

Summary
It has been proposed that the reaction cycle of ATP binding cassette (ABC) transporters is driven by dimerization of their ABC motor domains upon binding ATP at their mutual interface. However, no such ATP sandwich complex has been observed for an ABC from an ABC transporter. In this paper, we report the crystal structure of a stable dimer formed by the E171Q mutant of the MJ0796 ABC, which is hydrolytically inactive due to mutation of the catalytic base. The structure shows a symmetrical dimer in which two ATP molecules are each sandwiched between the Walker A motif in one subunit and the LSGGQ signature motif in the other subunit. These results establish the stereochemical basis of the power stroke of ABC transporter pumps.
Introduction
The ATP binding cassette (ABC) transporter superfamily consists of mechanochemically coupled integral membrane protein complexes in which ATP binding and hydrolysis drive solute transport against a cellular concentration gradient (Higgins, 1992; Paulsen et al., 1998; Holland and Blight, 1999; Davidson, 2002). These proteins play a causative role in a number of human diseases, notably cystic fibrosis (Riordan et al., 1989) and multidrug resistance by tumor cells and microbial pathogens (Gottesman et al., 1996). Genome sequence data suggests that ABC transporters comprise the most prevalent protein family in many organisms, representing 5% of the open reading frames in E. coli (Linton and Higgins, 1998; Dassa et al., 1999).
Functional ABC transporters contain four structural domains: two transmembrane (TM) domains that determine substrate specificity and trajectory and two stereotyped ATP binding cassettes (ABC’s) or ATPase motor domains that are peripherally associated with the cytoplasmic portion of the TM domains (Higgins, 1992; Gottesman et al., 1996; Holland and Blight, 1999). In prokaryotes, these domains are often expressed as separate polypeptides. However, in some prokaryotic transporters and most eukaryotic transporters, they are fused together in a single polypeptide. A low-resolution crystal structure has been determined for the MsbA integral membrane protein (Chang and Roth, 2001), and seven high-resolution crystal structures have been determined for six different ABC’s isolated from their cognate TM domains (Hung et al., 1998; Diederichs et al., 2000; Yuan et al., 2001; Karpowich et al., 2001; Gaudet and Wiley, 2001). These structures have shown that the ABC contains three subdomains (Karpowich et al., 2001): an F1-type ATP binding core containing the Walker A and Walker B ATPase motifs, an antiparallel β sheet subdomain that interacts with and positions the base and ribose of the nucleotide, and an α-helical subdomain that contains the “LSGGQ” signature motif that is the hallmark of the ABC transporters superfamily. Although the LSGGQ signature motif is solvent exposed and does not interact with the nucleotide in ABC monomers, mutations in this sequence impair the ATPase activity of ABC transporters (Koronakis et al., 1995; Schmees et al., 1999; Davidson, 2002).
A structural and thermodynamic model for the mechanochemistry of ABC transporters has been proposed based on experiments with Rad50, a remotely related mechanoenzyme involved in DNA repair (Hopfner et al., 2000). This model hypothesizes that ATP-driven dimerization of ABC’s represents the force-transducing “power stroke” of ABC transporter pumps. This model is based on the observation that Rad50 forms a stable dimer upon binding AMPPNP, a nonhydrolyzable analog of ATP. The crystal structure of the Rad50 dimer shows two AMPPNP molecules sandwiched between the Walker A motif in one subunit and the LSGGQ signature motif in the other subunit, i.e., between two sequence/structural motifs that are both rigorously conserved in true ABCs from ABC transporters. The nucleotide-mediated intersubunit interactions in this structure are similar to those predicted by Jones and George (1999) based on analysis of the structure of an ABC monomer.
However, there are four observations that have caused the Rad50 model for the mechanochemistry of ABC transporters to remain controversial. First, isolated wild-type ATP binding cassettes from ABC transporters have not been observed to dimerize upon binding either ATP or nonhydrolyzable analogs (Nikaido et al., 1997; Schneider et al., 1994; Greller et al., 1999). Second, no such nucleotide sandwich complex (i.e., ATP-bridged dimer) has been observed in any of the crystal structures of isolated ABCs that have been reported in various nucleotide-bound states (Hung et al., 1998; Diederichs et al., 2000; Yuan et al., 2001; Karpowich et al., 2001; Gaudet and Wiley, 2001). Third, the only dimer observed in the low-resolution crystal structure of the MsbA integral membrane protein positions the ABCs in a geometry where they are incapable of forming such a complex (Chang and Roth, 2001; Thomas and Hunt, 2001). And finally, although Rad50 shares an F1-type α/β ATP binding core with true ABCs, its structure has diverged quite considerably in the antiparallel β subdomain and the α-helical subdomain (Yuan et al., 2001). While Rad50 does possess the LSGGQ signature motif, its position is altered relative to that observed in true ABCs by a 5 residue expansion in the α helix that it caps. Furthermore, the other elements contacting the nucleotide in the symmetry-related subunit in the Rad50 dimer are derived from the regions where its structure diverges from that of true ABCs, i.e., the antiparallel β subdomain and the subdomain that replaces the α-helical subdomain. Considering these structural differences in combination with the failure to observe such a dimer of a true ABC, the Rad50 model for the structural mechanics of ABC transporters has remained controversial.
In this paper, we provide proof of the Rad50 model for ABC mechanochemistry by determining the crystal structure of a stable ATP sandwich dimer of a true ABC from an ABC transporter.
Results and Discussion
Mutation of the Hydrolytic Base in MJ0796 Leads to Stable Dimerization upon Binding ATP
A dimer of the MJ0796 ABC (Yuan et al., 2001) was obtained exploiting the observation of Thomas and coworkers that a glutamate to glutamine mutation in the hydrolytic base in the active site of an ABC produces a hydrolysis-deficient protein that forms a stable dimer upon binding ATP (Moody et al., 2002). The hydrolytic glutamate residue (171 in MJ0796) is located in the DEPT sequence that terminates the Walker B motif (Walker et al., 1982) (see Figure 3 below). The dimer formed upon ATP binding to the E171Q mutant of MJ0796 is sufficiently stable to remain associated during gel filtration chromatography in the absence of nucleotide, a result also observed for the equivalent mutant of the MJ1267 ABC (Moody et al., 2002). These results are consistent with those obtained when the hydrolytic bases in the ABCs of the multidrug resistance P glycoprotein were mutated in an equivalent manner (Urbatsch et al., 2000). In this case, both hydrolysis and nucleotide release were strongly inhibited, which would be consistent with sequestration of the nucleotides in a tightly locked nucleotide sandwich complex.
Figure 3. Nucleotide Contacts and Intersubunit Contacts in the MJ0796-E171Q Dimer.

(A) Contact diagram. The residues are color-coded according to subdomain of origin as in Figure 2A, with plain text or italics used to indicate residues from the subunit whose Walker A motif or LSGGQ signature motif, respectively, is interacting with the nucleotide. The putative hydrolytic water is shown in cyan. The outermost columns show the protein-protein contacts (CCP4, 1994) across the dimer interface. Van der Waals contacts (≤3.6 Å) are represented by thin black lines, while H bonds (≤3.3 Å) are represented by thick lines colored to correspond with the subdomain or molecule containing the donor atom. Only the strongest (i.e., shortest) van der Waals contact between any two groups is shown (so the number of contacts shown in this panel does not match the total number cited in the text). The contacts to the Na+ atom are represented by thick yellow lines, and the aromatic stacking interaction between Tyr11 and the adenine base is represented by the very thick dashed line. Although Gln171 acts as an H bond donor to the putative hydrolytic water, the glutamate at this position in the wild-type protein will be one of three competing acceptor groups within H bonding distance. The molecular contacts are nearly identical at the two active sites in the dimer.
(B) Sequence/structural alignment showing the residues making intersubunit contacts. The “Contacts” line above the sequence alignment indicates whether residues contact the nucleotide (n), the other protein subunit (p), or both (b), with plain text or italics used to indicate residues from the subunit whose Walker A motif or LSGGQ signature motif, respectively, is interacting with the nucleotide. The secondary structural elements in MJ0796 are color-coded according to subdomain of origin, with arrows, sinusoidal lines, and saw-toothed lines used to represent β strands, α helices, and 310-helices, respectively. The classification (Wilmot and Thornton, 1990) of well-defined β turns is indicated below the structural schematic, and the label “blg” indicates the positions of structurally conserved β bulges in the antiparallel β subdomain.
Figure 1 demonstrates the nucleotide-dependent dimerization of the E171Q mutant of MJ0796. ATP was titrated onto the Y11W variant of the MJ0796 ABC (at a 1 μM concentration) either with or without the E171Q mutation in a Mg-free buffer containing 200 mM NaCl, 1 mM Na-EDTA, 50 mM Tris-Cl (pH 7.5). The Y11W mutation allows tryptophan fluorescence anisotropy measurements to be used to characterize both the nucleotide binding and dimerization of this otherwise tryptophan-free ABC, and MJ0796 variants containing either Tyr or Trp at position 11 behave identically in gel filtration dimerization assays. The wild-type MalK ABC has a single tryptophan residue at the equivalent position, which makes stacking interactions with the adenine moeity in all nucleotide-bound ABC crystal structures (Hung et al., 1998; Yuan et al., 2001; Karpowich et al., 2001; Gaudet and Wiley, 2001). Previous experiments have shown that nucleotide binding to wild-type MalK produces approximately a 50% quenching in tryptophan fluorescence emission intensity (Schneider et al., 1994), and a similar effect is observed with the Y11W mutant of MJ0796 (bottom panel in Figure 1). These data show that the E171Q mutant of MJ0796 binds ATP with 14 μM affinity and a Hill coefficient of 1.9. The nucleotide is likely to be bound with Na+ substituted for the usual Mg2+ counterion cofactor, given the composition of the buffer used in this experiment. The corresponding anisotropy data (top panel in Figure 1) show a change in the rotational correlation time of the fluorophore during the Na-ATP titration, derived primarily from the slowing of the overall rate of molecular rotation upon dimerization. The midpoint of the anisotropy titration is slightly higher than that of the emission intensity titration, indicating that some population of nucleotide-bound monomers is present at low Na-ATP concentrations during the coupled nucleotide binding and dimerization reactions.
Figure 1. Na-ATP-Dependent Dimerization of the E171Q Mutant of MJ0796.

Tryptophan fluorescence anisotropy titrations were conducted at 24°C on the Y11W mutant of MJ0796 either with or without the hydrolytically deficient E171Q mutation. The protein was used at a 1 μM concentration. Relative total fluorescence values are shown in the bottom panel, while fluorescence anisotropy values from the same titrations are shown in the top panel. The variables “Kd” and “n” refer to the dissociation constant and the cooperativity, respectively, and were determined by nonlinear curve fitting (solid lines).
Notably, the ABC without the E171Q mutation neither binds Na-ATP nor dimerizes in the nucleotide concentration range shown in Figure 1. Titrations employing higher nucleotide concentrations in the absence of Mg2+ show this protein will bind Na-ATP with a Kd of ~800 μM (data not shown). However, it binds Mg-ATP with much higher affinity, exhibiting a dissociation constant similar to that of the E171Q mutant for Na-ATP (data not shown). Therefore, the E171Q mutation changes the counterion specificity of the active site and enhances the affinity of the ABC for Na-ATP by approximately two orders of magnitude. Experiments on the ATP-dependent dimerization of the equivalent E179Q mutant of the MJ1267 ABC suggest that its counterion specificity has been altered in a similar manner (Moody et al., 2002). These results indicate that the electrostatic balance at the active site of the ABC can produce dramatic modulation of intermolecular interactions.
Dimer Structure
The crystal structure of the stable ATP-bound MJ0796 dimer was determined by molecular replacement and refined to a working R factor of 18.5% and a free R factor of 25.1% at 1.9 Å resolution (Table 1). The structure shows a symmetrical ATP sandwich complex similar to that observed in Rad50 (Figure 2). The two LSGGQ signature motifs complete the ATPase active sites in the ABC dimer by capping the nucleotide triphosphates bound to the Walker A (P loop) (Walker et al., 1982) sequences in the symmetry-related subunits. Almost identical protein-nucleotide and protein-protein contacts are observed in the two active sites, even though they are related by noncrystallographic symmetry. A comprehensive description of the intermolecular contacts in a single active site is presented in Figure 3.
Table 1.
MJ0796-E171Q ATP Sandwich Dimer Refinement
| Space group C2221 | 80.9 × 106.3 × 116.9 Å (90°, 90°, 90°) at 100 K |
|
| |
| Refinement data quality
| |
| Resolution | 1.9 Å |
| Rsym | 11.4%a |
| Mean redundancy | 6.9 |
| Completeness | 100.0%b 89.3%c |
| Mean I/σI | 14.1 |
|
| |
| Crystallographic residuals (F ≥ σF)
| |
| Rfree | 25.1% |
| Rwork | 18.5% |
|
| |
| Model quality
| |
| Bond length deviation (rms) | 0.011 Å |
| Bond angle deviation (rms) | 1.5° |
|
| |
| Ramachandran distribution
| |
| Core | 93.1% |
| Allowed | 6.4% |
| Disallowed | 0.5% |
Model contents: 462 protein residues, 2 Na-ATP, 2 isopropanol, 417 water molecules. Standard definitions are used for all of the parameters (Drenth, 1994). The data collection statistics come from SCALEPACK (Otwinowski and Minor, 1997). The value of <I>/<σI> is 1.85 in the limiting resolution shell, which is 99.8% complete for all measured reflections and 39.5% complete for reflections with I ≥ 2σI. The refinement/geometric statistics come from X-PLOR (Brunger, 1992b), and the Ramachandran analysis was performed with PROCHECK (Laskowski et al., 1993).
I ≥ −σI for individual observations.
All measured reflections.
I ≥ σI for merged reflections.
Figure 2. Crystal Structure of the MJ0796-E171Q ATP Sandwich Dimer.

The stereo ribbon diagrams (Kraulis, 1991; Merritt and Bacon, 1997) are color coded according to subdomain organization, with the F1-type ATP binding core shown in orange, the antiparallel β subdomain in green, the α-helical subdomain in blue, and the γ-phosphate linker in red. This color scheme is equivalent to that used by Yuan et al. (2001), except for the presentation of the γ-phosphate linker in red. See Figure 3B for the location of the subdomains in the primary structure of the protein.
(A) Overall dimer structure. The bound Na-ATP molecules are shown in space-filling representations, with the nucleotides shown in magenta and the cation cofactors shown in yellow. Sidechains contacting either the nucleotides or the other subunit are shown in ball-and-stick representations, color-coded according to the subdomain of origin; see Figure 3A for a detailed contact diagram including the identity of all of these residues. As shown here, the proximal face of the ABC contacts the TM domains in the crystal structure of MsbA (Chang and Roth, 2001).
(B) Active site stereochemistry. Lighter shades are used to indicate segments coming from the “A” subunit whose Walker A motif is interacting with the nucleotide, while darker shades are used to indicate segments coming from the “B” subunit whose LSGGQ signature motif is interacting with the nucleotide. The dotted lines indicate H bonds and are colored to correspond to the donor group participating in the interaction. The yellow sphere indicates the Na+ cofactor of the nucleotide. The turquoise spheres indicate water molecules, with the rightmost one being the putative hydrolytic water.
Dimer formation buries 1100 Å2 of solvent-accessible surface area (Connolly, 1983; CCP4, 1994) per monomer, with just less than half of this surface coming from the two nucleotides (assuming that they are bound to the Walker A motif before dimerization, as observed in the previous ABC crystal structures). The shape-complementarity of the interface is 0.75, equivalent to the highest value typically observed for physiological protein-protein complexes (Lawrence and Colman, 1993). The intersubunit interaction (CCP4, 1994) involves 72 van der Waals contacts (≤3.6 Å) and 12 hydrogen bonds (H bonds, ≤3.3 Å) (counting the contacts at both nucleotide binding sites but excluding contacts to interfacial waters). Thirty-eight of the van der Waals contacts and 8 of the H bonds occur to one of the bound nucleotides, and 6 of the 13 residues making direct intersubunit protein-protein contacts also contact one of the nucleotides. While it is clear that the two nucleotides make major contributions to the intersubunit interactions, only eight van der Waals contacts and four H bonds in the dimer interface involve their γ-phosphates, even though these groups have a determining influence on the stability of the interface (Moody et al., 2002).
While the most important structural features of the Rad50 dimer (Hopfner et al., 2000) are conserved in the MJ0796 ATP sandwich dimer, the detailed stereochemistry of the intersubunit interaction is different. Closely similar contacts are made to the nucleotide triphosphates across the intersubunit interfaces in the two dimers. However, because of structural differences in the antiparallel β subdomain, the ribose and especially the adenine base of the nucleotide adopt a different conformation in Rad50 compared to true ABCs (Gaudet and Wiley, 2001; Yuan et al., 2001). Although the two closest contacts to these groups across the intersubunit interfaces (Figure 3A) are made by structurally homologous residues, the identity and conformation of the residue contacting the adenine is different (Gln145 in MJ0796 versus Phe791 in Rad50), and the secondary contacts to these groups are made by residues from divergent segments of the proteins. Moreover, of the 13 residues making intersubunit protein-protein contacts in MJ0796, only His204 and the 3 residues in the loop following the Walker B make stereochemically similar intersubunit interactions in Rad50 (data not shown). However, the largest difference between the dimers concerns the relative location and orientation of the two ATPase active sites caused by differences in the spatial separation between the Walker A motif and the LSGGQ signature motif in the different proteins. After aligning the active site in one of the subunits in the MJ0796 dimer with the equivalent region in one of the subunits of the Rad50 dimer (based on least-squares superposition of the β strands and Walker A helix in the ATP binding core), the ATP binding cores in the other subunits are rotated relative to one another by 28°, and the triphosphates of the nucleotides in the other active sites are displaced by 5 Å.
A Na+ Ion Cofactor Bound to the ATP
Biochemical (Figure 1) and crystallographic criteria indicate that the cation cofactors of the nucleotides in the MJ0796-E171Q dimer are Na+ rather than Mg2+. The MJ0796-E171Q dimer crystals grew in a Mg2+-free solution containing ≥50 mM Na+ and ≥0.5 mM EDTA. Refinement of the putative cations as water molecules yielded B factors of ~12 Å2 compared to values ranging from 18 to 27 Å2 for their four protein/nucleotide ligands. Refinement of these atoms as either Na+ or Mg2+ yielded B factors of ~23 Å2. However, the contact distances between the cation and its six ligands (2.2, 2.2, 2.5, and 2.8 Å for protein/nucleotide ligands and 2.1 and 2.1 Å for water ligands) were more consistent with those observed for Na+ in the small-molecule crystal structure of Na-ATP (Kennard et al., 1971) (~2.4 Å) than those observed for Mg2+ in high-resolution protein crystal structures containing Mg-ADP or Mg-AMPPNP (Karpowich et al., 2001; Hopfner et al., 2000) (~2.1 Å).
Active Site Stereochemistry
A water molecule is found in essentially ideal geometry for a hydrolytic attack on the γ-phosphate of the ATP in both active sites in the MJ0796-E179Q dimer (Figure 2B). An interaction between the Na+ cofactor and the oxygen on the amide sidechain of Gln90 positions the nitrogen of this amide to donate an H bond to this putative hydrolytic water. In MJ0796-E171Q, the sidechain amide of Gln171 (i.e., the mutated reside) donates a second H bond to this water, which in turn donates a bifurcated H bond to two oxygen atoms of the γ-phosphate and a second H bond to the backbone carbonyl of Ala175 in the dimer-related subunit. The fact that the H bonding potential of the hydrolytic water is comprehensively satisfied by strong H bonds (~2.8 Å) in nearly canonical geometry probably contributes to the high stability of the dimer. In the wild-type protein, the presence of Glu171 will result in four acceptors competing for two potential donor protons on this water molecule (i.e., only Gln90 will be able to donate an H bond, and all of the other H bonding partners shown in Figure 2B will be acceptors). Presumably, hydrolysis occurs when the hydrolytic water rotates so as to donate H bonds to the carbonyl of Ala175 and the acceptor carboxylate of Glu171 while directing a lone pair toward the γ-phosphate of ATP.
Comparison to the Mg-ADP-Bound Monomer Structure
The structural differences between the Mg-ADP-bound and Na-ATP-bound forms of the MJ0796 monomer (Figure 4) closely match those predicted to occur based on comparison of the Mg-ADP structure to the ATP-bound structure of the HisP ABC (Yuan et al., 2001; Karpowich et al., 2001). The sidechains of Glu/Gln171 and His204 undergo induced-fit rotamer changes to contact the hydrolytic water and the γ-phosphate, respectively. The amide sidechain of Gln90 at the N terminus of the γ-phosphate linker moves ~5 Å to contact the Na+ cofactor and putative hydrolytic water in the active site (Figure 3A). The LSGGQ signature motif moves ~7 Å in a similar direction based on a 17° rigid-body rotation of the α-helical subdomain and most of the γ-phosphate linker, which is mediated by dihedral angle changes in the residues flanking Gln90 (i.e., 88–92) in conjunction with pivoting of β strand 2 in the ATP binding core around its parallel H bonds to β strand 3.
Figure 4. Conformational Differences between ADP-Bound and ATP-Bound MJ0796 Monomers.

The crystal structure of Mg-ADP-bound wild-type MJ0796 (Yuan et al., 2001) (lighter colors) is superimposed on that of the Na-ATP-bound E171Q mutant (darker colors) based on least-squares alignment of 65 Cα atoms located in the α helix following the Walker A motif and the 6 β strands in the F1-type ATP binding core. The backbone and sidechains of each model are color coded according to subdomain organization as in Figure 2A. The crystal structure of the E171Q mutant of MJ0796 bound to Mg-ADP is essentially identical to that of the wild-type protein bound to the same ligand (data not shown). This view shows the molecular surface of the monomer facing the intersubunit interface in the nucleotide sandwich dimer.
Given the fact that the Na+ cofactor in the ATP-bound structure of MJ0796-E171Q is located at nearly the same position as the Mg2+ cofactor in the ADP-bound structure of the wild-type protein (Yuan et al., 2001), the interaction between Gln90 and the cation cofactor could occur in the Mg-ADP-bound ABC but is apparently not sufficient to stabilize the γ-phosphate linker and the α-helical subdomain in the ATP-bound orientation. In this context, the H bond between Gln90 and the well-ordered hydrolytic water (Figure 2B) presumably makes a significant contribution to driving the subdomain reorientation shown in Figure 4. However, additional cooperative stabilization of the ATP-bound orientation of the α-helical subdomain will be provided by the strong inter-subunit interactions between the LSGGQ signature motif and the γ-phosphates of the nucleotides in the symmetry-related subunits of the nucleotide sandwich dimer, which can only be made simultaneously in both subunits when the α-helical subdomains adopt the ATP-bound orientation.
Potential Mechanisms of Allosteric Control by the TM Domains
The reorientation of the α-helical subdomain could potentially be controlled by the conformation of the TM domain, given the fact that the MsbA crystal structure shows that it contacts the ABC in the region undergoing the conformational changes associated with the reorientation of the α-helical subdomain (Chang and Roth, 2001) (i.e., at the interface between the α-helical subdomain, the γ-phosphate linker, and β strand 2 in the ATP binding core). Therefore, substrate binding to the TM domains could potentially mediate reorientation of the α-helical subdomain relative to the ATP binding core if allosteric conformational changes in the segments of the TMs contacting the ABC were to trigger a conformational change similar to that shown in Figure 4. Driving the ABC into the ATP-bound conformation would simultaneously enhance the affinity for the nucleotide, as observed in the presence of the transport substrate in many ABC transporters (Davidson, 2002), and also facilitate formation of the nucleotide sandwich dimer.
The surface of the MJ0796-E171Q dimer that faces the TM domains contains strongly hydrophobic patches (Figure 5B) at the interface of the α-helical subdomain and the γ-phosphate linker in each of the two subunits (Figures 5A and 5B), i.e., in the region of the ABC that is observed to make direct contacts to the TM domain in the crystal structure of the MsbA transporter (Chang and Roth, 2001). The ATPase active sites are located very close to these patches on the membrane-proximal surface. The adenine base, the ribose, the triphosphates, and the cation cofactor of each bound nucleotide are partially solvent-exposed on this same surface in the isolated ABC dimer, i.e., in a region that is located in close proximity to segments of the TM domains in the intact transporter. In this context, residues from the TM domains could possibly make molecular contacts to the bound nucleotides and even participate in the catalytic reaction cycle. This possibility raises a second potential structural mechanism by which the substrate occupancy and conformational state of the TM domains could affect allosteric control of ATPase activity (Liu and Sharom, 1996; Davidson, 2002).
Figure 5. Surface Properties of the MJ0796-E171Q ATP-Bound Monomer and Dimer.
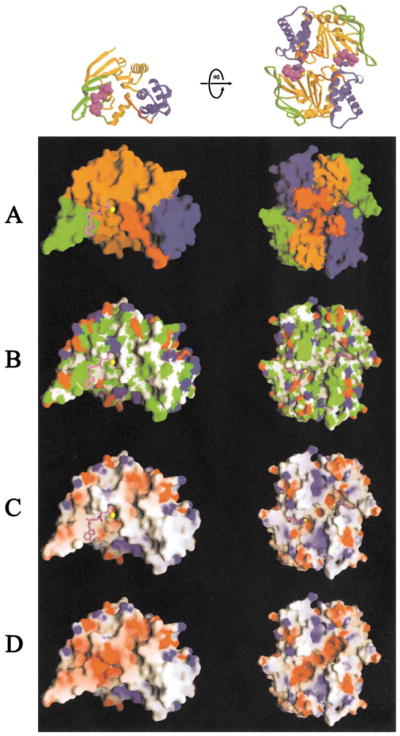
The molecular surface (Nicholls, 1992) of the monomer facing the intersubunit interface is shown on the left, while the molecular surface of the dimer facing the transmembrane domains is shown on the right. The Na-ATP was not included in the molecular surface calculations in (A)–(C), where it is shown in ball-and-stick representation colored as in Figure 2A. For reference, ribbon diagrams of the protein molecules are shown in equivalent orientations at the top.
(A) Color-coding according to subdomain organization with colors as in Figure 2A.
(B) Color-coding according to chemical polarity, with acidic functional groups colored red, basic functional groups (including the atoms other than Cβ and Cγ in histidine side-chains) colored blue, uncharged polar atoms colored white, and carbon atoms not in charged functional groups colored green.
(C and D) Color-coding according to electrostatic potential (Nicholls, 1992), assuming an ionic strength of 100 mM. Red and blue are used to represent negative and positive potentials, respectively, with fully saturated colors indicating a potential of magnitude ≥8 kT. The nucleotide was included in the calculation of both the molecular surface and the electrostatic potential in (D).
Electrostatic Charge Balance at the ATPase Active Site
The ATP binding site in the F1-type ATP binding core of the MJ0796 monomer is modestly acidic (Figure 5C) but becomes intensely acidic upon binding the nucleotide (Figures 5C and 5D). Upon dimerization, this highly charged surface binds to the LSGGQ signature motif in the apposing monomer (Figure 3A), involving a surface that is electrostatically neutral except for the sidechain of Arg153 (Figures 3A and 5D). Thus, the fully formed active site in the nucleotide sandwich dimer is characterized by a relatively intense concentration of negative charge.
Several lines of evidence suggest that the electrostatic charge balance at the ATPase active site plays a critical role in the mechanochemistry of ABCs. First, upon removal of a negative charge from the active site by the E171Q mutation in MJ0796 or the E179Q mutation in MJ1267, the formation of a stable nucleotide sandwich dimer is observed when a positive charge is removed from the ATP ligand by substitution of the usual Mg2+ cofactor by Na+. Second, the crystal structure of the ATP-bound HisP monomer (Hung et al., 1998) is consistent with biochemical data showing that wild-type ABCs do not dimerize upon binding ATP in the absence of the Mg2+ cofactor (Figure 1). Therefore, it is likely that a single surplus negative charge at each active site prevents formation of a tight nucleotide sandwich complex, given the fact that a monovalent cation generally occupies the cofactor site in the absence of Mg2+ (as shown here and in the small-molecule crystal structure of Na-ATP).
Upon nucleotide hydrolysis, an additional negative charge could form in the active site due to deprotonation of the inorganic phosphate product or the sidechain of the proton-accepting catalytic base, and this event could occur very rapidly, given the solvent-exposure of the active site. However, even in the absence of a such an increase in charge, the intense accumulation of negative charge associated with nucleotide binding is likely to cause electrostatic repulsion of the inorganic phosphate product, which will carry a net charge of at least −2 following hydrolysis. Given the fact that nucleotide sandwich dimer formation does not occur when ADP is bound to the Walker A/P loop site, the direct contacts between the LSGGQ signature motif and the γ-phosphate of the nucleotide (Figure 3A) are likely to be strong because they must make a significant contribution to the stability of the interface. The existence of such a strong interaction raises the possibility that the γ-phosphate could remain bound to the LSGGQ signature motif even after hydrolysis. Because the ADP product and the cation cofactor will remain bound to the Walker A/P loop site, as observed in the various ADP-bound ABC crystal structures, electrostatic repulsion of the inorganic phosphate product could push the ABCs apart if the phosphate transiently remains bound to the LSGGQ signature motif after hydrolysis.
Mechanistic Implications
Putting together the biochemical and structural observations on the nucleotide binding properties of ABCs, a model can be formulated for the thermodynamics and structural mechanics of the motor domains during the ATP-driven transport cycle in ABC transporters (Figure 6). ATP binding to empty ABCs triggers formation of a tight nucleotide sandwich dimer which moves the ABC into close apposition (Hopfner et al., 2000). This step represents one possibility for the power stroke of the pump, because the very high binding energy of ATP could be directly harnessed to do mechanical work if movements of the ABCs during dimer formation are coupled to appropriate displacements of the TM helices. The TM domains can mediate kinetic control of the ATP binding reaction in response to the binding of transport substrate if allosteric interactions with the ABC (Liu and Sharom, 1996) produce prealignment of its subdomains in the conformation found in the ATP sandwich dimer; such kinetic control would enhance the thermodynamic efficiency of the pump. The free energy gain upon ATP hydrolysis is used to drive the thermodynamic destabilization of the nucleotide sandwich dimer required to enable the pump to turn over. Following hydrolysis, electrostatic repulsion between the ADP product bound to the Walker A motif and the inorganic phosphate product bound to the LSGGQ signature motif in the apposing subunit drives separation of the ABCs (Davidson, 2002). This step represents a second possibility for the power stroke of the pump, because the electrostatic repulsion could also be harnessed to do mechanical work based once again on coupling of movements of the ABCs to displacements of the TM helices. In this case, hydrolysis would play a more direct role in driving transport, but the high energy of ATP binding would still be driving the overall reaction cycle because of its ability to produce a high concentration of negative electrostatic charge in the active site. Finally, the conformational reaction cycle is completed when nucleotide is released from the active site due to its reduced affinity for ADP compared to ATP.
Figure 6. The ATP-Driven Reaction Cycle of ABC Transporters.

The orange subdomain in the mechanistic schematic represents the antiparallel β subdomain combined with the F1-type ATP binding core, while the blue subdomain represents the γ-phosphate linker combined with the α-helical subdomain. The nucleotide is shown in magenta, with each circle representing one of its phosphate groups. The black trace in the free energy schematic represents the energy derived from the protein-nucleotide interactions, while the gray trace represents the energy of nucleotide hydrolysis. See text for additional information.
While this manuscript was coming to press, the crystal structure of the BtuCD integral membrane protein complex was reported (Locher et al., 2002). The homodimeric ABCs in this nucleotide-free structure are arranged in a geometry similar to that observed in the stable ATP sandwich dimer of MJ0796, but each is rotated by 14°, resulting in a substantial tilt at the intersubunit interface. Superimposing the ABCs in the BtuCD half-transporter on those in the MJ0796 dimer results in significant overlap of the TM domains. In this context, ATP binding to the BtuCD transporter seems likely to push the transmembrane domains towards one another, which could potentially lead to a wider opening at their periplasmic surface if they were to pivot about their interface near the cytoplasmic surface. Such an opening at the periplasmic surface could promote binding of the periplasmic binding protein while enabling vitamin B12, the transport substrate of the BtuCD pump, to enter the vestibule found between the two transmembrane domains in that structure. The resulting complex, with ATP bound at the active sites and the periplasmic binding protein bound at the opposite surface of the pump, would resemble that inferred to exist in the maltose transporter based on the biochemical studies of Davidson et al. (2002).
Experimental Procedures
Protein Expression and Purification
The E171Q mutant of MJ0706 was expressed as previously described (Yuan et al., 2001) except for the fact that cells were grown in conventional LB using E. coli strain BL21(DE3) harboring the pRIL rare tRNA plasmid (Stratagene, Cedar Creek, TX). Following cell harvesting and lysis as previously described, the supernatant from a 30 min centrifugation at 4°C in an SA600 rotor (Sorvall, Newtown, CT) was heat-shocked at 65°C for 10 min. Streptomycin sulfate (Sigma, St. Louis, MO) was added to the resulting suspension to a final concentration of 4% (w/v), and this mixture was incubated for 30 min at 4°C prior to centrifugation in the SA600 rotor for 30 min at 4°C. The resulting supernatant was purified on QEAE and butyl Sepharose-FF columns (Amersham-Pharmacia, Piscataway, NJ) as previously described. The pooled protein from the hydrophobic interaction column containing 50–100 mM (NH4)2SO4 was concentrated to about 1 mg/ml in a Centriprep-10 (Millipore, Bedford, MA), which resulted in the appearance of a protein precipitate. The protein was resolubilized by diluting the suspension 1:1 with 1 M arginine-HCl (Sigma) (pH 7.5) prior to adding Na-ATP and Na-EDTA to final concentrations of 20 mM and 1 mM, respectively. Although the resulting solution appeared clear, it was filtered through a 0.45 μm cellulose acetate filter (Corning-Costar, Acton, MA) after a 30 min incubation at 4°C. The filtrate was concentrated to 3 mg/ml using a Centriprep 10 prior to exchange into a buffer containing 20 mM Na-ATP, 1 mM Na-EDTA, 10% glycerol, 4 mM DTT, and 10 mM Tris-HCl (pH 8.0) using a HiPrep 26/10 desalting column (Amersham-Pharmacia). After concentration of the protein to about 5 mg/ml in this buffer, single-use aliquots were snap-frozen in liquid N2 and stored at −80°C.
Fluorescence Binding Experiments
The Y11W mutations were introduced into E171Q and wild-type MJ0796 using the Quick Change kit (Stratagene). T format fluorescence measurements were performed at 24°C using a QuantaMaster C-61 spectrofluorimeter (PTI, Lawrenceville, NJ) equipped with dual photomultiplier detectors and Glan-Thompson polarizers. Trp-specific 297 nm excitation was used with 4 nm slits on all monochromators. Emission intensity at 340 nm was averaged during a 60 s time interval after allowing samples to equilibrate for 30 s. Total fluorescence (|| + 2 × g × ⊥) and anisotropy r = ((|| − g × ⊥)/(|| + 2 × g × ⊥ were calculated from parallel and perpendicular emission channels after buffer subtraction. Values for Kd and cooperativity, n, were estimated using nonlinear curve fitting in the program Delta Graph (SPSS, Chicago, IL).
Crystallization
Crystals were grown by hanging drop vapor diffusion, mixing the purified protein 1:1 with a well solution containing 18% (w/v) PEG 4000 (Fluka, St. Louis, MO), 10% isopropanol (Amend, Irvington, NJ), 100 mM Na-HEPES (pH 7.0) (Fisher, Pittsburgh, PA). Crystals generally grew to full size (~100 μm3 ) within 12 hr. They were cryo-protected for 30 s in a solution containing 25% PEG 4000, 10% glycerol, 10% isopropanol, 100 mM sodium HEPES (pH 7.0) prior to freezing in liquid propane.
X-Ray Data Collection
Crystallographic data were measured on NSLS beamline X12C at Brookhaven National Lab using a wavelength of 0.97884 Å and the Brandeis-2K CCD detector. A 190°continuous sweep of diffraction data was collected using 1° oscillations and then scaled and reduced with DENZO and SCALEPACK, respectively (Otwinowski and Minor, 1997).
Structure Determination
Molecular replacement was preformed with normalized structure factors using the CCP4 v4.0 (CCP4, 1994) version of AMoRe (Navaza, 1994). The search model was derived from the refined model of Mg-ADP-bound MJ0796 (Yuan et al., 2001), but the α-helical subdomain (residues 97–147) was omitted. The rotation and translation functions were evaluated using data from 15–3.5 Å. Using an integration radius of 25 Å, the top solution in the rotation function was correct but gave only a 3.5% correlation coefficient on intensities. The best solution in the translation function for the first molecule gave a correlation coefficient of 13.8%, which improved to 14.4% after rigid body refinement. The translation function for the second molecule gave a correlation coefficient of 27.3%, which improved to 28.8% and yielded an R factor of 49.7% after rigid body refinement. The α-helical subdomain was then added to both monomers using the crystal structure of the ATP-bound HisP ABC from S. typhimurium (Hung et al., 1998) as a guide to establish its location. Once the antiparallel β subdomain and F1-type ATP binding core of HisP were aligned with the corresponding regions in MJ0796, the α-helical subdomain from MJ0796 was superimposed on the corresponding region of HisP, yielding a correct fit to the electron density.
Refinement
Model building was performed using O (Jones et al., 1991). The refinement started with a round of torsional angle dynamics at 6000 K in CNS (Brunger et al., 1998) in order to remove model bias. The refinement was completed using XPLOR 3.851 (Brunger, 1992b) with Engh and Huber parameters (Engh and Huber, 1991). Overall anisotropic B factors were refined for the structure, individual isotropic B factors were refined for all atoms, and a bulk solvent correction was applied. The Rfree set contained 8% of the reflections selected at random (Brunger, 1992a).
Acknowledgments
This work was supported by research grants from the March of Dimes and the Cystic Fibrosis Foundation to J.F.H. and by grants from the National Institutes of Health, the Cystic Fibrosis Foundation, and the Welch Foundation to P.J.T. The authors thank Anand Saxena for support during data collection at NSLS.
Footnotes
Accession Numbers
The model of the stable ATP sandwich dimer of the E171Q mutant of MJ0796 has been deposited in the PDB under accession code 1L2T.
References
- Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992a;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Brunger AT. X-PLOR Version 3.1: A System for Crystallography and NMR. New Haven, CT: Yale University Press; 1992b. [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Chang G, Roth CB. Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science. 2001;293:1793–1800. doi: 10.1126/science.293.5536.1793. [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project 4) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Connolly ML. Analytical molecular surface calculation. J Appl Crystallogr. 1983;16:548–558. [Google Scholar]
- Dassa E, Hofnung M, Paulsen IT, Saier MH. The escherichia coli ABC transporters: an update. Mol Microbiol. 1999;32:887–889. doi: 10.1046/j.1365-2958.1999.01392.x. [DOI] [PubMed] [Google Scholar]
- Davidson AL. Mechanism of coupling of transport to hydrolysis in bacterial ATP-binding cassette transporters. J Bacteriol. 2002;184:1225–1233. doi: 10.1128/JB.184.5.1225-1233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs K, Diez J, Greller G, Muller C, Breed J, Schnell C, Vonrhein C, Boos W, Welte W. Crystal structure of MalK, the ATPase subunit of the trehalose/maltose ABC transporter of the archaeon thermococcus litoralis. EMBO J. 2000;19:5951–5961. doi: 10.1093/emboj/19.22.5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth J. Principles of Protein X-ray Crystallography. New York: Springer-Verlag; 1994. [Google Scholar]
- Engh RA, Huber R. Accurate bond and angle parameters for x-ray protein structure refinement. Acta Crystallogr. 1991;A47:392–400. [Google Scholar]
- Gaudet R, Wiley DC. Structure of the ABC ATPase domain of human TAP1, the transporter associated with antigen processing. EMBO J. 2001;20:4964–4972. doi: 10.1093/emboj/20.17.4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM, Pastan I, Ambudkar SV. P-glycoprotein and multidrug resistance. Curr Opin Genet Dev. 1996;6:610–617. doi: 10.1016/s0959-437x(96)80091-8. [DOI] [PubMed] [Google Scholar]
- Greller G, Horlacher R, DiRuggiero J, Boos W. Molecular and biochemical analysis of MalK, the ATP-hydrolyzing subunit of the trehalose/maltose transport system of the hyperthermophilic archaeon Thermococcus litoralis. J Biol Chem. 1999;274:20259–20264. doi: 10.1074/jbc.274.29.20259. [DOI] [PubMed] [Google Scholar]
- Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- Holland IB, Blight MA. ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J Mol Biol. 1999;293:381–399. doi: 10.1006/jmbi.1999.2993. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 2000;101:789–800. doi: 10.1016/s0092-8674(00)80890-9. [DOI] [PubMed] [Google Scholar]
- Hung LW, Wang IX, Nikaido K, Liu PQ, Ames GF, Kim SH. Crystal structure of the ATP-binding subunit of an ABC transporter. Nature. 1998;396:703–707. doi: 10.1038/25393. [DOI] [PubMed] [Google Scholar]
- Jones PM, George AM. Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol Lett. 1999;179:187–202. doi: 10.1111/j.1574-6968.1999.tb08727.x. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Karpowich N, Martsinkevich O, Linda Millen L, Yuan YR, Dai PL, MacVey K, Thomas PJ, Hunt JF. Crystal structures of the MJ1267 ATP-binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure. 2001;9:571–586. doi: 10.1016/s0969-2126(01)00617-7. [DOI] [PubMed] [Google Scholar]
- Kennard O, Isaacs NW, Motherwell WDS, Coppola JC, Wampler DL, Larsen AC, Watson DG. The crystal structure of Na-ATP. Proc R Soc Lond Ser A. 1971;325:401. [Google Scholar]
- Koronakis E, Hughes C, Milisav I, Koronakis V. Protein exporter function and in vitro ATPase activity are correlated in ABC-domain mutants of HlyB. Mol Microbiol. 1995;16:87–96. doi: 10.1111/j.1365-2958.1995.tb02394.x. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of proteins. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- Linton KJ, Higgins CF. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol Microbiol. 1998;28:5–13. doi: 10.1046/j.1365-2958.1998.00764.x. [DOI] [PubMed] [Google Scholar]
- Liu R, Sharom FJ. Site-directed fluorescence labeling of P-glycoprotein on cysteine residues in the nucleotide binding domains. Biochemistry. 1996;35:11865–11873. doi: 10.1021/bi960823u. [DOI] [PubMed] [Google Scholar]
- Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- Merritt EA, Bacon DJ. Raster3D photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Moody J, Millen L, Binns D, Hunt JF, Thomas PJ. Cooperative, ATP-dependent dimerization of ATP-binding cassettes during the catalytic cycle of ABC transporters. J Biol Chem. 2002;277:21111–21114. doi: 10.1074/jbc.C200228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr. 1994;A50:157–163. [Google Scholar]
- Nicholls A. GRASP: Graphical Representation and Analysis Surface Properties. New York: Columbia University; 1992. [Google Scholar]
- Nikaido K, Liu PQ, Ames GF. Purification and characterization of HisP, the ATP-binding subunit of a traffic ATPase (ABC transporter), the histidine permease of Salmonella typhimurium. Solubility, dimerization, and ATPase activity. J Biol Chem. 1997;272:27745–27752. doi: 10.1074/jbc.272.44.27745. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Paulsen IT, Sliwinski MK, Saier MH. Microbial genome analyses: global comparisons of transport capabilities based on phylogenies, bioenergetics and substrate specificities. J Mol Biol. 1998;277:573–592. doi: 10.1006/jmbi.1998.1609. [DOI] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Schmees G, Stein A, Hunke S, Landmesser H, Schneider E. Functional consequences of mutations in the conserved ‘signature sequence’ of the ATP-binding-cassette protein MalK. Eur J Biochem. 1999;266:420–430. doi: 10.1046/j.1432-1327.1999.00871.x. [DOI] [PubMed] [Google Scholar]
- Schneider E, Wilken S, Schmid R. Nucleotide-induced conformational changes of MalK, a bacterial ATP binding cassette transporter protein. J Biol Chem. 1994;269:20456–20461. [PubMed] [Google Scholar]
- Thomas PJ, Hunt JF. A snapshot of Nature’s favorite pump. Nat Struct Biol. 2001;8:920–923. doi: 10.1038/nsb1101-920. [DOI] [PubMed] [Google Scholar]
- Urbatsch IL, Gimi K, Wilke-Mounts S, Senior AE. Investigation of the role of glutamine-471 and glutamine-1114 in the two catalytic sites of P-glycoprotein. Biochemistry. 2000;39:11921–11927. doi: 10.1021/bi001220s. [DOI] [PubMed] [Google Scholar]
- Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmot CM, Thornton JM. Beta-turns and their distortions: a proposed new nomenclature. Protein Eng. 1990;3:479–493. doi: 10.1093/protein/3.6.479. [DOI] [PubMed] [Google Scholar]
- Yuan YR, Blecker S, Martsinkevish O, Millen L, Thomas PJ, Hunt JF. The crystal structure of the MJ0796 ATP-binding cassette: implications for the structural consequences of ATP hydrolysis in the active site of an ABC transporter. J Biol Chem. 2001;276:32313–32321. doi: 10.1074/jbc.M100758200. [DOI] [PubMed] [Google Scholar]