM, Graham JL, Hatcher B, Griffen SC, Bremer AA, Berglund L, McGahan JP, Havel PJ, et al. Consumption of fructose-sweetened beverages for 10 weeks reduces net fat oxidation and energy expenditure in overweight/obese men and women. Eur J Clin Nutr. 2012;66:201–208. doi: 10.1038/ejcn.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maier IB, Stricker L, Ozel Y, Wagnerberger S, Bischoff SC, Bergheim I. A low fructose diet in the treatment of pediatric obesity: a pilot study. Pediatr Int. 2011;53:303–308. doi: 10.1111/j.1442-200X.2010.03248.x. [DOI] [PubMed] [Google Scholar]
- 19.Pollock NK, Bundy V, Kanto W, Davis CL, Bernard PJ, Zhu H, Gutin B, Dong Y. Greater fructose consumption is associated with cardiometabolic risk markers and visceral adiposity in adolescents. J Nutr. 2012;142:251–257. doi: 10.3945/jn.111.150219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010;74:213–220. doi: 10.1253/circj.cj-09-0706. [DOI] [PubMed] [Google Scholar]
- 21.Cox CL, Stanhope KL, Schwarz JM, Graham JL, Hatcher B, Griffen SC, Bremer AA, Berglund L, McGahan JP, Keim NL, et al. Circulating concentrations of monocyte chemoattractant protein-1, plasminogen activator inhibitor-1, and soluble leukocyte adhesion molecule-1 in overweight/obese men and women consuming fructose- or glucose-sweetened beverages for 10 weeks. J Clin Endocrinol Metab. 2011;96:E2034–E2038. doi: 10.1210/jc.2011-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown IJ, Stamler J, Van Horn L, Robertson CE, Chan Q, Dyer AR, Huang CC, Rodriguez BL, Zhao L, Daviglus ML, et al. Sugar-sweetened beverage, sugar intake of individuals, and their blood pressure: international study of macro/micronutrients and blood pressure. Hypertension. 2011;57:695–701. doi: 10.1161/HYPERTENSIONAHA.110.165456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Heaney AP. Refined fructose and cancer. Expert Opin Ther Targets. 2011;15:1049–1059. doi: 10.1517/14728222.2011.588208. [DOI] [PubMed] [Google Scholar]
- 24.Seneff S, Wainwright G, Mascitelli L. Nutrition and Alzheimer’s disease: the detrimental role of a high carbohydrate diet. Eur J Intern Med. 2011;22:134–140. doi: 10.1016/j.ejim.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 25.Friberg E, Wallin A, Wolk A. Sucrose, high-sugar foods, and risk of endometrial cancer--a population-based cohort study. Cancer Epidemiol Biomarkers Prev. 2011;20:1831–1837. doi: 10.1158/1055-9965.EPI-11-0402. [DOI] [PubMed] [Google Scholar]
- 26.Ye X, Gao X, Scott T, Tucker KL. Habitual sugar intake and cognitive function among middle-aged and older Puerto Ricans without diabetes. Br J Nutr. 2011;106:1423–1432. doi: 10.1017/S0007114511001760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruggeman EC, Li C, Ross AP, Doherty JM, Williams BF, Frantz KJ, Parent MB. A high fructose diet does not affect amphetamine self-administration or spatial water maze learning and memory in female rats. Pharmacol Biochem Behav. 2011;99:356–364. doi: 10.1016/j.pbb.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 28.Ross AP, Bruggeman EC, Kasumu AW, Mielke JG, Parent MB. Non-alcoholic fatty liver disease impairs hippocampal-dependent memory in male rats. Physiol Behav. 2012;106:133–141. doi: 10.1016/j.physbeh.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 29.Bouchard-Thomassin AA, Lachance D, Drolet MC, Couet J, Arsenault M. A high-fructose diet worsens eccentric left ventricular hypertrophy in experimental volume overload. Am J Physiol Heart Circ Physiol. 2011;300:H125–H134. doi: 10.1152/ajpheart.00199.2010. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Zhang BH, Yu YR, Tang CS, Qi YF. Adrenomedullin protects against fructose-induced insulin resistance and myocardial hypertrophy in rats. Peptides. 2011;32:1415–1421. doi: 10.1016/j.peptides.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 31.Zhou YB, Zhang J, Cai Y, Teng X, Duan XH, Song JQ, Du J, Tang CS, Qi YF. Insulin resistance induces medial artery calcification in fructose-fed rats. Exp Biol Med (Maywood) 2012;237:50–57. doi: 10.1258/ebm.2011.011252. [DOI] [PubMed] [Google Scholar]
- 32.Guo Q, Mori T, Jiang Y, Hu C, Ohsaki Y, Yoneki Y, Nakamichi T, Ogawa S, Sato H, Ito S. Losartan modulates muscular capillary density and reverses thiazide diuretic-exacerbated insulin resistance in fructose-fed rats. Hypertens Res. 2012;35:48–54. doi: 10.1038/hr.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hein GJ, Chicco A, Lombardo YB. Fish oil normalizes plasma glucose levels and improves liver carbohydrate metabolism in rats fed a sucrose-rich diet. Lipids. 2012;47:141–150. doi: 10.1007/s11745-011-3623-4. [DOI] [PubMed] [Google Scholar]
- 34.Silva RJ, Bernardes N, Brito Jde O, Sanches IC, Irigoyen MC, De Angelis K. Simvastatin-induced cardiac autonomic control improvement in fructose-fed female rats. Clinics (Sao Paulo) 2011;66:1793–1796. doi: 10.1590/S1807-59322011001000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vasudevan H, Yuen VG, McNeill JH. Testosterone-dependent increase in blood pressure is mediated by elevated Cyp4A expression in fructose-fed rats. Mol Cell Biochem. 2012;359:409–418. doi: 10.1007/s11010-011-1035-7. [DOI] [PubMed] [Google Scholar]
- 36.Chen L, Lan Z, Zhou Y, Li F, Zhang X, Zhang C, Yang Z, Li P. Astilbin attenuates hyperuricemia and ameliorates nephropathy in fructose-induced hyperuricemic rats. Planta Med. 2011;77:1769–1773. doi: 10.1055/s-0030-1271135. [DOI] [PubMed] [Google Scholar]
- 37.Kunde SS, Roede JR, Vos MB, Orr ML, Go YM, Park Y, Ziegler TR, Jones DP. Hepatic oxidative stress in fructose-induced fatty liver is not caused by sulfur amino acid insufficiency. Nutrients. 2011;3:987–1002. doi: 10.3390/nu3110987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roncal-Jimenez CA, Lanaspa MA, Rivard CJ, Nakagawa T, Sanchez-Lozada LG, Jalal D, Andres-Hernando A, Tanabe K, Madero M, Li N, et al. Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metabolism. 2011;60:1259–1270. doi: 10.1016/j.metabol.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen GC, Huang CY, Chang MY, Chen CH, Chen SW, Huang CJ, Chao PM. Two unhealthy dietary habits featuring a high fat content and a sucrose-containing beverage intake, alone or in combination, on inducing metabolic syndrome in Wistar rats and C57BL/6J mice. Metabolism. 2011;60:155–164. doi: 10.1016/j.metabol.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 40.London E, Castonguay TW. High fructose diets increase 11β-hydroxysteroid dehydrogenase type 1 in liver and visceral adipose in rats within 24-h exposure. Obesity (Silver Spring) 2011;19:925–932. doi: 10.1038/oby.2010.284. [DOI] [PubMed] [Google Scholar]
- 41.Morris M, Araujo IC, Pohlman RL, Marques MC, Rodwan NS, Farah VM. Timing of fructose intake: an important regulator of adiposity. Clin Exp Pharmacol Physiol. 2012;39:57–62. doi: 10.1111/j.1440-1681.2011.05636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spruss A, Kanuri G, Uebel K, Bischoff SC, Bergheim I. Role of the inducible nitric oxide synthase in the onset of fructose-induced steatosis in mice. Antioxid Redox Signal. 2011;14:2121–2135. doi: 10.1089/ars.2010.3263. [DOI] [PubMed] [Google Scholar]
- 43.Kanuri G, Spruss A, Wagnerberger S, Bischoff SC, Bergheim I. Role of tumor necrosis factor α (TNFα) in the onset of fructose-induced nonalcoholic fatty liver disease in mice. J Nutr Biochem. 2011;22:527–534. doi: 10.1016/j.jnutbio.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 44.Samuel VT. Fructose induced lipogenesis: from sugar to fat to insulin resistance. Trends Endocrinol Metab. 2011;22:60–65. doi: 10.1016/j.tem.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Tessari P, Coracina A, Cosma A, Tiengo A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2009;19:291–302. doi: 10.1016/j.numecd.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 46.Ahmed MH, Byrne CD. Current treatment of non-alcoholic fatty liver disease. Diabetes Obes Metab. 2009;11:188–195. doi: 10.1111/j.1463-1326.2008.00926.x. [DOI] [PubMed] [Google Scholar]
- 47.Browning JD. Statins and hepatic steatosis: perspectives from the Dallas Heart Study. Hepatology. 2006;44:466–471. doi: 10.1002/hep.21248. [DOI] [PubMed] [Google Scholar]
- 48.Kiyici M, Gulten M, Gurel S, Nak SG, Dolar E, Savci G, Adim SB, Yerci O, Memik F. Ursodeoxycholic acid and atorvastatin in the treatment of nonalcoholic steatohepatitis. Can J Gastroenterol. 2003;17:713–718. doi: 10.1155/2003/857869. [DOI] [PubMed] [Google Scholar]
- 49.Gómez-Domínguez E, Gisbert JP, Moreno-Monteagudo JA, García-Buey L, Moreno-Otero R. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment Pharmacol Ther. 2006;23:1643–1647. doi: 10.1111/j.1365-2036.2006.02926.x. [DOI] [PubMed] [Google Scholar]
- 50.Vilà L, Rebollo A, Ađalsteisson GS, Alegret M, Merlos M, Roglans N, Laguna JC. Reduction of liver fructokinase expression and improved hepatic inflammation and metabolism in liquid fructose-fed rats after atorvastatin treatment. Toxicol Appl Pharmacol. 2011;251:32–40. doi: 10.1016/j.taap.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 51.Ortego M, Bustos C, Hernández-Presa MA, Tuñón J, Díaz C, Hernández G, Egido J. Atorvastatin reduces NF-kappaB activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis. 1999;147:253–261. doi: 10.1016/s0021-9150(99)00193-8. [DOI] [PubMed] [Google Scholar]
- 52.Planavila A, Laguna JC, Vázquez-Carrera M. Atorvastatin improves peroxisome proliferator-activated receptor signaling in cardiac hypertrophy by preventing nuclear factor-kappa B activation. Biochim Biophys Acta. 2005;1687:76–83. doi: 10.1016/j.bbalip.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Korieh A, Crouzoulon G. Dietary regulation of fructose metabolism in the intestine and in the liver of the rat. Duration of the effects of a high fructose diet after the return to the standard diet. Arch Int Physiol Biochim Biophys. 1991;99:455–460. [PubMed] [Google Scholar]
- 54.Vilà L, Roglans N, Perna V, Sánchez RM, Vázquez-Carrera M, Alegret M, Laguna JC. Liver AMP/ATP ratio and fructokinase expression are related to gender differences in AMPK activity and glucose intolerance in rats ingesting liquid fructose. J Nutr Biochem. 2011;22:741–751. doi: 10.1016/j.jnutbio.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, Johnson RJ, Abdelmalek MF. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol. 2008;48:993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alegret M, Ferrando R, Vázquez M, Adzet T, Merlos M, Laguna JC. Relationship between plasma lipids and palmitoyl-CoA hydrolase and synthetase activities with peroxisomal proliferation in rats treated with fibrates. Br J Pharmacol. 1994;112:551–556. doi: 10.1111/j.1476-5381.1994.tb13109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roglans N, Vilà L, Farré M, Alegret M, Sánchez RM, Vázquez-Carrera M, Laguna JC. Impairment of hepatic Stat-3 activation and reduction of PPARalpha activity in fructose-fed rats. Hepatology. 2007;45:778–788. doi: 10.1002/hep.21499. [DOI] [PubMed] [Google Scholar]
- 58.Vilà L, Roglans N, Alegret M, Sánchez RM, Vázquez-Carrera M, Laguna JC. Suppressor of cytokine signaling-3 (SOCS-3) and a deficit of serine/threonine (Ser/Thr) phosphoproteins involved in leptin transduction mediate the effect of fructose on rat liver lipid metabolism. Hepatology. 2008;48:1506–1516. doi: 10.1002/hep.22523. [DOI] [PubMed] [Google Scholar]
- 59.Kim SK, Novak RF. The role of intracellular signaling in insulin-mediated regulation of drug metabolizing enzyme gene and protein expression. Pharmacol Ther. 2007;113:88–120. doi: 10.1016/j.pharmthera.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 61.Hanke N, Scheibe RJ, Manukjan G, Ewers D, Umeda PK, Chang KC, Kubis HP, Gros G, Meissner JD. Gene regulation mediating fiber-type transformation in skeletal muscle cells is partly glucose- and ChREBP-dependent. Biochim Biophys Acta. 2011;1813:377–389. doi: 10.1016/j.bbamcr.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 62.Volynets V, Spruss A, Kanuri G, Wagnerberger S, Bischoff SC, Bergheim I. Protective effect of bile acids on the onset of fructose-induced hepatic steatosis in mice. J Lipid Res. 2010;51:3414–3424. doi: 10.1194/jlr.M007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y, Inoki K, Vacratsis P, Guan KL. The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J Biol Chem. 2003;278:13663–13671. doi: 10.1074/jbc.M300862200. [DOI] [PubMed] [Google Scholar]
- 64.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 65.Guo S, Dunn SL, White MF. The reciprocal stability of FOXO1 and IRS2 creates a regulatory circuit that controls insulin signaling. Mol Endocrinol. 2006;20:3389–3399. doi: 10.1210/me.2006-0092. [DOI] [PubMed] [Google Scholar]
- 66.Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, Lee J, Fisher SJ, White MF, Biddinger SB, et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med. 2011;17:356–365. doi: 10.1038/nm.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pfaffenbach KT, Nivala AM, Reese L, Ellis F, Wang D, Wei Y, Pagliassotti MJ. Rapamycin inhibits postprandial-mediated X-box-binding protein-1 splicing in rat liver. J Nutr. 2010;140:879–884. doi: 10.3945/jn.109.119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee J, Sun C, Zhou Y, Lee J, Gokalp D, Herrema H, Park SW, Davis RJ, Ozcan U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat Med. 2011;17:1251–1260. doi: 10.1038/nm.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006;4:107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 70.Rodríguez-Calvo R, Barroso E, Serrano L, Coll T, Sánchez RM, Merlos M, Palomer X, Laguna JC, Vázquez-Carrera M. Atorvastatin prevents carbohydrate response element binding protein activation in the fructose-fed rat by activating protein kinase A. Hepatology. 2009;49:106–115. doi: 10.1002/hep.22570. [DOI] [PubMed] [Google Scholar]
- 71.Pashkov V, Huang J, Parameswara VK, Kedzierski W, Kurrasch DM, Tall GG, Esser V, Gerard RD, Uyeda K, Towle HC, et al. Regulator of G protein signaling (RGS16) inhibits hepatic fatty acid oxidation in a carbohydrate response element-binding protein (ChREBP)-dependent manner. J Biol Chem. 2011;286:15116–15125. doi: 10.1074/jbc.M110.216234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, Dentin R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest. 2010;120:4316–4331. doi: 10.1172/JCI41624. [DOI] [PMC free article] [PubMed] [Google Scholar]