

Abstract
Motor neuron diseases (MNDs) are progressive neurodegenerative disorders characterized by selective death of motor neurons leading to spasticity, muscle wasting and paralysis. Human VAMP-associated protein B (hVAPB) is the causative gene of a clinically diverse group of MNDs including amyotrophic lateral sclerosis (ALS), atypical ALS and late-onset spinal muscular atrophy. The pathogenic mutation is inherited in a dominant manner. Drosophila VAMP-associated protein of 33 kDa A (DVAP-33A) is the structural homologue of hVAPB and regulates synaptic remodeling by affecting the size and number of boutons at neuromuscular junctions. Associated with these structural alterations are compensatory changes in the physiology and ultrastructure of synapses, which maintain evoked responses within normal boundaries. DVAP-33A and hVAPB are functionally interchangeable and transgenic expression of mutant DVAP-33A in neurons recapitulates major hallmarks of the human diseases including locomotion defects, neuronal death and aggregate formation. Aggregate accumulation is accompanied by a depletion of the endogenous protein from its normal localization. These findings pinpoint to a possible role of hVAPB in synaptic homeostasis and emphasize the relevance of our fly model in elucidating the pathophysiology underlying motor neuron degeneration in humans.
INTRODUCTION
Motor neuron diseases (MNDs) encompass a group of inherited disorders characterized by the selective dysfunction and death of motor neurons leading to spasticity, hyperreflexia, generalized weakness, muscle atrophy and paralysis (1). The best characterized and the most common of these diseases is amyotrophic lateral sclerosis (ALS) with a prevalence of approximately 5/100 000 individuals. The majority of ALS cases are sporadic while only ~10% are familial, manifesting a variety of inheritance patterns with linkage to multiple independent chromosome loci (2). Among the familial cases, ~20% are caused by dominantly inherited mutations in the protein encoded by the gene Cu/Zn superoxide dismutase 1 (SOD1) (3).
In 2004, a genetic linkage study mapped the locus responsible for a group of MNDs to chromosomal region 20q13.3 (ALS8). The disease affects both sexes equally and the clinical onset occurs between the third and fifth decade. Most patients have lower motor neuron symptoms but some show bulbar involvement (4). Mutation screening led to the identification of a Proline to Serine substitution (P56S) at codon 56 in human VAMP-associated protein B (hVAPB) (5). In a branch of the same large family the P56S mutation has been shown to cause a lower motor neuron disorder accompanied by autonomic involvement and dyslipidemia (6). The mutated Proline is present in a stretch of amino acids that is very highly conserved from yeast to man in all VAP homologs.
hVAPB is a type II integral membrane protein that belongs to a highly conserved family of proteins. VAP proteins have been implicated in glucose transport trafficking, expression of phospholipid biosynthetic genes, regulation of synaptic growth, neurotransmitter release and ER-Golgi and intra-Golgi transport (7–11). These seemingly different functions have been investigated in different species and cell types and they are possibly mediated by different members of the same family.
The overall structure of all VAP proteins is similar and consists of a cytoplasmic N-terminal region and a trans-membrane domain at the C-terminus. The N-terminal domain shares a high degree of structural similarity with the Caenorhabditis elegans major sperm proteins (MSPs) (12). MSPs are highly abundant proteins expressed in the amoeboid nematode sperm. The movement of these cells is driven by the assembly of MSP proteins into fibrous networks (13). MSP proteins have also been shown to function as signaling molecules as they antagonize ephrin/Eph receptor signaling in order to promote oocyte meiotic maturation and ovarian muscle contractions in C. elegans (12).
DVAP-33A (Drosophila VAMP-associated protein of 33 kDa A) exhibits significant homology with hVAPB. DVAP-33A regulates bouton budding at larval neuromuscular junctions (NMJs) in a dosage-dependent manner. It is required for structural remodeling of synapses where it controls microtubule cytoskeleton dynamics. We have previously proposed that synapse formation is dependent on DVAP-33A in a process similar to budding in yeast (9). Recently, it has been shown that, MSP localizes to membranes and can generate the protrusive force necessary to induce vesicle budding from male germ cells in C. elegans (14).
To better understand the pathophysiology underlying VAP-induced MNDs in humans, we have undertaken a functional characterization of VAP proteins in flies. In Drosophila, structural remodeling induced by loss-of-function and overexpression of DVAP-33A is paralleled by functional and ultrastructural compensation at the synapse. We show that hVAPB and DVAP-33A are functionally interchangeable and that transgenic expression of mutant VAP in neurons recapitulates several hallmarks of the human disease including locomotion defects, neuronal apoptosis and aggregate deposition. Interestingly, aggregate accumulation is associated with a strong reduction in the abundance of the endogenous protein at its normal localization. Taken together these data underline a possible role for hVAPB in synaptic homeostasis and highlight the importance of this fly model in elucidating the pathomechanism of VAP-induced motor neuron degeneration in humans.
RESULTS
Homeostatic regulation of neurotransmitter release at NMJs with altered expression of DVAP-33A
Hypomorphic and null mutations in DVAP-33A cause a severe decrease in bouton number and an increase in bouton size. Conversely, overexpression of DVAP-33A in neurons induces a highly significant increase in the number of boutons with a concomitant decrease in their size (Supplementary Material, Fig. S1). Since the number and size of synaptic contacts between a neuron and its target may affect synaptic strength, we investigated whether these structural changes have any consequences on synaptic physiology. We focused our electrophysiological analysis on DVAP-33AΔ166 partial loss-of-function mutants as many more of them survive to the third instar larval stage than null mutants though both mutants exhibit very similar phenotypes. As shown in Figure 1A and B, the amplitude of the evoked junctional potentials (EJPs) is not significantly different (P > 0.05) in synaptic terminals exhibiting fewer and larger boutons (36 ± 2 mV) relative to controls (37 ± 2 mV). To determine whether other aspects of synaptic transmission are altered in mutants, we studied the properties of miniature EJPs (mEJPs). We found an increase in the mean frequency of mEJPs in mutants when compared with controls (3.80 ± 0.24 Hz versus 2.00 ± 0.11 Hz, P < 0.001) (Fig. 1E). In addition, as shown in Figure 1E and F, the mean mEJP amplitude is increased in partial loss-of-function mutants compared with controls (1.30 ± 0.02 mV in DVAP-33AΔ166 versus 0.80 ± 0.01 mV in controls). This difference in quantal size is significant (P < 0.001) and is also observed in null mutants (data not shown). We calculated the quantal content by dividing the mean EJP size by the mean of the mEJP size and found a decrease in quantal content of ~40% in mutants compared with controls (44.90 ± 0.9 in controls versus 26.91 ± 0.5 in mutants, P < 0.001).
Figure 1.

Electrophysiological analysis of larvae lacking and overexpressing DVAP-33A. (A) Examples of single traces showing EJPs in DVAP-33A mutants (Δ166) and controls (Revert); (B) summary of EJP amplitudes for DVAP-33AΔ166 mutants (n = 11) and controls (Rev. n = 10); (C) representative traces of EJPs in synaptic terminals overexpressing DVAP-33A (elav; UAS-DVAP) and controls (UAS-DVAP); (D) summary of EJP amplitudes for overexpression mutants (n = 12) and controls (n = 13). Note that evoked neurotransmitter release is normal in both DVAP-33A loss-of-function and overexpression mutants; (E) representative traces of mEJPs for DVAP-33A mutants and controls; (F) cumulative distribution of total mEJP amplitudes in DVAP-33A mutants and in controls. Histograms of mEJPs for Rev. and Δ166 are shown; (G) cumulative distribution and histograms of total mEJP amplitudes for terminals overexpressing DVAP-33A (elav; UAS-DVAP) and for controls (elav). DVAP-33A mutations significantly increase the frequency and the amplitude of mEJPs while DVAP-33A overexpressing mutants exhibit a significant decrease in mean mEJP amplitude. Error bars represent SEM.
In Drosophila, the UAS/GAL4 system allows the temporal and tissue-specific expression of a transgene by using a variety of GAL4 drivers (15). We overexpressed DVAP-33A in neurons by using the pan-neural driver elav-GAL4. In synaptic terminals overexpressing DVAP-33A, the EJP amplitude is not significantly changed compared with controls (32 ± 3 mV versus 33 ± 2 mV, P > 0.05) (Fig. 1C and D). Conversely, there is a significant decrease in quantal size in these mutants (0.59 ± 0.05 mV versus 0.90 ± 0.06 mV in controls, P < 0.05) (Fig. 1G). Interestingly, cumulative amplitude histograms indicate that the entire mEJP amplitude distribution is shifted towards larger values in mutants (Fig. 1F) and towards smaller values in animals overexpressing DVAP-33A (Fig. 1G). A significant increase in quantal content accompanies the increase in bouton number as the quantal content in DVAP-33A overexpressing larvae is 54.13 ± 1.5 compared with 36.66 ± 0.8 in controls (P < 0.05). Thus, changes in quantal size are responsible for maintaining normal synaptic transmission in loss-of-function and overexpression paradigms.
Ultrastructural remodeling at NMJs lacking and overexpressing DVAP-33A
To determine whether ultrastructural remodeling at the synapse accompanies the previously described functional compensation, we performed a serial section TEM (transmission EM) analysis of terminals lacking and overexpressing DVAP-33A. As shown in Figure 2, boutons from DVAP-33AΔ166 animals display an increase in the number of active zones: 2.0 ± 0.2 active zones per bouton cross-sectional area versus 0.8 ± 0.3 in controls (Fig. 2A–C and E). Although overexpression of DVAP-33A does not affect the number of active zones per surface area, other features clearly differentiate these boutons from wild-type (wt) terminals. As shown in Figure 2D, there are substantially more boutons which are significantly smaller in size when compared with wt boutons (Fig. 2A). In addition, these boutons appear to contain fewer vesicles. As reported in Figure 2F, in every small bouton resulting from DVAP-33A overexpression, more than 80% of the bouton cross-sectional area is devoid of synaptic vesicles, whereas in controls, numerous vesicles are packed in each bouton leaving only 40–50% of the bouton area devoid of synaptic vesicles. No change in the size of synaptic vesicles was observed in any of the genotypes (data not shown). In summary, analysis by TEM of DVAP-33AΔ166 mutant synapses, containing larger and fewer varicosities, reveals a compensatory increase in the number of active zones per bouton. In contrast, in synaptic terminals overexpressing DVAP-33A, which contain more boutons that are smaller in size, the density of the vesicles per bouton is decreased. These data indicate that synapses can undergo structural remodeling, whereby active zones are concentrated in a reduced number of boutons while the pool of vesicles can be diluted in an increased number of boutons to maintain functional and structural homeostasis.
Figure 2.

Nerve terminal ultrastructure of DVAP-33A mutants. (A and C) Electron micrographs of control NMJs: Canton S in (A) and Revertant in (C); (B) electron micrographs of DVAP-33AΔ166 hypomorphic mutant NMJs (Δ166); (D) electro-micrographs of DVAP-33A (elav; UAS-DVAP) overexpressing NMJs. Presynaptic active zones (arrow) and subsynaptic reticulum (SSR) are indicated. Arrow in (D) indicates synaptic vesicles; (E and F) morphometric analysis of Δ166 mutants and DVAP-33A overexpressing terminals. Nerve terminals were sectioned and analyzed for the number of active zones per bouton cross-sectional area (E), and for the bouton area devoid of synaptic vesicles (empty area) (F). DVAP-33A mutants exhibit an increase in the number of active zones per bouton, whereas in the overexpression the density of vesicles per bouton is decreased. At least 11 boutons per animals were analyzed. Error bars represent SEM.
DVAP-33A is the functional homolog of hVAPB
In Drosophila there are three proteins (CG33523, CG7919 also named farinelli and CG5014 which is DVAP-33A) with significant homology and structural similarity to hVAPB. Farinelli, expressed specifically in testes and in larval fat body, is required for male fertility. The proteins encoded by CG33523 and DVAP-33A are ubiquitously expressed but the CG33523 protein is only 34% similar while DVAP-33A is 62% similar to hVAPB. The degree of homology and the pattern of expression suggest that DVAP-33A is likely to be the Drosophila ortholog of hVAPB. DVAP-33A and hVAPB also share a common three-partite domain organization: an MSP homology domain (Supplementary Material, Fig. S2A and C) containing a stretch of 16 amino acids conserved from yeast to man (Supplementary Material, Fig. S2B), a coiled-coil domain (Supplementary Material, Fig. S2A and C), and a trans-membrane domain at the C-terminus (Supplementary Material, Fig. S2A and C).
Given the degree of homology between DVAP-33A and hVAPB, we tested whether the human gene can functionally substitute for the loss of DVAP-33A. We generated transgenic flies carrying the UAS-hVAPB cDNAs and tested several independent transgenic lines. We have previously shown that the synaptic bouton phenotype associated with DVAP-33A loss-of-function mutations can be rescued by driving the expression of the wt protein in neurons (9). We therefore used the same GAL4 drivers to test the ability of hVAPB to functionally replace DVAP-33A. The hVAPB gene was expressed using the C164-GAL4 (16) and D42-GAL4 (17) drivers in null (DVAP-33Δ448, DVAP-33Δ20) and hypomorphic (DVAP-33Δ166) mutant backgrounds. Although DVAP-33A is expressed ubiquitously and its zygotic loss results in larval lethality with rare adult escapers (~1%), the lethality associated with the loss or partial loss of DVAP-33A can be rescued with both drivers in combination with several UAS-hVAPB transgenes. We often obtained the expected Mendelian ratio. Rescued flies were fertile and did not show any obvious morphological or behavioral defects. We next investigated whether hVAPB can rescue the morphological and electrophysiological phenotypes associated with DVAP-33A loss-of-function mutations. We found that in synapses expressing hVAPB under the control of C164-Gal4 or D42-Gal4 in DVAP-33AΔ448 null background, the number of synaptic boutons is similar to controls (284 ± 11 boutons versus 278 ± 12 boutons in controls, P > 0.05, Fig. 3A and B). Moreover, electrophysiological analysis of the same synapses shows that the EJPs (36 ± 2 mV versus 37 ± 2 mV in controls; P > 0.05) and mEJPs (0.83 ± 0.02 mV versus 0.89 ± 0.03 mV in controls; P > 0.05) are both similar to controls (Fig. 3C and D). Hence, the lethality, aberrant NMJ morphology and increased mEJP amplitude associated with loss of DVAP-33A, are rescued by targeting the expression of hVAPB in neurons. These data indicate that the human and the Drosophila protein share a common structure and perform homologous functions.
Figure 3.

hVAPB and DVAP-33A are functional homologs. (A) Anti-HRP stainings of control NMJs (UAS-hVAP). (B) NMJs expressing hVAPB in DVAP-33A mutant background (DVAP/hVAP). In the inset, total number of boutons on muscles 12 and 13 for controls (blue) and NMJs expressing DVAP/hVAP (red). (C) Examples of single traces showing EJPs in controls and NMJs expressing DVAP/hVAP. In the inset, summary of EJP amplitudes for the corresponding genotypes. (D) Representative traces of mEJPs of control and DVAP/hVAP animals. In the inset, histograms of mEJP amplitudes for control and DVAP/hVAP NMJs are shown. Defects in synaptic function and morphology in DVAP-33A loss-of-function mutations are rescued by neuronal-specific expression of hVAPB. (E) Anti-HRP stainings of control NMJs (UAS-hVAP). (F) NMJs with transgenic expression of hVAPB in a wt background for DVAP-33A (elav; UAS-hVAP). In the inset, the total number of boutons on muscles 12 and 13 is reported for controls (blue) and elav; UAS-hVAP larvae (red). (G) Examples of traces of EJP amplitudes for control and elav; UAS-hVAP NMJs. In the inset, a summary of EJP amplitudes for the corresponding genotypes. (H) Representative traces of mEJP amplitudes for controls and elav; UAS-hVAP synapses. In the inset, histograms of mEJP amplitudes for controls and synapses expressing transgenic hVAPB are shown. Transgenic expression of hVAPB in neurons induces an increase in the number of smaller boutons, a small but significant decrease in the evoked response and a decrease in mini amplitude. UAS-hVAPB transgenic lines without the driver were used in all the control experiments reported in this figure. ‘*’ denotes statistically significant changes. n = 11 per genotype. Error bars are SEM. Scale bars = 10 μm.
Transgenic expression of hVAPB mimics DVAP-33A overexpression
As shown in Supplementary Material, Figure S1D, neuronal overexpression of DVAP-33A using the pan-neural elav-GAL4 driver causes a dramatic increase in the number of boutons as well as a decrease in their size. Despite these morphological changes, a homeostatic mechanism maintains muscle EJPs within normal values (Fig. 1C, D and G). To assess whether transgenic expression of the human protein in neurons has similar effects on synaptic structure and function, we used the same elav-Gal4 driver. As shown in Figure 3E and F, we observed a dramatic increase (P < 0.001) in the number of boutons (535 ± 16) with a concomitant decrease in bouton size when compared with controls (297 ± 7). Similar to the overexpression of DVAP-33A, expression of hVAPB in a wt background also causes a reduction in the average mEJP amplitude. In this case, however, a 50% reduction in the mEJP size (0.48 ± 0.01 mV versus 0.82 ± 0.01 mV in controls; P < 0.001, Fig. 3H) and about a 10% reduction in the EJP amplitude (29.0 ± 0.8 mV and 35.0 ± 0.7 mV in controls; P < 0.001, Fig. 3G) were observed. Although there is a small decrease in the amplitude of the evoked response, the decrease in quantal size allows a nearly normal post-synaptic response. Hence, the experiments in both loss-of-function and transgenic expression indicate that hVAPB and DVAP-33A can substitute for each other’s function.
Expression levels of VAP proteins affect the abundance of specific receptor subunits and the volume of post-synaptic receptor clusters
In DVAP-33A loss-of-function mutations, an increase in quantal size ensures functional homeostasis despite a significant decrease in bouton number (Supplementary Material, Fig. S1 and Fig. 1). This increased response to spontaneous release of neurotransmitter is usually due to changes in the composition or sensitivity of post-synaptic glutamate receptors. Neurotransmitter is released from presynaptic specializations termed active zones. In wt animals, glutamate receptors are clustered in puncta that lie opposite the presynaptic active zones, placing them in an ideal position to detect neurotransmitter released. To date, five ionotropic glutamate receptor subunits have been identified at the Drosophila NMJ: GluRIIA, GluRIIB (18,19), GluRIII (20,21), GluRIID and GluRIIE (21,22). We used previously characterized antibodies to assess glutamate receptor abundance and distribution in synapses lacking DVAP-33A. A significant increase in cluster count (P < 0.05) and a marked increase in the average cluster volume for GluRIIA were observed (P < 0.01, Fig. 4A–C). For all the other subunits a small but statistically significant decrease in cluster count was found (P < 0.05, Fig. 4C). GluRIIB and GluRIII also exhibit a marked reduction in the average cluster volume (30% reduction for GluRIIB, Fig. 4C; 46% for GluRIII, Fig. 4C–E, P < 0.001) while cluster size for GluRIID is similar to controls (Fig. 4C). The staining intensity of every subunit does not differ significantly between controls and mutants (data not shown).
Figure 4.

DVAP-33A loss-of-function mutations affect subunit abundance and cluster size of post-synaptic glutamate receptors. (A) Volume renderings of controls stained with anti-GluRIIA antibodies. (B) Mutant synapses for DVAP-33A (Δ448) stained with the same antibodies as in (A). (D) Control synapses stained with anti-GluRIII antibodies. (E) Δ448 synapses stained with the same antibodies as in (D). (C) Morphometric analysis of Δ448 NMJs reporting cluster count and mean cluster volume for every GluR subunit as percentages of control values. A striking increase in the average cluster volume for GluRIIA subunit is observed in DVAP-33A mutants. The revertant line generated by precise excision of the original P-element was used as a control in this experiment (9). Data in (C) are shown as mean ± SEM and n = 5 larvae for every analyzed genotype. Scale bar = 50 μm.
A striking physiological feature of transgenic expression of VAP proteins in neurons is a significant decrease in quantal size (Figs 1G and 3H). We investigated whether the decrease in quantal size was associated with changes in the post-synaptic glutamate receptors as well. We focused our analysis on synapses expressing transgenic hVAPB in neurons, which exhibit a greater reduction in quantal size (Fig. 3H). A marked decrease (P < 0.001) in GluRIIA abundance compared with controls was observed (Fig. 5A–G). This decrease was specific for GluRIIA as no difference in expression levels between controls and mutants was found for any other subunits (data not shown). Moreover, synapses expressing transgenic hVAPB exhibit a reduction in the average cluster volume for the subunit GluRIIA (P < 0.001 Fig. 5H–J), subunit GluRIIB and GluRIII (P < 0.05 in both cases, Fig. 5J). Cluster count on the other hand, does not change significantly (P > 0.05) except for the subunit GluRIII where a small but statistically significant increase is observed (P < 0.05, Fig. 5J). No significant changes (P > 0.05, Fig. 5J) in cluster count and average cluster volume were reported for the subunit GluRIID.
Figure 5.

Neuronal expression of VAP proteins affects post-synaptic glutamate receptor composition. (A–C) GluRIIA subunit localization (red) at control NMJs visualized by using the neuronal cell surface marker anti-HRP (green). (E–G) Synapses expressing transgenic hVAPB (elav; UAS-hVAP) stained using the same antibodies as in (A–C). (D) Quantification of the fluorescence intensity per volume unit of GluRIIA clusters in controls and in elav; UAS-hVAP synapses. (H and I) Synapses of relevant genotypes stained with anti-GluRIIA antibodies are shown as an example. Volume renderings of clusters immunoreactive to GluRIIA are presented irrespective of their signal intensity. (J) Morphometric analysis of cluster count and mean cluster volume for every GluR subunit in elav; UAS-hVAP synapses are presented as percentages of control values. Neuronal expression of VAP proteins induces a decrease in the expression levels of GluRIIA and a reduction in the receptor field size. NMJ of Canton S larvae were used as controls. Data in (D) and (J) are shown as mean ± SEM and in (D) the intensity is presented in arbitrary units. N = 5 larvae for every analyzed genotype. Scale bars = 50 μm.
Taken together, these data indicate that changes in the expression levels of VAP proteins regulate quantal size by shaping the post-synaptic glutamate receptor field and the abundance of specific subunits. Moreover, our data indicate that VAP proteins are components of a trans-synaptic signal as their presynaptic expression affects the post-synaptic sensitivity to neurotransmitters.
hVAPB carrying the ALS8 mutation rescues the DVAP-33A mutant phenotype
The Proline residue that is changed into a Serine in ALS8 patients is conserved and contained in a stretch of 16 amino acids that is virtually identical in all VAP proteins (5). Such a high degree of conservation suggests that this region plays a crucial role in the function of VAP proteins and mutations affecting this region are likely to have similar consequences in all VAP homologues. To help define the nature of the ALS8 mutation, we expressed mutant VAP proteins in a null background for DVAP-33A. We generated flies carrying UAS-hVAPBP56S (the human VAP mutant) transgene and flies carrying the UAS-DVAPP58S (the Drosophila mutant VAP) transgene. We tested the ability of these transgenes to rescue the mutant phenotypes associated with the loss of DVAP-33A. Lethality was rescued when the hVAPBP56S transgene was driven by the D42-Gal4 and C164-GAL4 drivers in a null background (DVAP-33AΔ20 and DVAP-33AΔ448, denoted as DVAP/hVAPmt in Fig. 6). In addition, as shown in Fig. 6A and B, the number of boutons is not significantly different in flies expressing the human mutant protein (C164-Gal4; UAS-hVAPmt; bouton number: 290 ± 11) compared with controls (UAS-hVAPmt; bouton number: 304 ± 11; P > 0.05). Moreover, no significant difference in EJPs was observed in flies expressing hVAPBP56S (36 ± 2 mV) compared with controls (34 ± 2 mV, P > 0.05, Fig. 6C). Finally, flies expressing hVAPBP56S exhibit quantal sizes (0.89 ± 0.02 mV) similar to those of control animals (0.83± 0.03 mV, P > 0.05, Fig. 6D). Similar data with respect to viability, morphological and electrophysiological properties were also observed with the UAS-DVAPP58S transgene expressed in DVAP-33A mutant background (data not shown). The fact that both the human and fly protein carrying the pathogenic mutation can substitute for the function of DVAP-33A indicates that the pathogenic allele retains wt properties.
Figure 6.

hVAPP56S rescues the mutant phenotypes associated with DVAP-33A mutations. (A) Anti-HRP stainings of control NMJs (UAS-hVAPmt). (B) NMJs expressing mutant hVAPB in DVAP-33A mutant background (DVAP/hVAPmt). In the inset, the total number of boutons on muscles 12 and 13 for the same genotypes is reported. (C) EJP traces are reported for control and DVAP/hVAPmt NMJs. In the inset, a summary of EJP amplitudes is reported for the respective genotypes. (D) Representative traces of mEJP amplitudes for control and DVAP/hVAPmt NMJs. In the inset, histograms of mEJP amplitudes are shown for the corresponding genotypes. Defects in synaptic function and morphology in DVAP-33A loss-of-functions are rescued by neuronal-specific expression of hVAPB carrying the ALS8 mutation. UAS-hVAPmt transgenic lines without the driver were used in the control experiments reported in this figure. n = 10 larvae for every genotype. Error bars are SEM. Scale bars = 10 μm.
Transgenic expression of the Drosophila mutant protein in neurons recapitulates several hallmarks of the human disease
Expression of hVAPB and hVAPBP56S in neurons rescues the lethality, morphological and electrophysiological phenotypes associated with DVAP-33A loss-of-function mutations. This suggests that the human and the fly proteins perform similar functions and that the pathogenic allele retains crucial properties of the normal protein. One of the most common features of VAP homologues is their ability to self-oligomerize (11,23,24). To test whether the fly and the human VAPs exhibit this ability, we employed the yeast two-hybrid system. We found that DVAP-33A as well as its human counterpart can form homodimers, supporting the data that the human protein and the fly protein are functionally interchangeable (Supplementary Material, Fig. S3). We also found that the human and the Drosophila proteins carrying the ALS8 mutation can self-oligomerize, supporting the evidence that both mutant proteins retain at least part of the functional properties of the wt protein (Supplementary Material, Fig. S3). Surprisingly, we observed that, although both the human and Drosophila protein can interact with their respective mutant protein, there was no significant interaction between proteins from different species (e.g. the human mutant protein does not interact with the wt Drosophila protein although the same protein strongly interacts with the human wt protein, Supplementary Material, Fig. S3B and C). This observation suggests that the best way to model the human disease inherited in a dominant manner is to use the Drosophila system where the Drosophila mutant protein will be expressed in the presence of the fly wt protein.
One of the earliest and most common symptoms of MND in humans is paralysis and impaired movements. Similarly, we found that transgenic larvae expressing DVAPP58S in neurons were sluggish and uncoordinated. We therefore performed a quantitative analysis of their locomotion behavior. Forward locomotion in larvae consists of contractions of the posterior end alternating with extension of the anterior body regions (25,26). Third instar larvae expressing transgenic DVAPP58S and control larvae reared in the same environmental conditions were observed on an agarose substrate for a period of at least 2 min. The frequency of strides in Hz (number of events per second) was calculated by dividing the number of strides by their duration.
Unexpectedly, transgenic larvae expressing DVAPP58S revealed significant heterogeneity in the mobility phenotype compared with controls. As shown in Figure 7A, 34% of the larvae are completely paralyzed or exhibit very few peristaltic contractions while the majority has a frequency of strides that is only 30% of the wt value. In controls, the frequency of strides is homogeneous (0.85 ± 0.01 Hz). The difference in frequency distribution between mutants and controls is statistically significant (P < 0.001, see legend of Fig. 7 and Materials and Methods for details).
Figure 7.
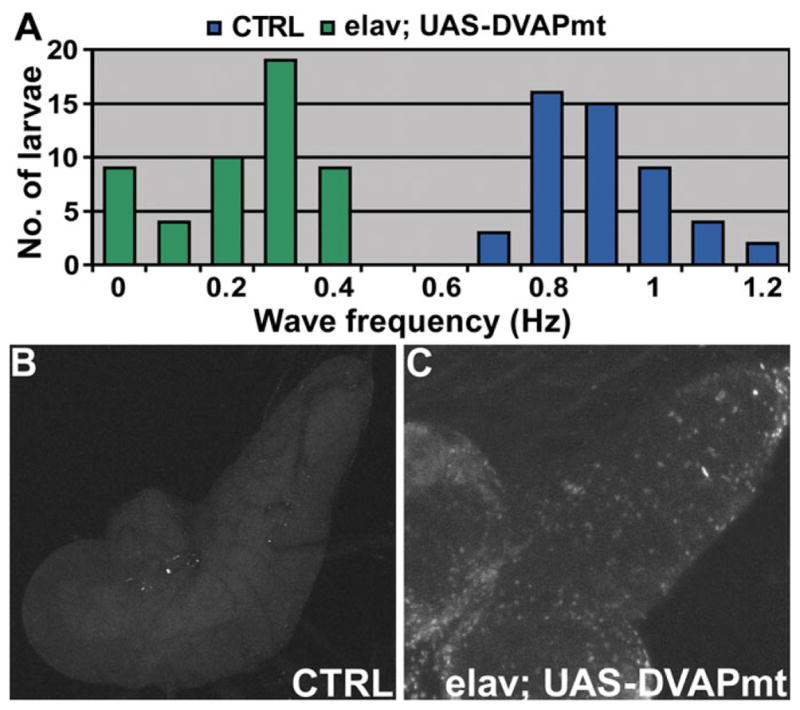
Transgenic expression of DVAPP58S in neurons induces locomotion defects and neuronal cell death. (A) Summary of the frequency of peristaltic waves for elav; UAS-DVAPmt larvae (green) and for controls (blue). n = 49 for controls and n = 51 for elav; UAS-DVAPmt. Differences between genotypes were highly significant (P < 0.001, accordingly to the non-parametric Mann–Whitney U test when the data sets relative to elav; UAS-DVAPmt were compared with controls. In this experiment, the UAS-DVAPP58S transgenic line without the driver was used as a control. (B) Neuronal cell death in UAS-DVAPP58S/+ control brains. (C) Neuronal cell death within larval brains expressing transgenic DVAPP58S.
Motor neuron death is one of the hallmarks of MND both in human patients and murine models (2). We therefore performed TUNEL analysis to assess whether transgenic expression of DVAPP58S causes neuronal apoptosis. Significantly enhanced neuronal death was observed in the central neurons of larvae expressing transgenic DVAPP58S compared with controls (Fig. 7B and C).
In mouse models for ALS expressing pathogenic SOD1, the paralytic phenotype and the neuronal cell death have been associated with the formation of aggregates that are strongly immunoreactive with SOD1 antibodies (27). Similarly, aggregates containing pathogenic SOD1 have been reported for sporadic and familial cases of ALS in humans (28). To test whether this feature is also common to VAP-induced MNDs, we performed Western analysis on whole tissue extracts of third instar larvae. Although DVAP-33A migrates exclusively as a monomer in SDS-PAGE of controls, in DVAPP58S transgenic lines an immunoreactive smear with a significant reduced mobility was observed (data not shown). The presence of these higher molecular weight species is suggestive of the ability of the protein to form aggregates. To identify where these aggregates accumulate and whether there are regional differences in the localization of the endogenous protein, we performed confocal analysis on larval brains, nerve fibers and neuromuscular synapses of DVAPP58S transgenic animals stained with antibodies specific for DVAP-33A. In control nerves, we observed faint but uniform staining while in the nerves of DVAPP58S transgenic larvae, highly immunoreactive aggregates of variable sizes were found (Fig. 8A and B). Large aggregates accumulate in the region of the nerves proximal to the brain and in their terminal tracts just before motor nerves sprout on the muscles to form the synaptic arbor (Fig. 8B). In between these regions, the deposition of aggregates was less prominent (Fig. 8C). In neuronal cell bodies of DVAPP58S larval brains, we also observed aggregate formation and the wt protein associated with the plasma membrane decreased to nearly undetectable levels (compare Figs 8E with D). Similarly, at neuromuscular synapses of DVAPP58S transgenic larvae, DVAP-33A positive immunoreactivity was virtually undetectable (less than 8% of the wt level, Figs 9A–C and G–J). This phenomenon was consistently observed in all five DVAPP58S transgenic lines examined. Conversely, in DVAP-33A overexpressing lines the protein is correctly targeted to the NMJ even when protein levels are 4-fold the wt level (Fig. 9A–F and J). No aggregates were found in the neuronal cell bodies and nerve fibers of DVAP-33A overexpressing lines (data not shown and compare arrow in Fig. 9E with arrow in Fig. 9H). Taken together, these data indicate that transgenic expression of DVAPP58S in neurons induces the formation of DVAP-33A immunoreactive aggregates and a depletion of the endogenous protein from its normal localization. At the Drosophila NMJ, decreasing the level of DVAP-33A induces a decrease in the number of boutons and an increase in their size (Supplementary Material, Fig. S1). As a consequence of the decreased synaptic level of DVAP-33A, the number of boutons at the DVAPP58S transgenic synapses was only 40% of the wt number (122 ± 3 as compared with 283 ± 12 in controls, P > 0.001, data not shown, but compare Fig. 9A with G). Although not directly quantified, an increase in bouton size was also observed (data not shown). A number of other abnormalities such as muscle wasting and synaptic degeneration were often observed at these NMJs. A detailed analysis of these phenotypes is reported in Supplementary Material, Fig. S4. Taken together these data indicate that neuronal expression of DVAPP58S in the presence of the wt protein recapitulates several hallmarks of the human disease including locomotion defects, neuronal cell death and aggregate formation. It is noteworthy that aggregate accumulation is associated with a depletion of the endogenous protein from its normal localization and a consequent decrease in its function.
Figure 8.

Aggregates strongly immunoreactive for DVAP-33A are markers of the disease in flies. Nerve fibers and brains of third instar larvae were stained with antibodies for DVAP-33A (red) and with antibodies for the neuronal cell surface marker anti-HRP (green). (A) Nerve fibers of control larvae. (B and C) Nerve fibers of larvae expressing transgenic DVAPP58S (elav; UAS-DVAPmt). In control nerves, a faint but uniform staining is observed (A). Conversely, in DVAPP58S transgenic nerves, large aggregates accumulate in the region of the nerves proximal to the brain and in their terminal tracts just before motor nerves contact the muscles (B). In between these regions, the deposition of aggregates was less prominent (C). (D) Brains of control larvae stained with anti-HRP (green) and anti DVAP-33A antibodies (red). (E) Brains of larvae expressing transgenic DVAPP58S (elav; UAS-DVAPmt) using the same antibodies. DVAP-33A associates mainly with the plasma membrane of neuronal cell bodies (D). On the contrary, in DVAPP58S transgenic brains, the DVAP-33A immunoreactivity is associated with intracellular aggregates of variable sizes (E). The anti DVAP-33A antibodies used in this report do not discriminate between the wt and the mutant protein. By western analysis, these antibodies recognize a band of similar size to the wt protein in protein extracts from NMJs expressing DVAPP58S in a null background for the endogenous protein (data not shown). In (D) and (E), single sections of confocal images are shown. Canton S larvae were used as controls in the experiments reported in this figure. Scale bars = 20 μm.
Figure 9.

Transgenic expression of DVAPP58S affects synaptic levels of the endogenous protein. NMJs were stained with antibodies specific for DVAP-33A (red, DVAP) and for anti-HRP (green) to visualize the synapses. (A–C) Control NMJs. (D–F) NMJs overexpressing DVAP-33A (elav; UAS-DVAP). (G–I) NMJs expressing transgenic DVAPP58S (elav; UAS-DVAPmt). (J) Quantification of synaptic DVAP-33A fluorescence intensity for the reported genotypes. Note that despite a 4× increase in the level of expression of DVAP-33A, the protein is located at the synapse and no aggregates are visible in the nerves of these transgenic lines expressing the wt protein (arrow in E). In DVAPP58S transgenic lines, aggregates are evident in the terminal part of the nerve (arrow in H) and the endogenous protein at the synapse is nearly undetectable (H and J). Canton S larvae were used as controls in the experiment reported in this figure. In (J) fluorescence intensity is presented in arbitrary units. Scale bar = 10 μm.
DISCUSSION
Recently, hVAPB has been shown to be the causative gene of late-onset autosomal dominant forms of motor neuron disorders, including typical and atypical ALS and late-onset spinal muscular atrophy (5, 6). The pathogenic mutation predicts a substitution of a Serine for a conserved Proline (P56). We decided to study the role of hVAPB in MNDs using Drosophila genetics.
One of the hallmarks associated with loss-of-function and neuronal overexpression of DVAP-33A is decreased and increased bouton formation at the NMJ, respectively. Despite this structural alteration, synaptic transmission is maintained within a wt range. At the mechanistic level, muscles respond to a decreased number of boutons and quantal content by upregulating quantal size; conversely muscles compensate an increase in number of boutons and quantal content by downregulating quantal size. Compensatory changes in quantal size during synaptic homeostasis are thought to be determined, largely, by the properties of transmitter receptors. At the Drosophila NMJ, there are two classes of glutamate receptors: one set containing the subunit IIA and another one containing the subunit IIB (29). In DVAP-33A loss-of-function mutations, the increase in quantal size is associated with an increase in the number and average cluster volume of subunit IIA. Conversely, the decrease in quantal size in the oversprouting mutants is accompanied by a decrease in the level of post-synaptic receptor subunit IIA and a reduction in the average cluster volume for several subunits. In agreement with our data, the IIA subunit receptors have been shown to affect quantal size and receptor channel open time (18,30). Similar to our oversprouting mutants, in synapses lacking the receptor subunit IIA, a homeostatic increase in neurotransmitter release compensates for the reduction in quantal size and the evoked response is maintained within normal values (18,30). These data indicate that expression levels of VAP proteins play a crucial role in synaptic homeostasis by coordinating structural remodeling and post-synaptic sensitivity to neurotransmitter to ensure synaptic efficacy.
Interestingly, expression of hVAPB in neurons rescues lethality, morphological and electrophysiological phenotypes associated with DVAP-33A loss-of-function mutations. Moreover, neuronal expression of hVAPB in a wt background induces phenotypes similar to the overexpression of DVAP-33A. These data clearly indicate that DVAP-33A and hVAPB perform homologous functions at the synapse and as a consequence, information gained by studying DVAP-33A is expected to be relevant for hVAPB function as well. Surprisingly, neuronal expression of mutant VAP proteins also rescues all phenotypes associated with mutations in DVAP-33A. Two alternative scenarios could be proposed to explain these data: the mutation is irrelevant for the ALS8 pathogenesis or the mutant allele has a pathogenic effect while retaining certain functional properties of the wt protein. We strongly favor the second hypothesis for the following reasons. First, the P56S mutation in hVAPB has been reported to be causative for an inherited form of MNDs in humans. This mutation affects nine related families totaling 1500 individuals of which 200 suffer from motor neuron disorders (31). Second, we have generated a genetic model for MNDs where the expression of the aberrant VAP recapitulates major hallmarks of the human disease, clearly indicating that the mutation has a pathogenic effect. Third, our data and data published by others (23) suggest that both the Drosophila and the human mutant proteins retain some functional wt properties such as the ability to self-oligomerize. However, neuronal expression of the pathogenic protein induces aggregate formation and depletes the wt protein from its normal localization. These effects are not observed when the wt protein is overexpressed, suggesting that the mutant protein has acquired a new, potentially toxic property.
Indeed, one of the most common features of MNDs and nearly all neurodegenerative diseases is the accumulation of aggregates that are intensively immunoreactive to disease-related proteins (2). Each disease, however, differs with respect to the anatomical location and morphology of the aggregates. The major component of the aggregates is usually the protein encoded by the gene mutated in the familial forms, which is also unique to each disease. Despite this diversity, a bulk of circumstantial evidence support the hypothesis that aggregates are typical hallmarks of neurodegenerative diseases and have a toxic effect on neurons (32). While no autopsy material is available for familial cases with the P56S mutation, SOD1-positive inclusions have been reported in human sporadic and familial ALS cases as well as in SOD1 mouse models (28). We found the presence of aggregates that are intensively immunoreactive for DVAP-33A both in neuronal cell bodies and in nerve fibers of our MND model. Interestingly, hVAPB carrying the pathogenic mutation has also been shown to undergo intracellular aggregation when expressed in a cell culture system (23). However, similarities between human disease and our fly model are not limited to aggregate formation as flies expressing transgenic VAP proteins carrying the ALS8 mutation, exhibit other hallmarks of the human disease such as neuronal cell death, muscle wasting and defective locomotion behavior.
Although it remains to be established whether the VAP protein in the aggregates represents the mutant protein, the endogenous protein or a mixture of both, we clearly observe a regional decrease in the level of the endogenous protein. The DVAP-33A protein that is normally associated with the plasma membrane in neuronal cell bodies and at the neuromuscular synapses is nearly undetectable in DVAPP58S transgenic animals. As a consequence of the decrease in synaptic levels of the endogenous protein, a decrease in the number of boutons is observed. We have previously shown that DVAP-33A regulates bouton formation at the synapse in a dosage-dependent manner (9). Despite these structural alterations a homeostatic mechanism is established to maintain synaptic efficacy within functional boundaries. We speculate that the depletion of the endogenous protein from its normal localization and the formation of aggregates would affect the homeostatic mechanism linking structural remodeling and synaptic efficacy controlled by DVAP-33A. Although not directly tested in our model, experiments in cell culture show that overexpression of mutant hVAPB induces formation of aggregates in which the endogenous wt protein is recruited (23,33). This would suggest that the pathogenic allele functions as a dominant negative. However, the depletion of the endogenous protein from its normal localization cannot be the principal mechanism of the disease as mutants lacking DVAP-33A do not develop MND. It is therefore possible that the pathogenic allele has acquired an abnormal, new toxic activity. Similar to what has been proposed for other neurodegenerative diseases, the formation of aggregates may directly interfere with critical cellular processes and/or compromise the ability of the system to keep up with the degradation of aggregated proteins (34).
Taken together these data offer experimental support to the hypothesis that VAP proteins play a conserved role in synaptic homeostasis and emphasize the relevance of this fly model in fostering our understanding of the molecular mechanisms underlying VAP-induced motor neuron degeneration in humans.
MATERIALS AND METHODS
Genetics and molecular techniques
DVAP-33AΔ166 is an hypomorphic mutation obtained by imprecise excision of P{ry+t7.2 = lArB}47, a P element inserted 600 bp upstream of the AUG (9). A revertant line generated by precise excision of the same P element was used as a control for loss-of-function mutations.
Site-directed mutagenesis on DVAP-33A and hVAPB cDNAs was performed using Quick Change Site Directed Mutagenesis Kit (Stratagene). All transgenic lines were established by following standard protocols (35). Basic molecular biology techniques were performed according to (36) and Western Blots on single dissected NMJ according to (9).
To test the ability of the hVAPB protein to rescue the lethality associated with DVAP-33A mutations, female flies, DVAP-33AΔ448/FM7; +/+; UAS-hVAPB/TM3, were mated to males contributing the C164-Gal4 or D42-Gal4 drivers. DVAP-33AΔ448/Y; C164-Gal4/+; UAS-hVAPB/+ adult, non-FM7, males were identified and counted. The specificity of the rescue was confirmed by the absence of DVAP-33AΔ448/Y; C164-Gal4/+; +/TM3 males. A similar protocol was used to test the ability of hVAPBP56S and DVAPP58S to rescue the lethality associated with DVAP-33A mutations. In all cases, the rescue was confirmed by using all DVAP-33A mutant alleles in combination with several transgenic lines expressing hVAPB, hVAPBP56S or DVAPP58S.
For the analysis of the morphological and physiological rescue, the following crosses were performed. yw/Y; C164-GAL4/C164-GAL4 males were crossed to DVAP-33AΔ448/FM7; +/+; UAS-hVAPB/UAS-hVAPB females. DVAP-33AΔ448/Y; C164-Gal4/+; UAS-hVAPB/+ males were identified as y+ third-instar larvae lacking the FM7 chromosome. A similar genetic scheme was applied to test the ability of Drosophila and human mutant proteins to rescue the morphological and physiological phenotypes. To characterize the transgenic expression phenotype, the Gal4 drivers were crossed with transgenic lines. Embryos were collected for 20–24 h and then transferred to a water-bath at 30°C.
Immunohistochemistry, imaging and morphometric analysis
Stainings of third instar larval NMJs and analysis of the morphological phenotype was performed as described in (9). For the phalloidin staining the NMJs were fixed in 4% paraformaldehyde and the phalloidin treatment was performed accordingly to the manufacturer’s instructions (Molecular Probes). NMJ stainings with antibodies specific for the glutamate receptor subunits were performed according to (20), except for the subunit IID where the protocol described in (21) was used. Larval NMJs were imaged using an Axiovert Zeiss Microscope. The same confocal gain settings were applied to control and mutant NMJs. A complete Z-stack was acquired for every NMJ and rendered on a 3D projection. For the morphometric analysis, images were initially trimmed using the Zeiss LSM Image Examiner 3.2.0.70 software (Carl Zeiss, 2002). Cluster counting and volume estimation were performed with the software package Imaris 4.7.2 (Bitplane AG, 2006). The minimum cluster radius was set to 0.4 μm and background object subtraction was used when applying the ‘spot detection’ function. The total cluster volume was found by fitting a 3D surface to the clusters with the iso-surface tool and no additional Gaussian smoothing or re-sampling steps were applied. The average volume of a single cluster was calculated by dividing the total cluster volume by the total number of clusters. Appropriate intensity thresholds were selected to properly identify clusters and ignore background intensities for both tools in the Imaris package. Statistical analysis was performed using a two-tailed Student’s t-test.
Electrophysiology
Intracellular recordings were performed in HL3 saline (37). Spontaneous mEJPs were obtained by intracellular recording from muscles bathed in HL3 saline containing low calcium concentration (0.3 mM) and 3 μM tetrodotoxin. For evoked transmitter release, 1 mM Ca2+ was added to the HL3 saline. Electrical signals were amplified through an Axoclamp 2B amplifier (Axon Instruments), digitized and recorded by a computer equipped with pClamp8 software (Axon Instruments). Quantal content was calculated by the method of dividing the size of the mean EJP with the size of the mean mEJP.
Analysis of the amplitude and frequency of mEJPs was performed using Mini Analysis (Synaptosoft, Inc.). mEJPs with slow time course arising from neighboring electrically coupled muscle cells were excluded from analysis (38). No significant differences were found in muscle resting potentials or muscle input resistance among different genotypes. The unpaired Student’s t-test was used for data statistics. Additionally, the Kolmogorov–Smirnov test was used when comparing quantal size analysis of different genotypes for the data reported in Figure 1 while one-way ANOVA (analysis of variance) was used for statistical analysis of different genotypes in the remaining figures.
Ultrastructural analysis
Body wall muscles were prepared for TEM as in (39). Synaptic boutons were serially sectioned and photographed at 10 000–30 000× using a JEOL 100S TEM. For morphometric analysis, the cross-section corresponding to the bouton midline (cross-section of largest diameter) was identified, the negative scanned at 60 000×, and used for quantification using NIH image (Version 1.62) as in (40). Number of active zones was determined by counting the number of T-bar structures (complete or partial) observed at the cross-sectional area. To measure the area of empty zones (devoid of synaptic vesicles) in the cross-sectional area of synaptic boutons, the boundary of synaptic vesicle pools was digitalized and the enclosed area was calculated by using the Measure function of NIH image. Serial sections taken from 11 synaptic boutons of two independent preparations for each genotype were used for EM analysis. Statistical analysis was performed using a two-tailed Student t-test.
Larval locomotion behavior
Wandering third instar larvae were collected from the vial and washed briefly in distilled water to remove traces of food. Each larva was transferred to the centre of a 9 cm Petri dish containing grape juice medium. The larva was then allowed to adjust to the Petri dish environment and the counting of the peristaltic waves was started only after observing the first wave of contractions. The contraction waves were counted for at least 2 min per larva and their number divided by the time in seconds to obtain the frequency of contractions expressed in Hz. The Lillefors test was applied to check for normality in the distribution of the different datasets. Since the data concerning the elav; UAS-DVAPmt failed to pass the test for normality, the non-parametric Mann–Whitney U test was used to compare the datasets.
Tunel staining for apoptosis detection
Larval brains were carefully dissected and fixed in 4% paraformaldehyde for 15 min. Detection of apoptotic neuronal cells was performed using the fluorescein cell death kit (Promega) following manufacturer’s instructions.
Supplementary Material
Acknowledgments
We thank H. Bellen, in whose laboratory this project was initiated, for helpful comments on a previous version of this manuscript. We are grateful to P. Brophy for his constant support and scientific advice. We thank C. O’Kane and R. Ribchester for their input and criticisms. We also thank A. DiAntonio and S. Sigrist for providing the glutamate receptor antibodies and T. Gillispie for the confocal analysis. The hVAPB cDNA was obtained from MRC, Gene Service, Cambridge (UK).
FUNDING
This research was supported by grants from the Wellcome Trust, the Scottish Motor Neuron Disease Association and the Royal Society to G.P., in part by a NIH grant (ES014441) to B.Z. and in part by a NIH grant (NS030072) to V.B. J.W. is supported by an EPSRC/MRC pre-doctoral fellowship at the School of Informatics and K.P. by a MRC pre-doctoral fellowship.
Footnotes
For Permissions, please email: journals.permissions@oxfordjournals.org
Supplementary Material is available at HMG Online.
Conflict of Interest statement. None declared.
References
- 1.Talbot K. Motor neurone disease. Postgrad Med J. 2002;78:513–519. doi: 10.1136/pmj.78.923.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 3.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 4.Nishimura AL, Mitne-Neto M, Silva HC, Oliveira JR, Vainzof M, Zatz M. A novel locus for late onset amyotrophic lateral sclerosis/motor neurone disease variant at 20q13. J Med Genet. 2004;41:315–320. doi: 10.1136/jmg.2003.013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marques VD, Barreira AA, Davis MB, Abou-Sleiman PM, Silva WA, Jr, Zago MA, Sobreira C, Fazan V, Marques W., Jr Expanding the phenotypes of the Pro56Ser VAPB mutation: proximal SMA with dysautonomia. Muscle Nerve. 2006;34:731–739. doi: 10.1002/mus.20657. [DOI] [PubMed] [Google Scholar]
- 7.Foster LJ, Weir ML, Lim DY, Liu Z, Trimble WS, Klip A. A functional role for VAP-33 in insulin-stimulated GLUT4 traffic. Traffic. 2000;1:512–521. doi: 10.1034/j.1600-0854.2000.010609.x. [DOI] [PubMed] [Google Scholar]
- 8.Kagiwada S, Hosaka K, Murata M, Nikawa J, Takatsuki A. The Saccharomyces cerevisiae SCS2 gene product, a homolog of a synaptobrevin-associated protein, is an integral membrane protein of the endoplasmic reticulum and is required for inositol metabolism. J Bacteriol. 1998;180:1700–1708. doi: 10.1128/jb.180.7.1700-1708.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pennetta G, Hiesinger PR, Fabian-Fine R, Meinertzhagen IA, Bellen HJ. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron. 2002;35:291–306. doi: 10.1016/s0896-6273(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 10.Skehel PA, Martin KC, Kandel ER, Bartsch D. A VAMP-binding protein from Aplysia required for neurotransmitter release. Science. 1995;269:1580–1583. doi: 10.1126/science.7667638. [DOI] [PubMed] [Google Scholar]
- 11.Soussan L, Burakov D, Daniels MP, Toister-Achituv M, Porat A, Yarden Y, Elazar Z. ERG30, a VAP-33-related protein, functions in protein transport mediated by COPI vesicles. J Cell Biol. 1999;146:301–311. doi: 10.1083/jcb.146.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuwabara PE. The multifaceted C. elegans major sperm protein: an ephrin signaling antagonist in oocyte maturation. Genes Dev. 2003;17:155–161. doi: 10.1101/gad.1061103. [DOI] [PubMed] [Google Scholar]
- 13.Roberts TM, Stewart M. Nematode sperm locomotion. Curr Opin Cell Biol. 1995;7:13–17. doi: 10.1016/0955-0674(95)80039-5. [DOI] [PubMed] [Google Scholar]
- 14.Kosinski M, McDonald K, Schwartz J, Yamamoto I, Greenstein D. C. elegans sperm bud vesicles to deliver a meiotic maturation signal to distant oocytes. Development. 2005;132:3357–3369. doi: 10.1242/dev.01916. [DOI] [PubMed] [Google Scholar]
- 15.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 16.Torroja L, Packard M, Gorczyca M, White K, Budnik V. The Drosophila beta-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci. 1999;19:7793–7803. doi: 10.1523/JNEUROSCI.19-18-07793.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elia AJ, Parkes TL, Kirby K, St George-Hyslop P, Boulianne GL, Phillips JP, Hilliker AJ. Expression of human FALS SOD in motorneurons of Drosophila. Free Radic Biol Med. 1999;26:1332–1338. doi: 10.1016/s0891-5849(98)00333-5. [DOI] [PubMed] [Google Scholar]
- 18.Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- 19.Schuster CM, Ultsch A, Schloss P, Cox JA, Schmitt B, Betz H. Molecular cloning of an invertebrate glutamate receptor subunit expressed in Drosophila muscle. Science. 1991;254:112–114. doi: 10.1126/science.1681587. [DOI] [PubMed] [Google Scholar]
- 20.Marrus SB, Portman SL, Allen MJ, Moffat KG, DiAntonio A. Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J Neurosci. 2004;24:1406–1415. doi: 10.1523/JNEUROSCI.1575-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin G, Schwarz T, Kittel RJ, Schmid A, Rasse TM, Kappei D, Ponimaskin E, Heckmann M, Sigrist SJ. Four different subunits are essential for expressing the synaptic glutamate receptor at neuromuscular junctions of Drosophila. J Neurosci. 2005;25:3209–3218. doi: 10.1523/JNEUROSCI.4194-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Featherstone DE, Rushton E, Rohrbough J, Liebl F, Karr J, Sheng Q, Rodesch CK, Broadie K. An essential Drosophila glutamate receptor subunit that functions in both central neuropil and neuromuscular junction. J Neurosci. 2005;25:3199–3208. doi: 10.1523/JNEUROSCI.4201-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanekura K, Nishimoto I, Aiso S, Matsuoka M. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8) J Biol Chem. 2006;281:30223–30233. doi: 10.1074/jbc.M605049200. [DOI] [PubMed] [Google Scholar]
- 24.Weir ML, Xie H, Klip A, Trimble WS. VAP-A binds promiscuously to both v- and tSNAREs. Biochem Biophys Res Commun. 2001;286:616–621. doi: 10.1006/bbrc.2001.5437. [DOI] [PubMed] [Google Scholar]
- 25.Fox LE, Soll DR, Wu CF. Coordination and modulation of locomotion pattern generators in Drosophila larvae: effects of altered biogenic amine levels by the tyramine beta hydroxlyase mutation. J Neurosci. 2006;26:1486–1498. doi: 10.1523/JNEUROSCI.4749-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang JW, Soll DR, Wu CF. Morphometric description of the wandering behavior in Drosophila larvae: a phenotypic analysis of K+ channel mutants. J Neurogenet. 2002;16:45–63. doi: 10.1080/01677060213106. [DOI] [PubMed] [Google Scholar]
- 27.Hart PJ. Pathogenic superoxide dismutase structure, folding, aggregation and turnover. Curr Opin Chem Biol. 2006;10:131–138. doi: 10.1016/j.cbpa.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 28.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 29.DiAntonio A. Glutamate receptors at the Drosophila neuromuscular junction. Int Rev Neurobiol. 2006;75:165–179. doi: 10.1016/S0074-7742(06)75008-5. [DOI] [PubMed] [Google Scholar]
- 30.DiAntonio A, Petersen SA, Heckmann M, Goodman CS. Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J Neurosci. 1999;19:3023–3032. doi: 10.1523/JNEUROSCI.19-08-03023.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishimura AL, Al-Chalabi A, Zatz M. A common founder for amyotrophic lateral sclerosis type 8 (ALS8) in the Brazilian population. Hum Genet. 2005;118:499–500. doi: 10.1007/s00439-005-0031-y. [DOI] [PubMed] [Google Scholar]
- 32.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 33.Teuling E, Ahmed S, Haasdijk E, Demmers J, Steinmetz MO, Akhmanova A, Jaarsma D, Hoogenraad CC. Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J Neurosci. 2007;27:9801–9815. doi: 10.1523/JNEUROSCI.2661-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao J, Diamond MI. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet. 2007;16:R115–R123. doi: 10.1093/hmg/ddm213. [DOI] [PubMed] [Google Scholar]
- 35.Spradling AC, Rubin GM. Transposition of cloned P elements into Drosophila germ line chromosomes. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- 36.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith AJ, Struhl K. Current Protocols in Molecular Biology. John Wiley & Son; New York: 1998. [Google Scholar]
- 37.Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol [A] 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- 38.Zhang B, Koh YH, Beckstead RB, Budnik V, Ganetzky B, Bellen HJ. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron. 1998;21:1465–1475. doi: 10.1016/s0896-6273(00)80664-9. [DOI] [PubMed] [Google Scholar]
- 39.Koh YH, Popova E, Thomas U, Griffith LC, Budnik V. Regulation of DLG localization at synapses by CaMKII-dependent phosphorylation. Cell. 1999;98:353–363. doi: 10.1016/s0092-8674(00)81964-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Budnik V, Koh YH, Guan B, Hartmann B, Hough C, Woods D, Gorczyca M. Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron. 1996;17:627–640. doi: 10.1016/s0896-6273(00)80196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.