

Abstract
Similar to human CLL, the de novo NZB mouse model has a genetically determined age-associated increase in malignant B-1 clones and decreased expression of microRNAs miR-15a and miR-16 in B-1 cells. In the present study, lentiviral vectors were employed in vivo to restore miR-15a/16, and both the short-term single injection and long-term multiple injection effects of this delivery were observed in NZB. Control lentivirus without the mir-15a/16 sequence was used for comparison. We found that in vivo lentiviral delivery of mir-15a/16 increased miR-15a/16 expression in cells that were transduced (detected by GFP expression) and sera when compared to control lentivirus treatment. More importantly, mice treated with the miR- expressing lentivirus had decreased disease. The lentivirus had little systemic toxicity while preferentially targeting B-1 cells. Short-term effects on B-1 cells were direct effects and only malignant B-1 cells transduced with miR-15a/16 lentivirus had decreased viability. In contrast, long-term studies suggested both direct and indirect effects resulting from miR-15a/16 lentivirus treatment. A decrease in B-1 cells was found in both the transduced and non-transduced populations. Our data support the potential use of systemic lentiviral delivery of miR-15a/16 to ameliorate disease manifestations of CLL.
Keywords: chronic lymphocytic leukemia, lentivirus, microRNA, mouse models
Introduction
Chronic lymphocytic leukemia (CLL), the most common leukemia to affect adults in the Western world, is an age-associated malignancy characterized by the expansion of CD5+ B-1 cells (1). Over 50% of CLL patients have a deletion within the 13q14 chromosomal region (2) containing the DLEU2 gene (3, 4), a non-coding RNA which contains the microRNA locus for mir-15a/16-1 within an intronic region (5). MicroRNAs (miRNAs) are small, evolutionarily conserved, non-coding single-stranded RNAs that regulate gene expression by binding with an RNA-Induced Silencing Complex (RISC complex) to the 3′ untranslated region (UTR) of target mRNA (6–8). Mutations and alterations, resulting in the loss or amplification of miRNAs affect the regulation of cell cycle and survival mechanisms, and have been linked to many human cancers and leukemias (5, 9–12). Many miRNAs were found to be located within genomic ‘fragile sites’ associated with malignant transformation such as regions of amplification, deletion, loss of heterozygosity, and breakpoint regions near oncogenes and tumor suppressor genes. (13, 14). miRNAs mir-15a and mir-16-1 are located in the frequently deleted 13q14 region, and are also associated with decreased levels of mature miR-15a and miR-16 in a subpopulation of patients with B cell chronic lymphocytic leukemia (B-CLL) (15, 16).
The New Zealand Black (NZB) mouse, in contrast to all other available CLL murine models, is a de novo model for both autoimmunity (17) and CLL (18, 19). Similar to CLL, the NZB develop an age-associated expansion of polyreactive, CD5 expressing, malignant B-1 cells, with clones often possessing chromosomal abnormalities resulting in aneuploidy (18–21). At 9 mo of age, all NZB mice have expanded B-1 populations, however, approximately 10% of the NZB mice that live beyond 17 months of age develop T cell clones with elevated IFN gamma production leading to an eventual decrease in B-1 cells at 17 months of age (22). NZB mice also exhibit a T -> A germline point mutation 6 bases downstream from pre-mir-16-1 on chromosome 14 (23), similar to the C -> T point mutation reported in human CLL (24), as well as decreased miR-15a and miR-16 expression.
We have previously reported the exogenous addition of miR-15a/16 in vitro to an NZB-derived malignant B-1 cell line to lead to a significant accumulation of cells in G1 and decrease in cyclin D1 protein levels (25). In this report, mir-15a/16-1 was systemically delivered to NZB mice with CLL via in vivo lentiviral delivery of a vector expressing both GFP and the wild-type mir-15a/16-1 sequence (mir-15a/16). We proposed that restoration of miR-15a/16 to malignant B cells in vivo would have similar effects as in vitro, particularly growth arrest and eventual death, resulting in disease reduction. Because transduced malignant B-1 cells were found to secrete the exogenously-delivered microRNA into the circulation, a subpopulation of lentivirus-injected NZB mice were re-injected at Day 24 [a time approaching the half-life of lymphoyctes (26)] and analyzed 4 days later to increase the likelihood of finding viable GFP+ cells. The peritoneum, spleen, blood, and liver of treated mice were evaluated 8–9 days (single injection, short-term) and 28–29 days (two injections, long-term) post-injection for the presence of malignant B-1 clones, the extent of organ involvement, and toxicity. Lentiviral delivery of mir-15a/16-1 to NZB mice resulted in a reduction of malignant B-1 cells and decreased splenic and hepatic involvement. Our data support the potential use of systemic lentiviral delivery of miR-15a/16 to ameliorate disease manifestations of CLL.
Methods
Mice
NZB/BlNJ, C57Bl/6J, and DBA/2J were purchased from Jackson Laboratory (Bar Harbor, ME) and housed under standard pathogen-free conditions at the research animal facility at UMDNJ – New Jersey Medical School, Newark, NJ. All non-NZB strains were used as control strains that do not develop CLL disease.
In vivo lentiviral delivery of miR-15a/16
Aged NZB mice (9–17mo) with disease were injected with lentivirus containing either a control GFP-expressing vector (control-lenti,) or a GFP and mir-15a/16-1 expressing vector (mir-lenti,). Four separate in vivo experiments were performed. For the first two experiments (terminated on Day 8–9 post single injection and Day 28 post two injections) the lentiviruses containing the mir-15a/16 loci and GFP or solely GFP were generated as previously described (27). The lentivirus was introduced iv for a systemic effect and ip for a local effect since the target B-1 cells in NZB mice are located in the blood, peritoneum and spleen. NZB mice were injected (ip.) with 100ul and i (i.p.) with 100ul lentivirus solution, containing 1×108 TU in media, 4ug/ml polybrene, and 1× PBS. Mice were either sacrificed at 8 and 9 days post-injection (control n=5, mir-15a/16, n=8), or mice were re-injected i.p. on day 24, and sacrificed at 28 and 29 days post-initial-injection (control n=4, mir-15a/16, n=4). In the third repeat experiment, NZB mice (9mo) were injected with lentiviral constructs containing the microRNAs miR15a/16 and GFP obtained from Systems Biosciences (SBI, Mountain View, CA, USA) and the packaging vectors were obtained from Addgene (Cambridge, MA, USA)). Mice were injected ip with 100ul and iv with 100ul lentivirus solution, containing 5×107 TU in media, 4ug/ml polybrene, and 1× PBS. Mice were sacrificed at 8 days post-injection (control-lenti n=3, mir-15a/16, n=3). The fourth repeat experiment was performed with the same SBI lentiviruses injection sites, doses and duration as for the third experiment with the exception that the NZB mice were all 12 months of age (control-lenti n=4, mir-15a/16, n=4). Mice were terminally bled prior to euthanasia and animal and spleen weight determined. For sera levels of microRNAs, EDTA was employed as an anticoagulant.
Detection of B-1 cells via flow cytometry
For identification of malignant B-1 cells, single-cell suspensions were made from spleen, peritoneal wash cells (PWC), and red blood cell lysed peripheral blood (PB) and surface stained with anti-mouse IgM allophycocyanin (APC, Caltag Invitrogen) and anti-mouse CD5 phycoerythrin (PE, Caltag, Invitrogen) and in some cases anti-CD5-APC, anti-B220-PE-Cy7 (RA36B2 clone) both from Becton Dickinson. Twenty-thousand events were acquired on a FACSCalibur and data was analyzed using CELLQUEST software (Becton Dickinson).
Analysis of DNA content
Cells from spleens, PWC, and PB were stained with hypotonic PI (0.05mg/ml PI, 0.1% Triton X-100, 0.1% sodium citrate) at time of sacrifice and acquired on Becton Dickinson FACSCalibur using CELLQUEST software (Becton Dickinson) and analyzed using ModFit LT V3.1 software (Verity Software House).
Cell sorting
Cells from spleens of NZB injected with either control-GFP lentivirus (control) or miR-15a/16-GFP-expressing lentivirus (mir-15a/16) were stained with IgM PE. Cells were then sorted on a FACSVantage (Becton Dickinson) on the basis of their GFP and IgM expression into GFP+/IgM+ and GFP−/IgM+ populations. In additional sorts, cells were stained with anti-CD5-PE and anti-B220-PE/Cy5 and sorted into transduced and non-transduced B-1 or B-2 cells (GFP+/ B220dull/CD5dull = transduced B-1, GFP−/ B220dull/CD5dull = non-transduced B-1, GFP+/ B220+/CD5− = transduced B-2, GFP−/ B220+/CD5− = non-transduced B-2)
MicroRNA extraction and quantification
Total RNA, including miRNA, was extracted from sorted splenic cells according to the Trizol (Invitrogen) manufacturer’s protocol. Quantitative real time PCR (qPCR) was used to quantitate mature miR-16 expression in sorted cells using the TaqMan microRNA Reverse Transcription and TaqMan miRNA hsa-miR-16 Assay Kit (Applied Biosystems, Foster City, CA). The qPCR reaction was run on the Applied Biosystems 7500 Real-Time PCR Systems for 40 cycles at 60°C. The miR-16 relative quantification (RQ) values of GFP+ cells from control-treated NZB compared to miR-16 from cells of miR-16-treated NZB were determined using the standard 2−ΔΔ CT method according to the manufacturer’s protocol. The total amount of input RNA was normalized to Taqman U6 snRNA (Applied Biosystems). Circulating levels of microRNA were performed by analysis of 20 μl of plasma (EDTA) from individual NZB mice 9 mo of age (control-lenti or mIR-15a/16 groups, n=3) obtained 8 days post in vivo lentivirus injection. Plasma was diluted to a final volume of 200 ul in RNase-free water and TRIzol® LS Reagent (Life Technologies) RNA isolation method was followed as per manufacturer’s instructions. The final RNA pellet was resuspended in 20 μl of RNase-free water and subsequently reverse transcribed (TaqMan Universal PCR Master Mix No AmpErase®UNG, Applied Biosystems), followed by TaqMan®MicroRNA quantitative RTPCR for miR15a and miR30c (endogenous control). Individual RQ values were generated in triplicate and then the group average RQ determined and graphed.
Histopathology
Formalin-fixed tissue sections of liver and spleen from control and TW-mir-15a/16 NZB mice were stained with hematoxylin and eosin and analyzed using an Olympus BX40 microscope with 10x, 20x and 40x objective lenses. Digital images were taken.
Liver chemistry
Sera and plasma collected in heparinized tubes were analyzed by Ani Lytics Inc. (Gaithersburg, MD) for aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), bilirubin, and albumin levels. Untreated NZB (11–12 mo and 17mo) were used as noninjected controls.
Statistical analysis
All experiments performed were at least in triplicate to obtain standard deviations and to calculate the SEM. Student’s t test was used where appropriate to determine statistical significance, p ≤ 0.05.
Results
NZB as a de novo model for chronic lymphocytic leukemia
The New Zealand Black (NZB) mouse model is a de novo model of CLL (18). Similar to CLL, and in contrast to other normal strain mice, NZB mice develop a malignant expansion of hyperdiploid IgM+ CD5dull B220 dull CD11b dull B-1 clones with age (19, 21, 28). Spleens were isolated from NZB mice at different ages: young (1–6 months), mid-aged (6–10 months) and old (11–15 months), (at least 3 mice per group), and compared to normal strain mice. Splenic single cell suspensions were then analyzed for DNA content and for the presence of IgM+ CD5dull B220 dull B-1 cells. Flow cytometric analysis revealed a lack of hyperdiploidy and minimal presence of an IgM+ CD5dull B-1 population in the spleens of normal non-NZB strain mice (Figure 1A) and of young NZB mice (Figure 1B). However, NZB mice were shown to develop an expansion of hyperdiploid B-1 clones with age (IgM+ CD5dull B220 dull, only IgM and CD5 expression shown). The disease in most cases progresses from a pauciclonal state (see middle panel with two hyperdiploid peaks) to CLL with a single dominant clone characterized as IgM+CD5dull B220 dull (Figure 1B). Middle-aged NZB mice exhibit multiple malignant hyperdiploid clones, as seen in reported cases of Monoclonal B-cell Lymphocytosis (MBL) (29) (Figure 1B), demonstrating a precursor state to the development of CLL in old NZB. Aging NZB spleens had significantly higher levels of B-1 cells when compared to the spleen cells from age-matched non-NZB normal strain mice (Figure 1C). In addition to an expansion of hyperdiploid malignant B-1 clones, NZB mice also possess a germline point mutation in the mir-15a/16-1 locus on chromosome 14 (23), similar to the point mutation reported in a CLL patient (24). This mutation is correlated with a decrease in mature miR-16 levels in the spleen (23). Levels of miR-16 were measured in the spleens of NZB mice at the aforementioned ages and compared to age-matched non-NZB normal strain mice with wild-type mir-15a/16-1. At all ages, NZB are found to have a decrease in miR-16 levels when compared to age-matched non-NZB mice, with significantly lower levels in young and old NZB mice (Figure 1D). The miR-16 levels in NZB seem to be constant throughout their lifetime; however, levels in non-NZB strain mice seem to fluctuate throughout the aging process, although remaining higher than levels in the NZB (Figure 1D). Subsequent experiments were carried out using middle-aged and old NZB mice with disease.
Figure 1. Age-associated disease of the NZB de novo mouse model of CLL.

A, Top: Representative analysis of DNA content from old (15mo) disease-free C57Bl/6 (non-NZB) spleen. The peak represents diploid G1 (peaks shown are computer modeled). Bottom: Flow cytometric analysis of surface staining of non-NZB splenic cells (lymphoid gate) expressing IgM (x-axis) and CD5 (y-axis). B, Top: Representative analysis of DNA content from young (1–6mo) disease-free, middle-aged (6–10mo), and old (11–15mo) NZB spleens. Bottom: Flow cytometric analysis of surface staining of NZB splenic cells (from spleen samples above) for IgM and CD5 (arrows indicate location of malignant B-1 clones). C, Columns represent the percentage of IgM+ CD5dull cells (B-1 cells) in non-NZB (black) and NZB (white) spleens. (#) indicates statistical significance between percentage of B-1 cells in NZB and non-NZB. (t) indicates statistical significant difference among aging NZB. Error bars represent the SEM for 3 independent analyses. D, Columns represent the normalized RQ 2−ΔΔ CT value of miR-16 in spleens of non-NZB (black) and NZB (white) mice. (#) indicates significant difference in amount of miR-16 between non-NZB and NZB strains. In B–D, (*) indicates the ages at which NZB were treated for subsequent experiments. Statistical significance = p<0.05, student’s t test, two-tailed.
In vivo analysis of cells transduced with mir-15a/16 lentivirus have increased miR-16 production
Aged NZB mice with disease were injected with lentivirus containing either a control GFP-expressing vector (control-lenti) or a GFP and mir-15a/16-1 expressing vector (mir--lenti). Mice were sacrificed at 8–9 days (short-term) after the initial injection and analyzed for miR-16 levels, B-1 number, % of transduced cells and apoptosis. The post 8–9 timepoint for study was based on our observation that in the NZB B cell line, delivery of the lentivirus in vitro led to apoptosis in 3 days (25) and in vivo injection most likely would take longer for the lentivirus to circulate, enter the tissue, bind to the cell, integrate and express. Sera was analyzed for the levels of miR-16 by realtime PCR and sera obtained from NZB mice injected with lentivirus containing the mir-15a/16 loci had significantly elevated levels of miR-16 (Fig 2A) compared to sera from the control lentivirus injected NZB. Spleens from lentivirus injected NZB were analyzed for GFP and surface marker expression and sorted into both B-1 and B-2 transduced and non-transduced cells (representative sorts shown in Fig 2D control-lenti and Fig 2E mir-lenti mice). The sorted GFP+ B-1 cells from the spleens of miR-lenti treated mice had significantly elevated miR-16 levels when compared to the miR-16 levels in the GFP+ B-1 cells from the control-lenti treated group (Fig 2B). PWC were also analyzed for the presence of GFP+ cells, and similar to the spleen, the B-1 cells preferentially expressed GFP relative to B-2 cells or non-B cells (Supplemental Figure 1). Overall, there was a decrease in total B-1 percentages in the spleens of mice injected with miR-lentivirus compared to the B-1 percentage in control-lenti spleens (Fig 2C). Furthermore, analysis of the lenti-virus treated mice compared to uninjected NZB demonstrated a significant decrease in splenic B-1 cells only in the mir-15a/16-lenti treated group (Supplemental Fig 2). Percentages of total live cells (R1 gate, Figures 3A and 4A) were analyzed from mice injected with mir-15a/16 lentivirus compared to control-lenti. Live cells were then gated on GFP and further analyzed for IgM and CD5 expression (Figures 3B and 4B). (The majority of GFP+ cells were also IgM+, 93.7% ± 1.07 (short-term mice) and 97.7% ± 1.36 (long-term mice)). The percentage of live GFP+ B-1 (IgM+/CD5+/dull) in the spleen, PWC, and PB decreased in short-term NZB injected with mir-15a/16-lenti (Figure 3B, C,) when compared to control-lenti. Over a 40% decrease in GFP+ B-1 (IgM+/CD5+/dull) in the spleen and PWC was detected in long-term NZB injected with mir-15a/16-lenti (Figure 4C) when compared to NZB injected with control lentivirus. Unexpectedly, an increase in the percentage of GFP+ B-1 cells was seen in the PB (Figure 4C), although this may be due to a splenic flush.
Figure 2. miR-16 levels 8–9 days following in vivo lentivirus injections.
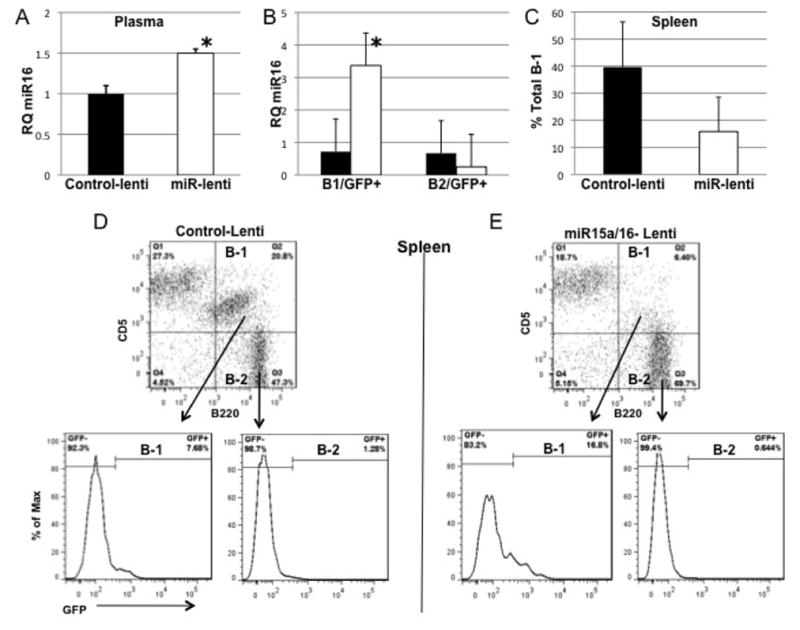
A. Effect of Lentiviral injection on miR-16 levels in the plasma of NZB mice (9 mo, n = 3 control-lenti, n=3 miR-lenti) 8 days post injection. miR-16 was determined by quantitative RT/PCR of RNA extracted from each sample using the standard 2−ΔΔCT method. miR-16 expression analysis in each sample is reported relative to the expression in a control-Lenti NZB (control set as 1). Black columns are control-lenti values and open columns are miR-lenti-values. Columns represent the mean and the bars are the SEM, * = statistical significance between miR-16 levels of NZB injected with mir15a/16-Lenti group compared with control-Lenti injected group (p < 0.01, student’s t test, two tailed test). B. Spleens from NZB treated either with mir-15a/16-lentivirus or control-lentivirus were stained with CD5, B220 and sorted into B-1(CD5dull, B220+) and B-2 (CD5−, B220+) and further sorted on the basis of GFP expression. Sorted cells were analyzed for the levels of miR-16 expression and columns represent the mean RQ values plus SEM. * = statistical significance between miR15a levels of GFP+ cells from NZB injected with miR15a/16-Lenti group compared with control-Lenti injected group (p < 0.05, student’s t test, two tailed test, n=3 for both NZB groups). C. Total B-1(CD5dull, B220+) cells in the spleen of short-term lenti-treated mice as a percentage of the total cells in the lymphoid gate. Columns are mean percent ± SD, n=3 for both groups. D and E. Representative sorting results (at least three individual spleens were sorted). The percent of cells in each population is indicated on the histograms.
Figure 3. Short-term: Flow analysis of the fate of B-1 cells.
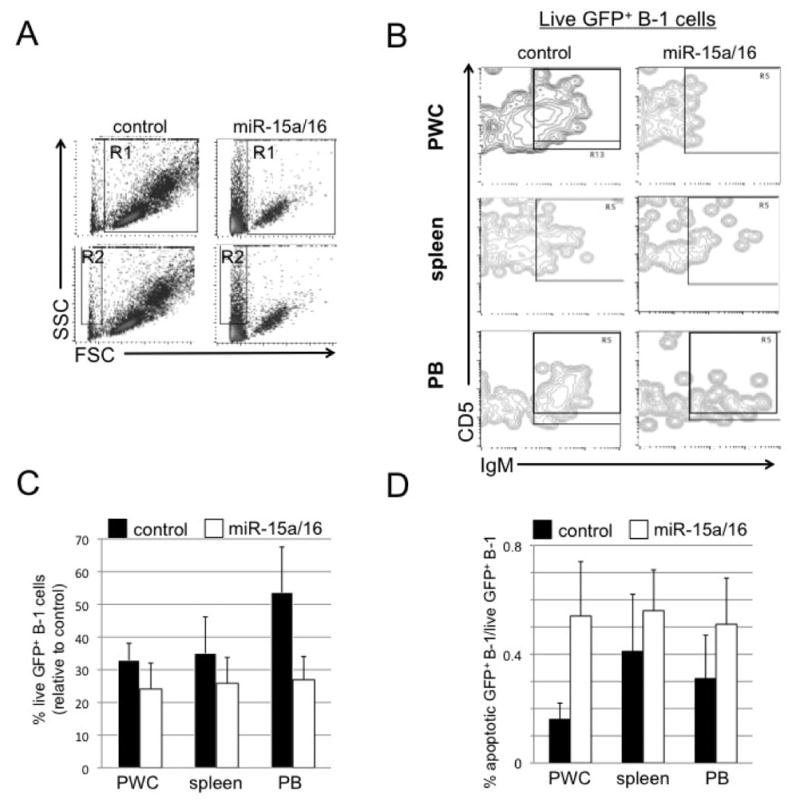
Spleen, PWC, and blood (PB) were obtained from NZB 8–9 days post-lentivirus treatment. A, Representative analysis of forward and side scatter (FSC and SSC) gating strategy. R1 contains live cells, R2 contains dying, apoptotic cells. Shown is PWC following treatment with control lentiviral vector (left) or miR-15a/16 (right) lentivirus. B, Representative flow analysis of live GFP+ cells (gated on R1 and GFP) that are analyzed for IgM and CD5 expression. The boxed area contain cells that are GFP+/IgM+/CD5+/dull C, Results from flow cytometric analysis of percentage of live cells (R1) gated on GFP and further analyzed for B-1 cells from miR-15a/16-injected NZB relative to NZB injected with control lentivirus. D, Flow cytometric analysis of the ratio of the percentage of apoptotic (R2) GFP+ B-1 cells to live GFP+ B-1 (R1) cells is shown. Black columns = NZB injected with control lentivirus and white = NZB injected with miR-15a/16-expressing lentivirus. Error bars represent SEM (control, n = 5, and miR-15a/16 n = 8).
Figure 4. Long-term: Flow analysis of the fate of B-1 cells.

A, Representative analysis of forward and side scatter (FSC and SSC) gating strategy. R1 contains live cells, R2 contains dying, apoptotic cells. Shown is PWC following treatment with control lentivirus (left) or miR-15a/16 (right) lentivirus. B, Representative flow analysis of live GFP+ cells (gated on R1 and GFP) that are analyzed for IgM and CD5 expression. The boxed area contain cells that are GFP+/IgM+/CD5+/dull C, Results from flow cytometric analysis of percentage of live cells (R1) gated on GFP and further analyzed for B-1 cells from miR-15a/16-injected NZB relative to NZB injected with control lentivirus. (*) indicates statistical significance (student’s t-test, p<0.05). D, Flow cytometric analysis of the ratio of the percentage of apoptotic (R2) GFP+ B-1 cells to live GFP+ B-1 (R1) cells is shown. Black columns = NZB injected with control lentivirus and white = NZB injected with miR-15a/16-expressing lentivirus. Error bars represent SEM (n = 4 for each group).
The percentage of GFP+ B-1 cells was also analyzed in the apoptotic gate (R2 gate Figures 3A and 4A). The percentage of apoptotic GFP+ B-1 cells was compared to the percentage of live GFP+ B-1 cells. The short-term NZB injected with mir-15a/16-lenti had a higher ratio of apoptotic to live GFP+ B-1 cells, particularly in the PWC (Figure 3D). This suggests that B-1 cells that picked up the miR-15a/16-expressing lentivirus underwent more apoptosis than in the same population in control-lenti treated NZB. The long-term NZB injected with mir-15a/16-lenti had a higher ratio of apoptotic to live GFP+ B-1 cells in the PWC and spleen (Figure 4D).
Genomic instability is a feature of CLL(30) and NZB mice demonstrate progressive aneuploidy in the B-1 clones as they age (31). A decrease in the overall presence of malignant aneuploid cells was detected from long-term NZB injected with mir-15a/16-lenti when compared to NZB treated with control empty vector (Supplement Fig 2).
Histopathological analysis of NZB treated with miR-15a/16 lentivirus indicates decreased disease
Histopathological analysis of short-term treated NZB mice found that three of the four control Tween-treated NZB exhibited bulk disease in the spleen, with malignant lymphoid cells (characterized by a dark, compact nucleus and scant cytoplasm) invading both red and white pulp, resulting in the total loss of splenic architecture. Four of five mice treated with TW-miR-15a/16 exhibited a decrease in splenic involvement. Less malignant lymphoid cells were present, and splenic architecture (red/white pulp and germinal centers) was more defined in the miR-15a/16 treated mice. Three of the four mice treated with control lentivirus exhibited liver involvement of endogenous disease, marked by large foci forming around the blood vessels in the liver. Four of the five mice treated with miR-15a/16 exhibited a decrease in liver involvement, with a marked decrease in foci by the blood vessels. Representative histology is shown in Figure 5.
Figure 5. Histopathology of NZB treated with lentivirus.
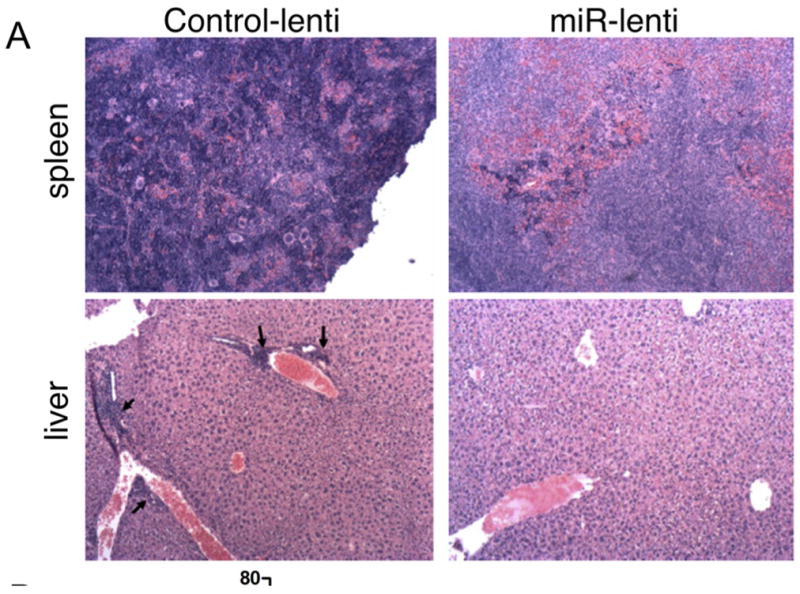
Representative H&E staining (200x magnification) of spleen (top) and liver (bottom) sections from short-term NZB treated with control vector (left) or miR-15a/16 (right) lentivirus. Top: The control spleen has bulk endogenous disease with malignant lymphocytes (dark purple cells) invading the red and white pulps, along with a total loss in splenic architecture. This is markedly decreased in the miR-15a/16 treated mice. Bottom: The control vector-treated liver shows disease involvement with large foci forming around the blood vessels (indicated by arrows). These foci are lost in the miR-15a/16 treated mice.
Lack of toxicity following lentiviral delivery of miR-15a/16
Animal weight and liver function were analyzed in lentivirus treated animals. The following functional liver enzymes were measured in the serum or plasma of lentiviral treated and untreated NZB: aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), bilirubin, and albumin. AST and ALT are involved in catabolizing amino acids, and ALP in bile production. Bilirubin is the resulting product of hemolysis and is excreted in bile, and albumin is a protein made by the liver. Abnormal levels of liver enzymes are indicative of improper liver function and damage. Injury to the liver can cause hepatocytes to release their enzymes into the bloodstream, thereby raising serum concentrations of AST and ALT, for example (32). NZB have naturally low levels of ALT, which remained consistent throughout treatment with either the control Tween lentiviral vector or miR-15a/16 vector (TW-mir-15a/16). AST levels remained within normal range throughout short-term treatment; however, AST levels elevated slightly above normal in long-term NZB treated with the mir-15a/16 lentiviral vector, though this increase was not significant. ALP levels were low after treatment with either control vector or mir15a/16-lenti (Figure 6A). Bilirubin levels were normal after treatment with either control- lenti or mir15a/16 lentiviral vector for both short-term and long-term mice. Likewise animal weight was similar in both lenti-treatment groups and not different from untreated NZB. Albumin levels were slightly lower than normal in the short-term control vector treated mice and in the long-term control vector and miR-15a/16 treated mice (Figure 6B).
Figure 6. Liver functional enzyme levels.
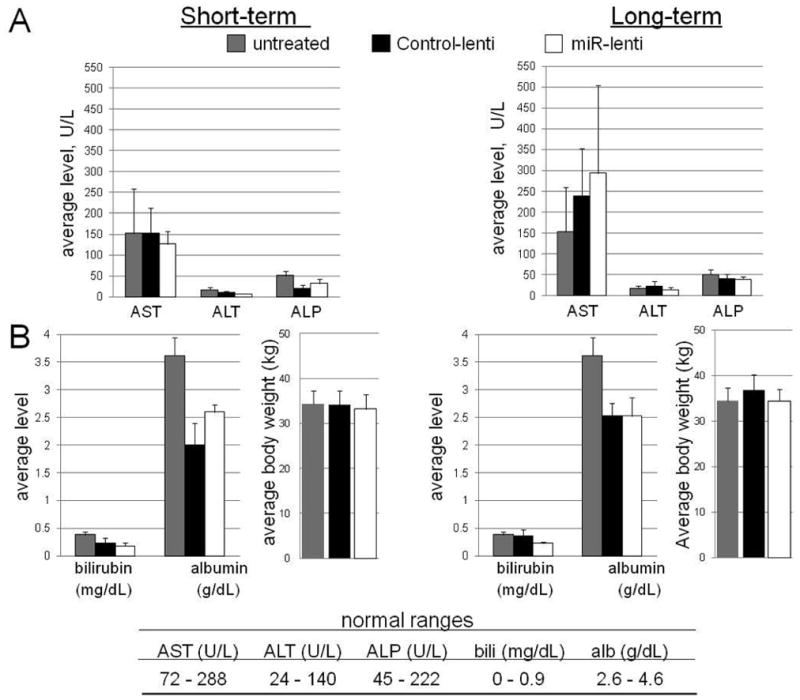
Untreated aged NZB (9–17mo) and NZB treated with control vector or miR-15a/16-expressing lentivirus were bled at 8–9 days (short-term) or 28–29 days (long-term) and either serum or plasma was evaluated for levels of liver enzymes aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), as well as bilirubin (bili), and albumin (alb). A, Average levels of AST, ALT, ALP in U/L. B, Average levels of bilirubin and albumin in mg/dL and g/dL, respectively and average animal weight(g). Gray columns are untreated NZB, black columns are NZB injected with control lentiviral vector, and white columns are NZB injected with miR-15a/16 lentiviral vector. Error bars represent the SEM for at least 4 different mice for each group. Normal ranges for each enzyme and protein is indicated in the table at the bottom.
Discussion
In this report we employed the NZB mouse model of CLL, which similar to human CLL, have an age-associated increase in malignant B-1 clones and decreased expression of miR-15a/16 in B-1 cells. We found that in vivo lentiviral delivery of miR-15a/16 significantly increased miR-15a/16 expression in cells that were transduced (GFP+) and significantly increased the serum levels of miR-16. More importantly, mice treated with the miR-expressing lentivirus had decreased disease. The lentivirus had little systemic toxicity while preferentially targeting B-1 cells as evidenced by decreased proliferation and increased apoptosis in these cells. Previously published reports have supported the therapeutic uses of miRNAs in cancers and other diseases using tumor cell lines and xenograft animal models. Non-viral and viral delivery methods of a deficient miRNA to cancer models and cell lines has been shown to reduce malignancy and tumor load in xenograft models (33–36). Reconstitution of miR-15a/16 expression also resulted in a reduction in prostate tumors engrafted into a mouse model for prostate cancer (27). These models restore miRNA levels in vitro to tumor cell lines that are then transplanted into immuno-compromised animal models or deliver virus directly into tumor mass, demonstrating the lack of ability of the miRNA-reconstituted malignant cells to form bulky tumors. Few have reported systemic delivery of miRNA, either in the form of synthetic mimics (37) or produced and delivered virally, and shown to be effective and well-tolerated (36, 38). In this report, we demonstrate the effects of systemically restoring miR-15a/16 in vivo to a naturally-diseased mouse model, the NZB mouse model of CLL, and that this restoration is also both effective and well-tolerated. Lentiviral vectors are advantageous in treatment of CLL in that they transduce non-dividing cells and hematopoietic cells (39, 40). Integrase-defective lentiviral vectors are viewed to be less hazardous in that they do not integrate into the host genome (41). These vectors can also be used in CLL therapy in that once miR-15a/16 is delivered to the deficient malignant cells, their proliferation is ceased or they undergo apoptosis and replication of the lentivirus is not needed. Aside from being used as diagnostic and prognostic markers (42), miRNAs also have potential therapeutic uses, as supported by our present data. Restoration of miR-15a/16 has also been shown to enhance drug sensitivity of malignant cells while having little to no effect on normal cells (25), further supporting the use of miR-15a/16 delivery as potential therapeutic use for CLL.
The most recently engineered mouse model of CLL demonstrates that a lack of mir-15a/16-1 leads to development of a B-1 cell proliferative disorder (43). Our data show that in vivo restoration of miR-15a/16, delivered via lentivirus, to the NZB model of CLL has a negative effect on malignant B-1 cells. Short-term (8–9 days post single lentivirus treatment) and long-term (28–29 days post initial lentivirus injection) effects of endogenous miR-15a/16 addition were evaluated in the NZB. The overall effect of lentiviral delivery of miR15a/16, was an increase in level of these microRNAs in the sera and a decrease in the percentage of B-1 cells. The variability in the efficiency of transduction and the inherent variability between mice diminishes the statistical significance between the control-lenti and miR-lenti treated groups. Despite this, repeat experiments also supported the conclusion that B-1 cell number is decreased in the miR-lenti treated group and the decrease in B-1 cells was statistically significant. Evaluation of long-term mice treated with mir-15a/16 lentivirus exhibited a decrease in aneuploid cells in the PWC, spleen and PB when compared to those injected with the control lentivirus. In order for miR-15a/16 restoration to result in a decrease in the total percentage of aneuploid malignant B-1 cells, longer than a week’s time is required and/or multiple injections of miRNA are needed. This report demonstrates the potential therapeutic value of restoring miR-15a/16 levels in CLL. However, these are initial studies and many unanswered questions remain. Formal pharmacokinetics need to be established and the effect of sustained elevation of miR-15a/16 levels in vivo over time in the NZB analyzed. The ultimate goal of miR-15a/16 restoration would be a significant increase in lifespan despite the initial presence of CLL. Although these present studies are preliminary, there is a preferential uptake of the lentivirus into B-1 cells.
The malignant cells in both the NZB and in human CLL are IgM+/CD5+/dull B-1 cells. The percentage of live B-1 cells was evaluated in the control and miR-15a/16 treated mice. The percentage of live B-1 cells that integrated the mir-15a/16 lentivirus (GFP+/IgM+/CD5+/dull) was decreased in both short and long term groups. Evaluation of GFP+ B-1 cells in short term treatment revealed a decrease of greater than 40% in the spleen, PWC and blood of mir-15a/16-lenti injected NZB compared to the percentage of live GFP+ B-1 cells in the control NZB.
Long-term effects of the lentivirus also revealed a decrease in GFP+ B-1 cells in the PWC and spleen. The spleen had only trace amounts of GFP+ apoptotic cells, suggesting that the immune system may have cleared the dead cells in the spleen prior to evaluation of the long-term mice. The PWC and spleen of mir-15a/16-lenti injected NZB had more dead GFP+ B-1 cells (a higher ratio in the percentage of apoptotic to live GFP+ B-1 cells), suggesting that elevation of miR-15a/16 specifically kills malignant B-1 cells. The blood, however, did not show a decrease, suggesting that perhaps the surviving malignant B-1 cells exited the spleen and entered the peripheral circulation.
The majority of cells that integrated the lentivirus are expressing IgM, suggesting that the lentivirus is preferentially incorporated by mature B cells. However, the effects of miR-15a/16 restoration leads to cell death preferentially in B-1 cells. The long-term treated mice exhibited a decrease in live B-1 cells regardless of whether the cells incorporated the miR-expressing lentivirus, suggesting an indirect effect on the remaining malignant B-1 cells. This indirect effect could be the secretion of miR-15a/16 either directly in the extracellular environment, via cell-cell interactions or via exosomes containing the miRNAs (44) to cells that did not incorporate the miR-15a/16-expressing lentivirus. Recent reports have shown that exosomes play important roles in intercellular communication. These cargo containing nano-vesicles (30–100 nm) are secreted by numerous cell types and proteomic studies have shown that they harbor an abundance of micro RNAs, mRNAs and proteins characteristic of their particular cellular origin (reviewed in (45)). Several reports have shown that secreted exosomes can act as important vehicles to horizontally transfer biological information to another cell (46, 47). This recently identified mechanism of exosome mediated cell-cell transfer has been shown to activate cell signaling mechanisms as well as exert immunomodulatory influences on surrounding cells (reviewed in (48)). Indeed, in CLL there is evidence for the increased presence of microvesicles in the serum when compared to control individuals(49). In this report the lenti-viral transduced NZB B cells may potentially release miR15a/16-1 enriched exosomes that are taken up by other cells. This in turn, could contribute to a more robust effect than what can be accounted for by miR15a/16-1 lenti-viral transduced cells only. Alternatively, the decrease in GFP- B-1 cells may be due to a decrease in supporting cells (perhaps Bregs, Tregs, or other B-1 cells) which have picked up the lentivirus and have reduced proliferation and/or production of growth factors (perhaps IL-10) which aid in the in vivo expansion of the malignant B-1 clone (See Fig 7). The finding of reduced growth of malignant B-1 cells in NZB mice treated in vivo with a lentivirus, which can integrate and lead to increased miR-15a/16 expression, is consistent with our reported in vitro results (25). This data suggests that lentiviral miR-15a/16 restoration does have a negative effect on the cells into which it integrates, particularly IgM+ B cells. Restoration induces apoptosis of malignant B-1 cells, and multiple injections may be needed to see a more pronounced effect and reduction in overall disease. In addition, lentiviral delivery of microRNAs may also result in indirect effects negatively impacting malignant cell survival by targeting cancer-supporting cells or by intercellular delivery of miRNAs to malignant cells.
Figure 7. Model of effects of miR-15a/16 restoration on malignant B-1 cells in vivo.
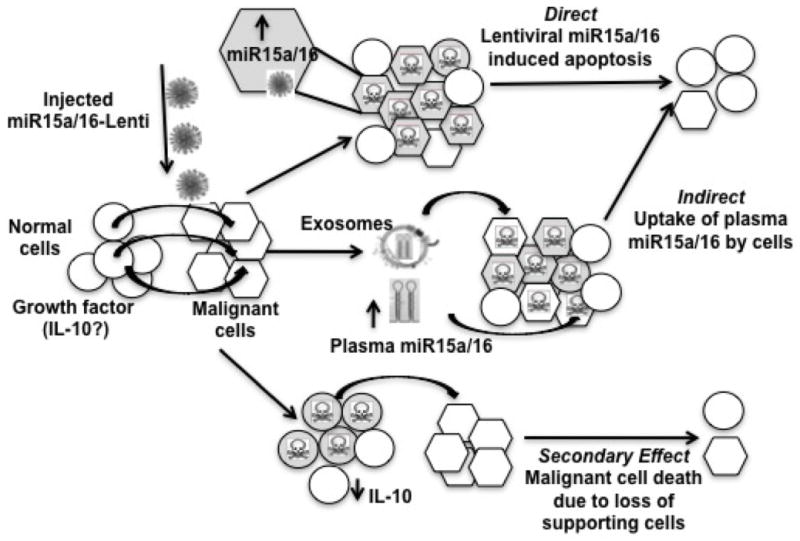
A, DIRECT EFFECTS: The lentiviral vector expressing the wild-type mir-15a/16-1 sequence integrated into a portion of B-1 cells resulting in increased miR-15a/16 expression and the direct death or decreased proliferation of the transduced B-1 cells due to loss of miR-15a/16 target gene expression (bcl-2, cyclin D1), minimal effect on other cell types. B, INDIRECT EFFECTS: Following mir-15a/16-lentivirus delivery, there was a decrease in both B-1 cells that did and did not integrate the miR-15a/16 lentiviral vector. This may be due to miR15a/16 secretion by the transduced malignant cells which can then be taken up by the non-transduced B-1 cells and subsequently mediate negative effects.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
FINANCIAL SUPPORT
This research was funded in part by NIH R01CA129826 (ER) and the NJ Commission on Cancer Research predoctoral fellowship 09-1255-CCR-E0 (ES).
We would like to thank the UMDNJ-NJMS Flow Cytometry Core.
References
- 1.Chiorazzi N, Rai K, Ferrarini M. Mechanisms of Disease: Chronic Lymphocytic Leukemia. New Eng J Med. 2005;352:804–15. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 2.Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–6. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 3.Bullrich F, Fujii H, Calin G, et al. Characterization of the 13q14 tumor suppressor locus in CLL: identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer Res. 2001;61:6640–8. [PubMed] [Google Scholar]
- 4.Liu Y, Corcoran M, Rasool O, et al. Cloning of two candidate tumor suppressor genes within a 10 kb region on chromosome 13q14, frequently deleted in chronic lymphocytic leukemia. Oncogene. 1997;15:2463–73. doi: 10.1038/sj.onc.1201643. [DOI] [PubMed] [Google Scholar]
- 5.Lerner M, Harada M, Loven J, et al. DLEU2, frequently deleted in malignancy, functions as a critical host gene of the cell cycle inhibitory microRNAs miR-15a and miR-16-1. Exp Cell Res. 2009;315:2941–52. doi: 10.1016/j.yexcr.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–85. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 8.Newman MA, Hammond SM. Emerging paradigms of regulated microRNA processing. Genes Dev. 2010;24:1086–92. doi: 10.1101/gad.1919710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calin GA, Garzon R, Cimmino A, Fabbri M, Croce CM. MicroRNAs and leukemias: how strong is the connection? Leuk Res. 2006;30:653–5. doi: 10.1016/j.leukres.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 10.Navarro F, Lieberman J. Small RNAs guide hematopoietic cell differentiation and function. J Immunol. 2010;184:5939–47. doi: 10.4049/jimmunol.0902567. [DOI] [PubMed] [Google Scholar]
- 11.Sevignani C, Calin GA, Siracusa LD, Croce CM. Mammalian microRNAs: a small world for fine-tuning gene expression. Mamm Genome. 2006;17:189–202. doi: 10.1007/s00335-005-0066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao H, Wang D, Du W, Gu D, Yang R. MicroRNA and leukemia: tiny molecule, great function. Crit Rev Oncol Hematol. 2010;74:149–55. doi: 10.1016/j.critrevonc.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Fabbri M, Garzon R, Andreeff M, Kantarjian HM, Garcia-Manero G, Calin GA. MicroRNAs and noncoding RNAs in hematological malignancies: molecular, clinical and therapeutic implications. Leukemia. 2008;22:1095–105. doi: 10.1038/leu.2008.30. [DOI] [PubMed] [Google Scholar]
- 14.Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicoloso MS, Kipps TJ, Croce CM, Calin GA. MicroRNAs in the pathogeny of chronic lymphocytic leukaemia. Br J Haematol. 2007;139:709–16. doi: 10.1111/j.1365-2141.2007.06868.x. [DOI] [PubMed] [Google Scholar]
- 17.Theofilopoulos AN. Genetics of systemic autoimmunity. Journal of Autoimmunity. 1996;9:207–10. doi: 10.1006/jaut.1996.0025. [DOI] [PubMed] [Google Scholar]
- 18.Phillips JA, Mehta K, Fernandez C, Raveche ES. The NZB mouse as a model for chronic lymphocytic leukemia. Cancer Res. 1992;52:437–43. [PubMed] [Google Scholar]
- 19.Scaglione BJ, Salerno E, Balan M, et al. Murine models of chronic lymphocytic leukaemia: role of microRNA-16 in the New Zealand Black mouse model. Br J Haematol. 2007;139:645–57. doi: 10.1111/j.1365-2141.2007.06851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foster MH. Relevance of systemic lupus erythematosus nephritis animal models to human disease. Semin Nephrol. 1999;19:12–24. [PubMed] [Google Scholar]
- 21.Dang AM, Phillips JA, Lin T, Raveche ES. Altered CD45 expression in malignant B-1 cells. Cell Immunol. 1996;169:196–207. doi: 10.1006/cimm.1996.0110. [DOI] [PubMed] [Google Scholar]
- 22.Raveche E, Fernandes H, Ong H, Peng B. Regulatory role of T cells in a murine model of lymphoproliferative disease. Cell Immunol. 1998;187:67–75. doi: 10.1006/cimm.1998.1319. [DOI] [PubMed] [Google Scholar]
- 23.Raveche ES, Salerno E, Scaglione BJ, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109:5079–86. doi: 10.1182/blood-2007-02-071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 25.Salerno E, Scaglione BJ, Coffman FD, et al. Correcting miR-15a/16 genetic defect in New Zealand Black mouse model of CLL enhances drug sensitivity. Mol Cancer Ther. 2009;8:2684–92. doi: 10.1158/1535-7163.MCT-09-0127. [DOI] [PubMed] [Google Scholar]
- 26.Young AJ, Hay JB. Rapid turnover of the recirculating lymphocyte pool in vivo. Int Immunol. 1995;7:1607–15. doi: 10.1093/intimm/7.10.1607. [DOI] [PubMed] [Google Scholar]
- 27.Bonci D, Coppola V, Musumeci M, et al. The miR-15a-miR-16–1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008 doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- 28.Caligaris-Cappio F, Ghia P. The normal counterpart to the chronic lymphocytic leukemia B cell. Best Pract Res Clin Haematol. 2007;20:385–97. doi: 10.1016/j.beha.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Lanasa MC, Allgood SD, Volkheimer AD, et al. Single-cell analysis reveals oligoclonality among ‘low-count’ monoclonal B-cell lymphocytosis. Leukemia. 2010;24:133–40. doi: 10.1038/leu.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin TT, Letsolo BT, Jones RE, et al. Telomere dysfunction and fusion during the progression of chronic lymphocytic leukemia: evidence for a telomere crisis. Blood. 116:1899–907. doi: 10.1182/blood-2010-02-272104. [DOI] [PubMed] [Google Scholar]
- 31.Salerno E, Yuan Y, Scaglione BJ, et al. The New Zealand black mouse as a model for the development and progression of chronic lymphocytic leukemia. Cytometry B Clin Cytom. 78(Suppl 1):S98–109. doi: 10.1002/cyto.b.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giannini EG, Testa R, Savarino V. Liver enzyme alteration: a guide for clinicians. CMAJ. 2005;172:367–79. doi: 10.1503/cmaj.1040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takamizawa J, Konishi H, Yanagisawa K, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–6. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 34.Calin GA, Cimmino A, Fabbri M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–71. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trang P, Medina PP, Wiggins JF, et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2010;29:1580–7. doi: 10.1038/onc.2009.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–17. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fumitaka Takeshita LP, Osaki Mitsuhiko, Takahashi Ryou-u, Yamamoto Yusuke, Kosaka Nobuyoshi, Kawamata Masaki, Kelnar Kevin, Bader Andreas G, Brown David, Ochiya Takahiro. Systemic Delivery of Synthetic MicroRNA-16 Inhibits the Growth of Metastatic Prostate Tumors via Downregulation of Multiple Cell-cycle. Genes Molecular Therapy. 2010;18:181–7. doi: 10.1038/mt.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiggins JF, Ruffino L, Kelnar K, et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010;70:5923–30. doi: 10.1158/0008-5472.CAN-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naldini L, Blomer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 40.VandenDriessche T, Thorrez L, Naldini L, et al. Lentiviral vectors containing the human immunodeficiency virus type-1 central polypurine tract can efficiently transduce nondividing hepatocytes and antigen-presenting cells in vivo. Blood. 2002;100:813–22. doi: 10.1182/blood.v100.3.813. [DOI] [PubMed] [Google Scholar]
- 41.Vargas J, Jr, Gusella GL, Najfeld V, Klotman ME, Cara A. Novel integrase-defective lentiviral episomal vectors for gene transfer. Hum Gene Ther. 2004;15:361–72. doi: 10.1089/104303404322959515. [DOI] [PubMed] [Google Scholar]
- 42.Fabbri M. miRNAs as molecular biomarkers of cancer. Expert Rev Mol Diagn. 2010;10:435–44. doi: 10.1586/erm.10.27. [DOI] [PubMed] [Google Scholar]
- 43.Klein U, Lia M, Crespo M, et al. The DLEU2/miR-15a/16–1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 44.Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A. 2010;107:6328–33. doi: 10.1073/pnas.0914843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simpson RJ, Jensen SS, Lim JW. Proteomic profiling of exosomes: current perspectives. Proteomics. 2008;8:4083–99. doi: 10.1002/pmic.200800109. [DOI] [PubMed] [Google Scholar]
- 46.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 47.Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2:282. doi: 10.1038/ncomms1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 49.Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood. 115:1755–64. doi: 10.1182/blood-2009-09-242719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.